Introduction
Standard therapy, CCRT (concomitant chemotherapy and
radiation therapy), is the most effective method and the only
curable therapy for glioblastoma multiforme (GBM) except surgery
(1,2). This method is composed of radiation
and temozolomide (TMZ) treatment for GBM patients. Radiation has
been an effective therapeutic method for various cancer types for a
long time; however, repeated radiation treatment often induces
radioresistance (3,4). Although TMZ is the only active
medicine available for GBM patients, it is not beneficial for every
patient. TMZ response varies in each patient because of MGMT
[O-(6)-methylguanine-DNA
methyltransferase] enzyme expression, which induces DNA repairment
of the damaged DNA after TMZ treatment. Methylation status of MGMT
promoter is important in enzyme expression (5). Patients with unmethylated MGMT
promoter show resistance to TMZ treatment and long-term treatment
also induces resistance to TMZ as seen in radioresistance (6,7). To
overcome such obstacle, many groups have studied radiation/TMZ
resistance mechanism and developed substitute treatment methods
(8–12).
shRNA (short hairpin RNA) library screening method
for targeting genome has been used for discovering new therapeutic
targets or tumor associated genes due to the shRNA ability to
induce deprived gene functions (13,14).
ShRNA induction experiments are conducted to identify specific
genes that are associated with drug treatment, metastasis, and
transcriptional activity. Several tumor suppressor genes have been
discovered by using such screening method (14–16).
In this study, we discovered the radiation/TMZ resistance related
gene DDX6, using in vivo shRNA screening and demonstrated
that suppression of DDX6 induces resistance to radiation/TMZ
treatment. DDX6 is a RNA helicase and regulates mRNA translation
and storage in P-bodies. Recently, Chen et al reported CNOT1
(CCR4-NOT complex 1) interacts with DDX6 for mRNA decapping in
human cells and CNOT1 complex regulates mRNA translation in breast
cancer and acute lymphoblastic leukemia (17). Although mRNA regulation and
indirection with other tumor-associated complex of DDX6 have been
reported, direct tumor-associated actions still remain unknown.
Herein, we present for the first time the resistant mechanism of
DDX6 to antitumor treatment.
Materials and methods
Patient-derived cell and sphere
culture
According to the Institutional Review Boards,
specimens were obtained from glioblastoma patients after surgery.
Patient-derived cells were cultured in Neurobasal-A medium (Thermo
Fisher Scientific, Waltham, MA, USA) supplemented with N2 and B27
supplements (0.5× respectively, Gibco) and human recombinant bFGF
(20 ng/ml, R&D systems, MN, USA) and EGF (20 ng/ml, R&D
Systems) (18,19). All used patient-derived cells were
maintained under 20 passages in vitro.
Viral production
293FT cells (Invitrogen) were transfected with
target vectors and viral package vectors (pCMV-PAX2 and pCMV-VSVG)
by using CalPhos™ (Clontech Laboratories, Inc., Mountain View, CA,
USA) according to the manufacturer's protocol. There are target
vectors for pGIPZ for shRNA screening and DDX6 shRNA vectors
(MISSON® shRNA, Sigma, St. Louis, MO, USA) for gene
validation. All vectors have puromycin selection markers. The
manufactured lentiviral supernatants were filtered through a 0.45
μm filter to remove cell debris and centrifuge at 20,000 × g for 2
h at 4°C to concentrate the virus supernatant.
Pooled shRNA screening
shRNA pool was generated using pGIPZ system (GE
System, CO, USA) and information of pGIPZ vector was based on the
website (http://dharmacon.gelifesciences.com). Target genes for
shRNA were chosen randomly. The 827 patient-derived cells were
infected with the shRNA library and selected by puromycin (0.5
μg/ml) for 3 days. After 3 days, selected cells were sorted by
fluorescence-activated cell sorter (FACSAria™, BD Biosciences,
Franklin Lakes, NJ, USA) for mouse implantation. The control cells
(before mouse implantation) and mass were harvested at the mouse
survival end point for genetic analysis. shRNA barcodes were
PCR-recovered from genomic samples and analyzed through next
generation sequencing (Illumina High-Seq 2000, San Diego, CA, USA).
shRNA level were normalized to its whole population and relative
alteration of shRNA expression were measured. shRNAs presented in 2
or more replicates were selected for next experiment.
Generation of candidate gene knockdown
cells
Five shRNAs specifically targeting the candidate
gene DDX6, was used to reduce gene expression. The sequences of
shRNAs were derived from the DDX6 coding region, but the control
shRNA did not depress the DDX6 expression (pLKO, Sigma). Transduced
cells were harvested for western blotting and successfully
depressed DDX6 clone was selected. Among five shRNAs, only one
shRNA was accepted, and used for functional validation.
Orthotopic xenograft model
Six-week-old male BALB/c nude mice (Orient Bio Inc.,
Seoul, South Korea) were used for pooled shRNA screening and gene
validation. Patient-derived GBM cells (5×104/mouse) were
intracranially injected in mouse brain by stereotactic instrument
(AP +0.5 mm, ML +1.7 mm, DV −3.2 mm from the bregma). Radiation or
temozolomide (TMZ) treatment were performed at time two thirds of
the control group median survival. Each mouse was sacrificed when
unusual conditions (cachexia, lethargy and seizures) or 20% body
weight loss were observed. For survival analysis, 5 mice were used
per groups. All mouse experiments were performed according to the
Association for Assessment and Accreditation of Laboratory Animal
Care-accredited guidelines of Samsung Medical Institute's Animal
Use and Care Committee (Permit number: K-B2-035).
Western blot assay
Cell lysis was performed in RIPA buffer supplemented
with protease inhibitor cocktail tablets (Roche, Basel,
Switzerland). Total proteins (10 μg/lane) were separated in
SDS-PAGE gel and transferred to PVDF membranes (Millipore). The
membranes were blocked for 2 h in room temperature by 5–10% skim
milk solution and were incubated with primary antibodies (rabbit
anti-DDX6 (Abcam, Cambridge, UK) and mouse anti-β-catenin (Santa
Cruz, Biotechnology, Inc., Dallas, TX, USA) overnight at 4°C. After
washing with TBST (Tris-Buffered Saline, 0.1% Tween-20), the
membranes were incubated with HRP-conjugated secondary antibodies
for 1 h at 4°C. Development of membrane was performed using the
chemiluminescence method (ECL, GE Healthcare, Pittsburgh, PA,
USA).
