Introduction
During cancer development, cell proliferation rates
exceed blood vessel formation, leading to hypoxia in solid tumor
microenvironment, which is a hallmark of highly proliferative tumor
cells (1). Proliferating cancer
cells proliferate using ‘the Warburg effect’, which consists in
increased glucose uptake and metabolism through anaerobic
glycolysis instead of oxidative phosphorylation (2), a process in part controlled by the
AMP-activated protein kinase (AMPK), a master regulator of cellular
energy pools (3). In addition,
AMPK is tightly linked to cancer by its ability to induce the
phosphorylation of the tumor suppressor p53, leading to DNA
synthesis inhibition (4) and cell
cycle arrest through inhibition of the mammalian target of
rapamycin (mTOR) (5).
Hypoxic exposure leads to an increase in
hypoxia-inducible factor 1α (HIF-1α), a transcription factor
essential in cell adaptation/survival (6). Whereas the HIF-1α protein is rapidly
degraded under normal (21%) oxygen conditions, hypoxia increases
HIF-1α levels and transcriptional activity, which then promotes the
expression of a number of genes necessary for cancer cell survival
(7).
Metastasis-associated lung adenocarcinoma transcript
1 (Malat1), also named nuclear-enriched abundant transcript 2, is a
conserved long non-coding RNA (lncRNA) ubiquitously expressed
(8). High levels of Malat1 were
initially associated to the severity of lung metastasis (8), but this observation has since been
extended to many other types of tumors (9–13).
Malat1 appears to stimulate cell proliferation at the expense of
differentiation and senescence (13–15).
Malat1−/− mouse xenografts show a nearly 80% lower tumor
development in vivo (10),
possibly through modification of serine/arginine splicing factors
(16).
Genetic loss of Malat1 does not affect mouse
viability (17); however, Malat1
has been suggested to modulate angiogenesis in vivo
(18). Interestingly, hypoxia
upregulates Malat1 in vitro (18,19),
and mice directly exposed to hypoxia also show increased Malat1
expression levels in specific tissues such as proximal tubules
(19). Based on these findings, it
is thus likely that the increase in Malat1 expression is part of an
adaptive response to hypoxia. The aim of this study was thus to
mechanistically investigate the pathways that contribute to the
transcriptional stimulation in Malat1 levels in cells under
hypoxia. Our results show that AMPK, through its upstream
calcium/calmodulin-dependent protein kinase kinase (CaMKK), is a
major node triggering Malat1 transcription upon hypoxia in a
HIF-1α-dependent manner.
Materials and methods
Cell culture and reagents
HeLa and HEK293T cells were from ATCC (Manassas, VA,
USA). Cells were grown in DMEM containing 1 g/l glucose, 10% FBS, 2
mM glutamine, and 1% penicillin-streptomycin. Compounds
(Sigma-Aldrich, Oakville, ON, USA) were suspended in the
appropriate vehicle (DMSO or medium without FBS). A pCDNA3
expression plasmid containing the human HIF-1α cDNA was purchased
from Addgene (Cambridge, MA, USA).
Oxygen conditions
Hypoxic conditions were maintained in a humidified
variable aerobic workstation (Coy Laboratory) at 37°C. To induce
hypoxia, oxygen concentrations were reduced from 21 to 1.5%, while
carbon dioxide remained at 5%. Oxygen sensor continuously monitored
and adjusted the oxygen level during experiments.
Cloning of the human Malat1 promoter
The Malat1 promoter was amplified from human genomic
DNA by PCR using primers 5′-TGTGGGAGCTTTTCAGTATTC-3′ and
5′-CTGGAATGGCCAGCCTATAA-3′, effectively resulting in a fragment
containing a sequence of 5.6 kb directly upstream of the Malat1
gene initiation site. The fragment was first subcloned in
TOPO®XL vector (Thermofisher) according to the
manufacturer’s instructions, and then cloned in
KpnI/XhoI-digested pGL3 luciferase reporter vector
(Promega). Validity of this construct was confirmed by
sequencing.
Gene reporter assays
HEK293T cells were seeded in 24-well plates at 80%
confluence. Six hours later, cells were transfected for 12 h with
250 ng of the reporter vector (hMALAT1_prom-pGL3 or
p(HA)HIF-1α-pCDNA3) and 50 ng of a β-galactosidase expression
vector as described previously (20). Transfected cells were FBS-starved
for 2 h before any pharmacological treatment. Luciferase activity
and β-galactosidase activity were measured as described previously
(20) using a Luminoskan™ Ascent
microplate luminometer or a Multiskan Spectrum (Thermo Scientific),
respectively. Luciferase activity levels were normalized against
β-galactosidase activity levels. The figures represent the mean
fold activation ± SEM of at least three independent gene reporter
experiments.
RNA isolation and quantitative PCR
Whole cell RNA extracts were prepared as recommended
by the manufacturer (GE Healthcare). DNA reverse transcription was
prepared with 0.5 μg of total RNA using qScript™ cDNA Synthesis kit
(Quanta Biosciences). Quantitative PCR were performed on a 7900HT
Applied Biosystems, using Sybr™-Green detection (Sigma-Aldrich),
normalized to a housekeeping gene. The following nucleotide pairs
were used to amplify Malat1: forward, GTAATGGAAAGTAAAGCCCTGAAC and
reverse, CCCCGGAACTTTTAAAATACCTCT.
Western blot analysis
Whole cell proteins were extracted by lysis with an
extraction buffer containing NP40 0.04%, Tween 0.02%, sodium
orthovanadate 1.5 mM and 10% protease inhibitors (Roche), and
incubated 10 min on ice. After centrifugation, the supernatant was
collected and considered as whole cell extract. Protein
concentrations were determined with DC™ Protein Assay Reagent
(Bio-Rad). Proteins (50 μg) per lane were loaded onto a 7.5%
SDS-polyacrylamide gel, and then blotted with
Trans-Blot® Turbo™ (Bio-Rad) on PVDF membranes.
Membranes were saturated in fat-free dry milk for 1 h, and then
incubated with primary antibodies overnight at 4°C in recommended
buffer at recommended dilutions (anti-HIF-1α from R&D,
anti-phospho-Thr172 AMPK and anti-AMPK total from Cell Signaling,
anti-β-actin from Millipore). Membranes were then incubated with
secondary antibodies coupled with HRP for 1 h (from Santa-Cruz for
anti-goat-HRP, from GE Healthcare for anti-mouse-HRP and
anti-rabbit-HRP). Signals were detected with ECL™ Western Blotting
Detection Reagents (GE Healthcare) on Kodak film.
Statistical analyses
Data are presented as mean ± SEM of at least three
independent experiments performed in triplicate. Data were analyzed
by one or two-way ANOVA as appropriate. A value of p<0.05 was
considered statistically significant.
Results
Hypoxia induces Malat1 expression through
AMPK