Cell proliferation and viability
test
Cell proliferation and viability were performed
using EZ-cytox cell viability kit (DAEIL Lab, South Korea)
according to the manufacturer's protocol. For cell proliferation,
0.5–1×103 cells were seeded per well in 96-well plate
and each sample was in triplicate. After 3–6 days, Ez-cytox was
added into each well and incubated for 2 h. The absorbance at 450
nm was measured for incubated plates. Cell viability were also
performed according to the manufacturer's protocol with treatment
schedule. Radiation or temozolomide (TMZ) treatment was
administered within 6 h of seeding.
Sphere limiting dilution assay (SLDA) and
sphere counting
Sphere limiting dilution assay (SLDA) was performed
according to published method (20). Briefly, each of the cells was
plated at densities range from 500 to 2 cells in a 96-well plate
with 6 replicates for each dilution and evaluated after several
days in culture. We scored each well for the absence (−) or
presence (+) of sphere growth to determine the fraction of negative
wells. The plot shows percentage for the fraction of non-responding
wells (y-axis) versus plating density (x-axis). In addition, sphere
counting was performed when each of the cells had similar
clonogenic ability in SLDA.
Establishment of radiation/TMZ resistant
cell lines
We established radiation/TMZ resistant cell lines by
modifying pre-published method (21,22).
Briefly, radiation or TMZ treatment was performed for each cell at
lethal dose and these cells were incubated several days to recover
their population. For competent resistant cell lines, repeated
treatments were conducted several times and these cells were
confirmed by toxicity test using EZ-cytox cell viability kit.
Statistics
All statistical analysis was conducted using
Student's t-test to determine the significance of results
(P<0.05, P<0.01, P<0.001). Overall survival curves were
plotted according to the Kaplan-Meier method.
Results
Pooled shRNA screening for CCRT
resistance
Screening using a shRNA library was used to identify
CCRT resistance-related genes. Patient-derived cells treated with
the various shRNAs were implanted in mouse brains. The mice
received a standard treatment of GBM, radiation and/or TMZ, and the
remaining tumor mass was harvested from each mouse at the survival
end-point for analysis of shRNA expression change (Fig. 1A). Survival increased in all
treatment groups as compared with control group. However, survival
gain in the combination therapy group (GIV, ILS=56%) was similar to
the radiation therapy group (GII, ILS=59%) (Fig. 1B). These data resulted from
different responses of the 827 cell samples to radiation therapy
(RT) and TMZ. Since the 827 samples were sensitive to RT and
already resistant to TMZ, no combination-related survival gain was
evident. The next experiments were done to demonstrate these
characteristics in vitro (Figs.
5 and 6). Next generation
sequencing analysis of the remaining tumor mass revealed shRNAs
with increased expression, which enabled selection of the targeted
genes in commonly expressing genes of three references to ensure
that the genetic change noted in the 827 cells was correct. The
approach revealed the abundant expression of DDX6 in all treatment
groups (Fig. 1C). These cells
survived the radiation/TMZ treatment, which indicated DDX6 as a
candidate gene regulating the resistance to radiation and/or TMZ
treatment in GBM.
Functional confirmation of decreased DDX6
in the xenograft model
For functional validation of DDX6, a stable DDX6
knock-down cell line was established using lentiviral shRNA in two
patient-derived cell lines (827 and 578). Since the 827 cells were
resistant to TMZ treatment (Fig.
1B), TMZ sensitive cells were needed to confirm TMZ resistance
in cells with abrogated expression of DDX6. Because 827 cells were
used in shRNA screening and 578 cells were sensitive to TMZ
treatment, these cell types were judged suitable. Analysis of
protein level confirmed reduced DDX6 expression in both cell types
in established DDX6 knock-down cells against non-target cells
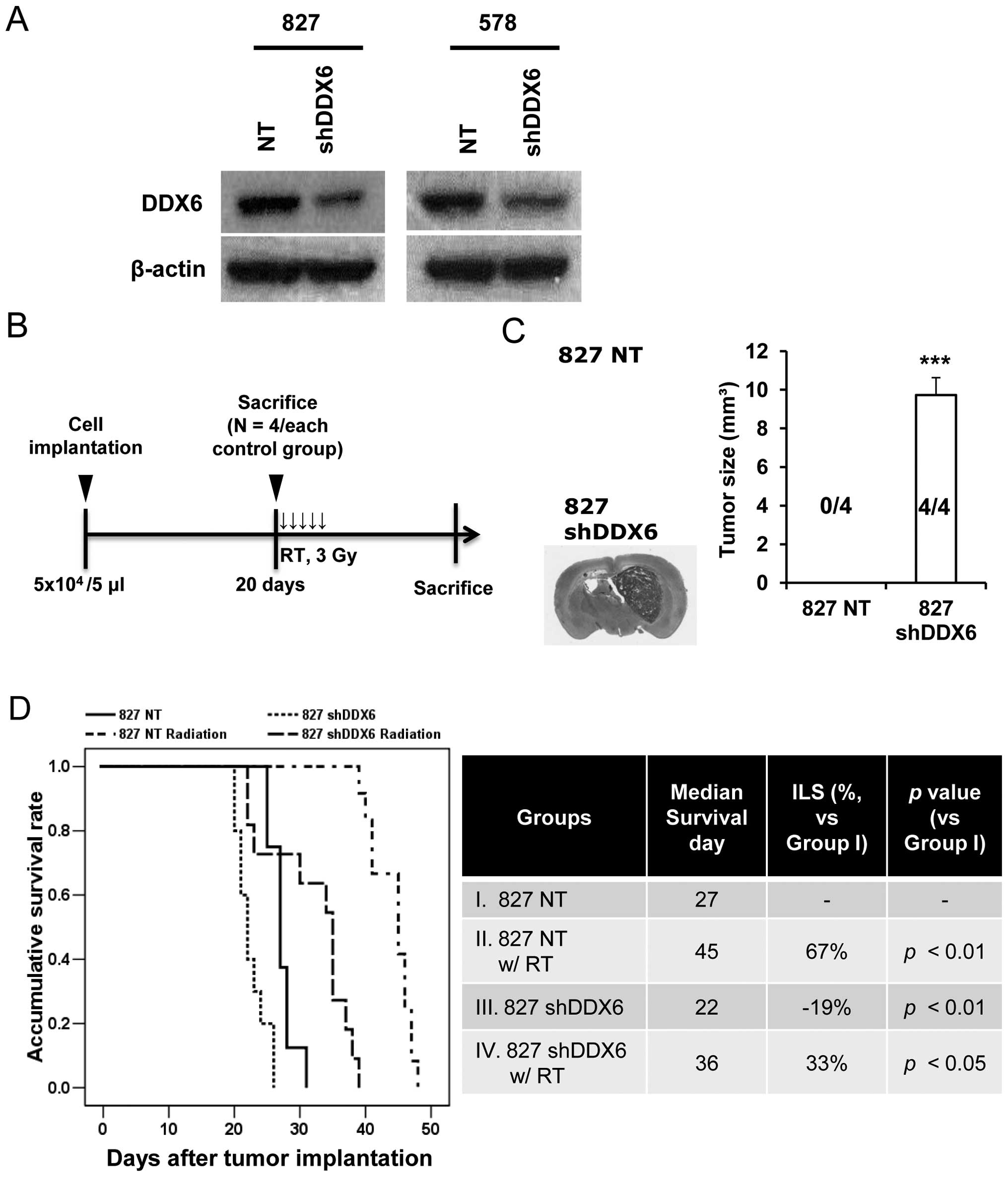
(Fig. 2A). Use of 827 cells
allowed the establishment of a xenograft model that confirmed
functional activity of DDX6 in radiation treatment, as evident in
the first screening experiment (Fig.
2B). Non-targeted cells and shDDX6 cells were implanted in the
mouse brain. Mice were sacrificed 20 days after cell implantation
(n=4 in each control group) to verify tumor incidence. Tumor
incidence and tumor size significantly improved (P<0.001) in the
827 shDDX6 cell implantation group compared to the NT cell group
(Fig. 2C). No tumor mass was
detected in NT implanted mouse brain. All shDDX6 implanted mice had
tumor masses. Other mice were sacrificed when their body weight
loss rate exceeded 20% or when abnormal behaviors (cachexia,
lethargy and seizures) were displayed. The sacrifice day
represented the survival end date for measuring median survival
(Fig. 2D). The shDDX6s implanted
mice (GIII) survived significantly shorter than NT implanted mice
(GI) without radiation treatment. The median survival of all RT
groups (NT-GII, shDDX6-GIV) was increased compared to untreated
mice. The median survival was slightly but significantly changed in
radiation treated mice by decreased DDX6 (GII vs. GIV, P<0.01).
Moreover, the range of survival end points within group was totally
different between RT groups (GII, 39–48 days; GIV, 22–39 days).
These results explained the earlier survival end point of the
shDDX6 implanted mice compared to the NT group against RT. The
results confirmed that decreased DDX6 increased tumorigenicity and
induced a resistant response to radiation in vivo.
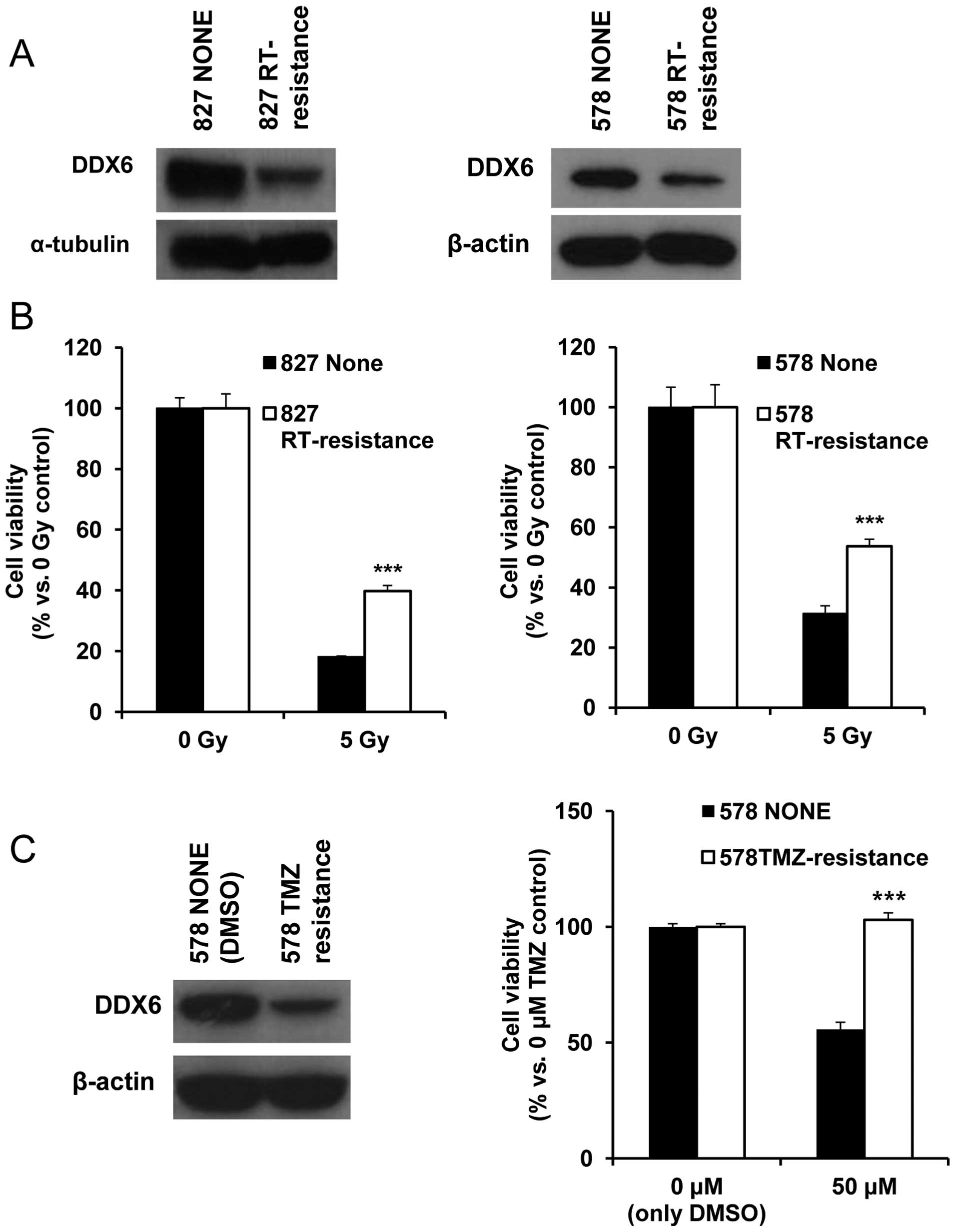
Establishment of RT/TMZ resistance cells
to confirm alteration of DDX6 in RT/TMZ treatment
For confirmation of substantial DDX6 alteration
against RT or TMZ treatment, we established RT/TMZ resistant cells
by repeated treatment. Similar to DDX6 K/D cells, we used 827 and
578 patient-derived cell types. Different protocols for the cell
types reflected their different RT/TMZ sensitivity. The 827 cell
type received 2 Gy radiation and the 578 cells received 1 Gy
radiation to isolate RT resistant cells. The resistant response to
radiation was confirmed by comparing post-treatment viability and
altered DDX6 production (Fig. 3A and
B). DDX6 protein expression decreased in both RT resistant cell
types. Since 827 cells were resistant to TMZ treatment, TMZ
sensitive cells were necessary to define TMZ resistance. In 578
cells, response to TMZ treatment was more sensitive and the
response to RT was similar to that of 827 cells (data not shown;
confirmed in Figs. 5A and 6A). Therefore, 578 cells were judged
suitable to define TMZ resistance. These cells were treated with 5
μM TMZ and their viability compared to control 578 cells treated
with DMSO, which was the TMZ solvent. Like radioresistant cells,
TMZ resistant 578 cells also displayed diminished expression of
DDX6 protein compared to control cells (Fig. 3C). This was evidence of decreased
DDX6-induced resistance to radiation and TMZ.
Confirmation of tumor associated ability
in 827 and 578 DDX6 knock-down cells
Next, alteration of tumor progression, proliferation
and clonogenicity were assessed in DDX6 K/D cells. Although
proliferation was unchanged, shDDX6s cells displayed significantly
more frequent tumor initiation than non-target (NT) cells in both
cell types (Fig. 4A and B). Since
altered clonogenic potential had been demonstrated in the 827
xenograft model (Fig. 2C), the
results suggested that decreased DDX6 can influence the increased
clonogenicity in vitro and in vivo.
Cell decreased DDX6 expression acquired
resistance against radiation and temozolomide treatment
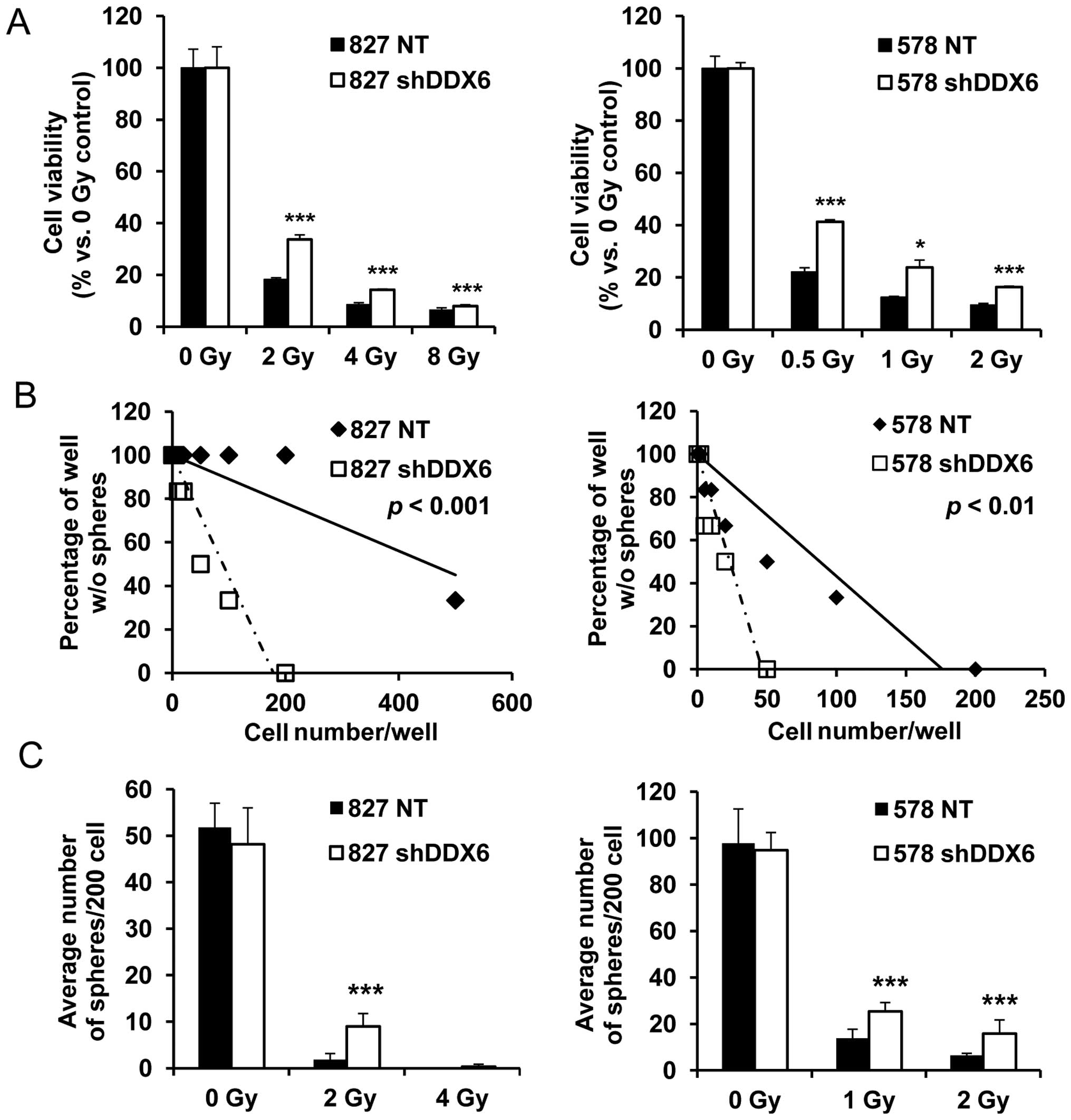
Irradiated 827 and 578 shDDX6s cells displayed
significantly better survival than their corresponding NTs
regardless of increased radiation intensity (Fig. 5A). Sphere formation was enhanced in
untreated 827 and 578 shDDX6s cells. Therefore, clonogenic
alteration due to the irradiation-mediated decreased DDX6 content
was confirmed using two sphere formation assays (Fig. 5B and C). In the sphere limiting
dilution assay (SLDA), irradiated shDDX6s formed spheres with fewer
cells compared to NT 827 and 578 cells (Fig. 5B). Consistently, irradiated shDDX6s
generated significantly more spheres than NT cells when cells were
seeded with similar clonogenic potential (Fig. 5C). On the other hand, the
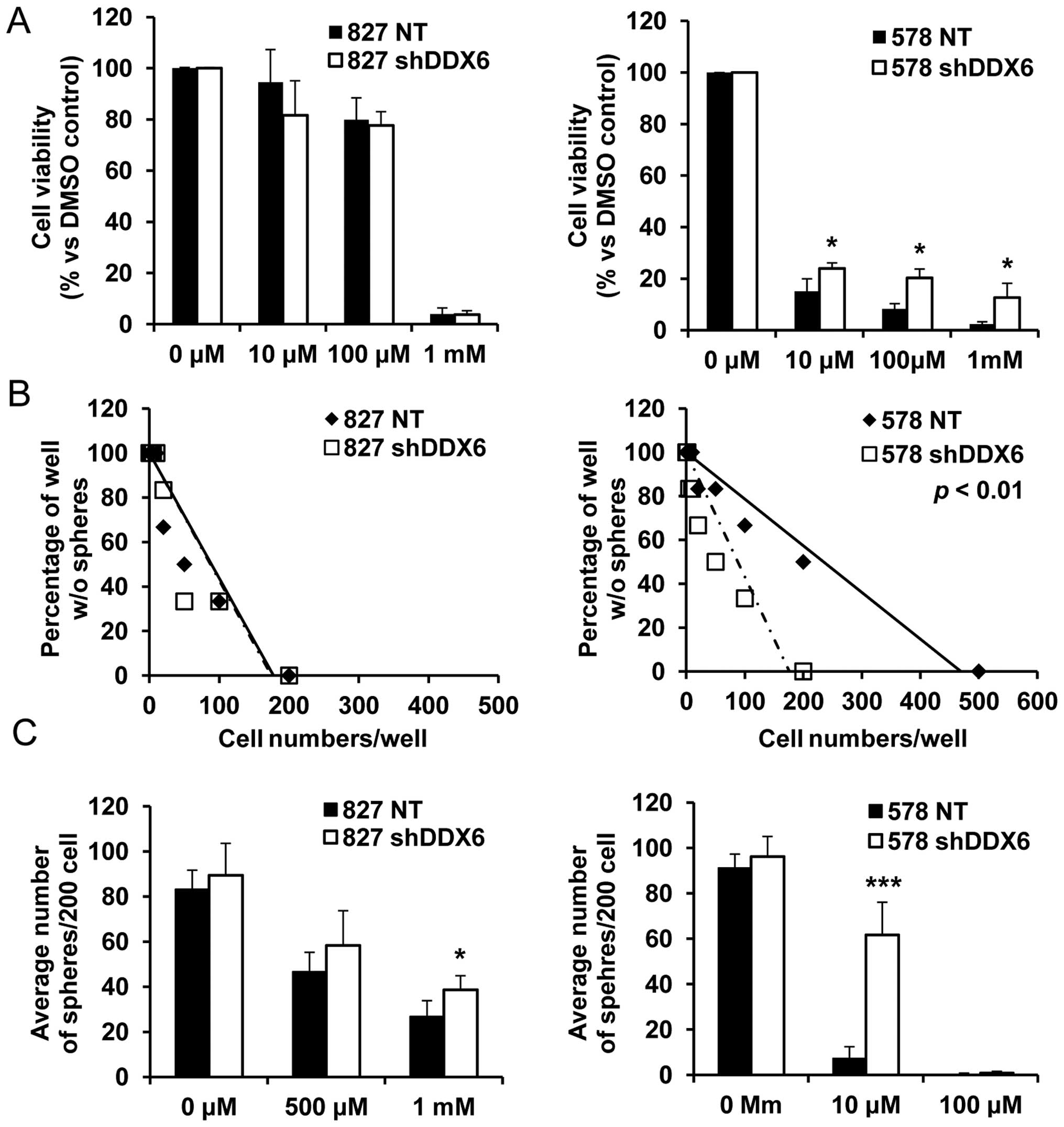
observation that cells harboring shRNA targeting DDX6 were enriched
after TMZ and combination treatment confirmed TMZ resistance due to
decreased DDX6 expression. In an experiment with 827 cells,
decreased DDX6 expression had no influence on cell viability and
sphere forming ability after TMZ treatment (Fig. 6). Although the limiting dilution
assay showed no influence by decreased DDX6 expression (Fig. 6B), average sphere numbers were
slightly increased at a high concentration of TMZ (1 mM) with
similar sphere forming capacity evident (Fig. 6C). Resistance to TMZ by decreased
DDX6 was clearly evident in 578 cells. Viability and sphere forming
ability were significantly improved after TMZ treatment in cells
with decreased DDX6 expression (Fig.
6A and B), and the average sphere number with similar
clonogenic capacity significantly increased in 578 shDDX6 cells, by
~12 times compared to NT cells (Fig.
6C).
Moreover, there were several primary-recurrent
paired samples from the same patients in our patient sample bank,
and several samples were cultured in vitro. As with shRNA
screening (Fig. 1), we collected
primary-recurrent paired samples to confirm the relationship
between DDX6 expression and recurrence against CCRT including TMZ
treatments. The DDX6 protein level was confirmed in two paired
samples. In these samples, the DDX6 protein level of recurrent
tumor cells were lower than in primary tumor cells (Fig. 7). These results supported previous
results using DDX6 K/D cells in which cells harboring shRNA
survived better after radiation/TMZ treatment, and, in vivo,
resistant or recurrent tumors remaining after anticancer therapy
were associated with a poor DDX6 expression. All the above
suggested that decreased DDX6 expression induces radiation/TMZ
resistance and influences prognosis.
Discussion
The present study was aimed at discovering potential
genes involved in tumorigenic ability using shRNA screening
techniques (14–16,23–25).
Previous studies have utilized a pool of less than 1,000 shRNAs as
it was difficult to select a single meaningful gene from a
large-scale shRNA pool. Therefore, they conducted preliminary
studies using a large-scale pool for more accurate results
(26–28). Using a large-scale pool, they
discovered a gene cluster that are involved in a particular pathway
rather than identifying a single meaningful gene. In this study, we
discovered DDX6 by treating shRNA-integrated cells with two
different types of individual cellular stress despite using a
large-scale pool.
Patient-derived xenograft models were used in our
previously reported models and well representing tumor
microenvironment and parental genomic characteristics compared to
the in vitro models (19,29,30).
Because of its advantages, we performed shRNA screening using
patient-derived xenograft models for identifying a particular gene
responsible for CCRT resistance. Screening with approximately 8,000
shRNAs has previously identified several targets including
transcriptional factors (RFX7 and ZNF 649), neurogenic factor
(PRTG), tumor suppressor (BTG3), carcinogen (MYD88) and other
cancer-related genes (31–36). Of those, 11 genes were selected in
the treatment groups. Among them, BTG3 and MYD88 are known to be
associated with cancer regulation and treatment resistance.
Collectively, identification of the above genes supports the
fidelity of our RNAi screen. Within the group, DDX6 was the only
gene that was identified as a candidate hit in all three different
treatment groups. Therefore, we sought to functionally validate
DDX6.
DDX6 is an RNA helicase that regulates RNA
modification (37). However, the
functional activity of DDX6 remains unknown in cancer including
GBM. Noteworthy, it has been reported that DDX6 upregulates
tumorigenicity in colon cancer. Moreover, upregulation of DDX6
decreases miR-145 expression and induces tumorigenicity in GBM
(38,39). Yet, our studies show that DDX6
induces RT/TMZ resistance rather than tumorigenic ability or miRNA
modulation. Since DDX6 was the only gene that was identified in the
treatment-resistant tumors and the results of our validation
experiments were reliable, we suggest that DDX6 acts to regulate
RT/TMZ resistance in GBM. Although 827 DDX6 K/D cells showed
unsettled response to TMZ, we reasoned that DDX6 protein level in
827 cells is lower than 578 cells and the level of 827 DDX6 K/D
cells was not sufficient to present any phenotypic changes to TMZ
resistance (data not shown). Therefore, we validated DDX6 function
to TMZ resistance using 578 DDX6 K/D cells. Our data showed DDX6
playing a critical role in determining the sensitivity to CCRT in
GBM patients. Therefore, DDX6 could be implemented as a therapeutic
target in the clinic to predict a proficient response to CCRT
treatment and to propose an alternative treatment.
To utilize DDX6 as a biomarker for patient
prognosis, more evidence including functional activity of DDX6 to
CCRT resistance need to be presented. Cancer stem cells (CSCs) are
reported as vital factor for treatment resistance in cancer therapy
(40–43). However, we found no reliable
changes of CSC markers in our DDX6 K/D cells (data not shown, in 8
CSC markers of GBM). Although we need more supplementary data for
the mechanism of action, we consider miRNA could be involved in
treatment resistance by DDX6 except CSC. miRNAs regulate many
molecular actions in cells (44–46).
In particular, several miRNAs have been reported for their role in
treatment resistance. Reported miRNAs consists of miRNA-221/222,
miRNA-181, miRNA-21, miRNA-195 (47–50).
We need more studies for interaction between DDX6 and miRNAs to
explain how DDX6 could regulate miRNAs in treatment resistance.
Furthermore, DDX6 could be a putative tumor suppressor because of
increased tumorigenicity of DDX6 K/D cells in vitro and
in vivo. Further research using DDX6 overexpressed
patient-derived cells is rneeded to confirm the tumor suppressor
role of DDX6. Investigation of the roles of other candidate genes
in response to CCRT resistance is also required.
Acknowledgements
This study was supported by a grant of the Korea
Health Technology R&D Project through the Korea Health Industry
Development Institute (KHIDI), funded by the Ministry of Health and
Welfare, Republic of Korea (HI14C3418) and the Global Frontier
Project grant (NRF-2012M3A6A-2010-0029781) of National Research
Foundation funded by the Ministry of Science, ICT and Future
Planning (MSIP) of Korea.
References
|
1
|
Nishikawa R: Standard therapy for
glioblastoma - a review of where we are. Neurol Med Chir (Tokyo).
50:713–719. 2010. View Article : Google Scholar
|
|
2
|
Kong D-S, Kim ST, Kim E-H, Lim DH, Kim WS,
Suh YL, Lee JI, Park K, Kim JH and Nam DH: Diagnostic dilemma of
pseudo-progression in the treatment of newly diagnosed
glioblastomas: The role of assessing relative cerebral blood flow
volume and oxygen-6-methylguanine-DNA methyltransferase promoter
methylation status. AJNR Am J Neuroradiol. 32:382–387. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Locke VL, Davey RA and Davey MW:
Modulation of drug and radiation resistance in small cell lung
cancer cells by paclitaxel. Anticancer Drugs. 14:523–531. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pires IM, Olcina MM, Anbalagan S, Pollard
JR, Reaper PM, Charlton PA, McKenna WG and Hammond EM: Targeting
radiation-resistant hypoxic tumour cells through ATR inhibition. Br
J Cancer. 107:291–299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hegi ME, Liu L, Herman JG, Stupp R, Wick
W, Weller M, Mehta MP and Gilbert MR: Correlation of
O6-methylguanine methyltransferase (MGMT) promoter methylation with
clinical outcomes in glioblastoma and clinical strategies to
modulate MGMT activity. J Clin Oncol. 26:4189–4199. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Happold C, Roth P, Wick W, Schmidt N,
Florea AM, Silginer M, Reifenberger G and Weller M: Distinct
molecular mechanisms of acquired resistance to temozolomide in
glioblastoma cells. J Neurochem. 122:444–455. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goellner EM, Grimme B, Brown AR, Lin YC,
Wang XH, Sugrue KF, Mitchell L, Trivedi RN, Tang JB and Sobol RW:
Overcoming temozolomide resistance in glioblastoma via dual
inhibition of NAD+ biosynthesis and base excision
repair. Cancer Res. 71:2308–2317. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ramirez YP, Weatherbee JL, Wheelhouse RT
and Ross AH: Glioblastoma multiforme therapy and mechanisms of
resistance. Pharmaceuticals (Basel). 6:1475–1506. 2013. View Article : Google Scholar
|
|
9
|
Hehlgans S and Cordes N: Caveolin-1: An
essential modulator of cancer cell radio-and chemoresistance. Am J
Cancer Res. 1:521–530. 2011.PubMed/NCBI
|
|
10
|
Kim Y, Kim KH, Lee J, Lee YA, Kim M, Lee
SJ, Park K, Yang H, Jin J, Joo KM, et al: Wnt activation is
implicated in glioblastoma radioresistance. Lab Invest. 92:466–473.
2012. View Article : Google Scholar
|
|
11
|
Fan Q-W and Weiss WA: Targeting the
RTK-PI3K-mTOR axis in malignant glioma: Overcoming resistance. Curr
Top Microbiol Immunol. 347:279–296. 2010.PubMed/NCBI
|
|
12
|
Huang H, Lin H, Zhang X and Li J:
Resveratrol reverses temozolomide resistance by downregulation of
MGMT in T98G glioblastoma cells by the NF-κB-dependent pathway.
Oncol Rep. 27:2050–2056. 2012.PubMed/NCBI
|
|
13
|
Lizardi PM, Forloni M and Wajapeyee N:
Genome-wide approaches for cancer gene discovery. Trends
Biotechnol. 29:558–568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rudalska R, Dauch D, Longerich T, McJunkin
K, Wuestefeld T, Kang TW, Hohmeyer A, Pesic M, Leibold J, von Thun
A, et al: In vivo RNAi screening identifies a mechanism of
sorafenib resistance in liver cancer. Nat Med. 20:1138–1146. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu H, Sun H, Zhang H, Liu J, Fan F, Li Y,
Ning X, Sun Y, Dai S, Liu B, et al: An shRNA based genetic screen
identified Sesn2 as a potential tumor suppressor in lung cancer via
suppression of Akt-mTOR-p70S6K signaling. PLoS One.
10:e01240332015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sroczynska P, Cruickshank VA, Bukowski
J-P, Miyagi S, Bagger FO, Walfridsson J, Schuster MB, Porse B and
Helin K: shRNA screening identifies JMJD1C as being required for
leukemia maintenance. Blood. 123:1870–1882. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Y, Boland A, Kuzuoğlu-Öztürk D,
Bawankar P, Loh B, Chang CT, Weichenrieder O and Izaurralde E: A
DDX6-CNOT1 complex and W-binding pockets in CNOT9 reveal direct
links between miRNA target recognition and silencing. Mol Cell.
54:737–750. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee J, Kotliarova S, Kotliarov Y, Li A, Su
Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al:
Tumor stem cells derived from glioblastomas cultured in bFGF and
EGF more closely mirror the phenotype and genotype of primary
tumors than do serum-cultured cell lines. Cancer Cell. 9:391–403.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Joo KM, Kim J, Jin J, Kim M, Seol HJ,
Muradov J, Yang H, Choi YL, Park WY, Kong DS, et al:
Patient-specific orthotopic glioblastoma xenograft models
recapitulate the histopathology and biology of human glioblastomas
in situ. Cell Rep. 3:260–273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rota LM, Lazzarino DA, Ziegler AN, LeRoith
D and Wood TL: Determining mammosphere-forming potential:
Application of the limiting dilution analysis. J Mammary Gland Biol
Neoplasia. 17:119–123. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wei QCSL, Shen L, Zheng S and Zhu YL:
Isolation and characterization of radiation-resistant lung cancer
D6-R cell line. Biomed Environ Sci. 21:339–344. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gomez-Casal R, Bhattacharya C, Ganesh N,
Bailey L, Basse P, Gibson M, Epperly M and Levina V: Non-small cell
lung cancer cells survived ionizing radiation treatment display
cancer stem cell and epithelial-mesenchymal transition phenotypes.
Mol Cancer. 12:942013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mullenders J and Bernards R:
Loss-of-function genetic screens as a tool to improve the diagnosis
and treatment of cancer. Oncogene. 28:4409–4420. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou P, Shaffer DR, Alvarez Arias DA,
Nakazaki Y, Pos W, Torres AJ, Cremasco V, Dougan SK, Cowley GS,
Elpek K, et al: In vivo discovery of immunotherapy targets in the
tumour microenvironment. Nature. 506:52–57. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hitomi J, Christofferson DE, Ng A, Yao J,
Degterev A, Xavier RJ and Yuan J: Identification of a molecular
signaling network that regulates a cellular necrotic cell death
pathway. Cell. 135:1311–1323. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wajapeyee N, Serra RW, Zhu X, Mahalingam M
and Green MR: Oncogenic BRAF induces senescence and apoptosis
through pathways mediated by the secreted protein IGFBP7. Cell.
132:363–374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen S, Blank JL, Peters T, Liu XJ,
Rappoli DM, Pickard MD, Menon S, Yu J, Driscoll DL, Lingaraj T, et
al: Genome-wide siRNA screen for modulators of cell death induced
by proteasome inhibitor bortezomib. Cancer Res. 70:4318–4326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mendes-Pereira AM, Sims D, Dexter T,
Fenwick K, Assiotis I, Kozarewa I, Mitsopoulos C, Hakas J, Zvelebil
M, Lord CJ, et al: Genome-wide functional screen identifies a
compendium of genes affecting sensitivity to tamoxifen. Proc Natl
Acad Sci USA. 109:2730–2735. 2012. View Article : Google Scholar :
|
|
29
|
Cho YB, Hong HK, Choi Y-L, Oh E, Joo KM,
Jin J, Nam DH, Ko YH and Lee WY: Colorectal cancer patient-derived
xenografted tumors maintain characteristic features of the original
tumors. J Surg Res. 187:502–509. 2014. View Article : Google Scholar
|
|
30
|
Lee HW, Lee JI, Lee SJ, Cho HJ, Song HJ,
Jeong E, Seo YJ, Shin S, Joung JG, Kwon YJ, et al: Patient-derived
xenografts from non-small cell lung cancer brain metastases are
valuable translational platforms for the development of
personalized targeted therapy. Clin Cancer Res. 21:1172–1182. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Amin S, Kumar A, Nilchi L, Wright K and
Kozlowski M: Breast cancer cells proliferation is regulated by
tyrosine phosphatase SHP1 through c-jun N-terminal kinase and
cooperative induction of RFX-1 and AP-4 transcription factors. Mol
Cancer Res. 9:1112–1125. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Severson PL, Tokar EJ, Vrba L, Waalkes MP
and Futscher BW: Coordinate H3K9 and DNA methylation silencing of
ZNFs in toxicant-induced malignant transformation. Epigenetics.
8:1080–1088. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wong Y-H, Lu A-C, Wang Y-C, Cheng HC,
Chang C, Chen PH, Yu JY and Fann MJ: Protogenin defines a
transition stage during embryonic neurogenesis and prevents
precocious neuronal differentiation. J Neurosci. 30:4428–4439.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang YC, Juan HC, Wong YH, Kuo WC, Lu YL,
Lin SF, Lu CJ and Fann MJ: Protogenin prevents premature apoptosis
of rostral cephalic neural crest cells by activating the
α5β1-integrin. Cell Death Dis. 4:e6512013. View Article : Google Scholar
|
|
35
|
Ou Y-H, Chung P-H, Hsu F-F, Sun T-P, Chang
W-Y and Shieh S-Y: The candidate tumor suppressor BTG3 is a
transcriptional target of p53 that inhibits E2F1. EMBO J.
26:3968–3980. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kfoury A, Le Corf K, El Sabeh R, Journeaux
A, Badran B, Hussein N, Lebecque S, Manié S, Renno T and Coste I:
MyD88 in DNA repair and cancer cell resistance to genotoxic drugs.
J Natl Cancer Inst. 105:937–946. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Weston A and Sommerville J: Xp54 and
related (DDX6-like) RNA helicases: Roles in messenger RNP assembly,
translation regulation and RNA degradation. Nucleic Acids Res.
34:3082–3094. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang Y-P, Chien Y, Chiou G-Y, Cherng JY,
Wang ML, Lo WL, Chang YL, Huang PI, Chen YW, Shih YH, et al:
Inhibition of cancer stem cell-like properties and reduced
chemoradio-resistance of glioblastoma using microRNA145 with
cationic polyurethane-short branch PEI. Biomaterials. 33:1462–1476.
2012. View Article : Google Scholar
|
|
39
|
Iio A, Takagi T, Miki K, Naoe T, Nakayama
A and Akao Y: DDX6 post-transcriptionally down-regulates
miR-143/145 expression through host gene NCR143/145 in cancer
cells. Biochim Biophys Acta. 1829:1102–1110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dean M, Fojo T and Bates S: Tumour stem
cells and drug resistance. Nat Rev Cancer. 5:275–284. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Baumann M, Krause M and Hill R: Exploring
the role of cancer stem cells in radioresistance. Nat Rev Cancer.
8:545–554. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rycaj K and Tang DG: Cancer stem cells and
radioresistance. Int J Radiat Biol. 90:615–621. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sotiropoulou PA, Christodoulou MS, Silvani
A, Herold-Mende C and Passarella D: Chemical approaches to
targeting drug resistance in cancer stem cells. Drug Discov Today.
19:1547–1562. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hayes J, Peruzzi PP and Lawler S:
MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol
Med. 20:460–469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lin S and Gregory RI: MicroRNA biogenesis
pathways in cancer. Nat Rev Cancer. 15:321–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Miller TE, Ghoshal K, Ramaswamy B, Roy S,
Datta J, Shapiro CL, Jacob S and Majumder S: MicroRNA-221/222
confers tamoxifen resistance in breast cancer by targeting p27Kip1.
J Biol Chem. 283:29897–29903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen G, Zhu W, Shi D, Lv L, Zhang C, Liu P
and Hu W: MicroRNA-181a sensitizes human malignant glioma U87MG
cells to radiation by targeting Bcl-2. Oncol Rep. 23:997–1003.
2010.PubMed/NCBI
|
|
49
|
Wong ST, Zhang XQ, Zhuang JT, Chan HL, Li
CH and Leung GK: MicroRNA-21 inhibition enhances in vitro
chemosensitivity of temozolomide-resistant glioblastoma cells.
Anticancer Res. 32:2835–2841. 2012.PubMed/NCBI
|
|
50
|
Ujifuku K, Mitsutake N, Takakura S,
Matsuse M, Saenko V, Suzuki K, Hayashi K, Matsuo T, Kamada K,
Nagata I, et al: miR-195, miR-455-3p and miR-10a(*) are implicated
in acquired temozolomide resistance in glioblastoma multiforme
cells. Cancer Lett. 296:241–248. 2010. View Article : Google Scholar : PubMed/NCBI
|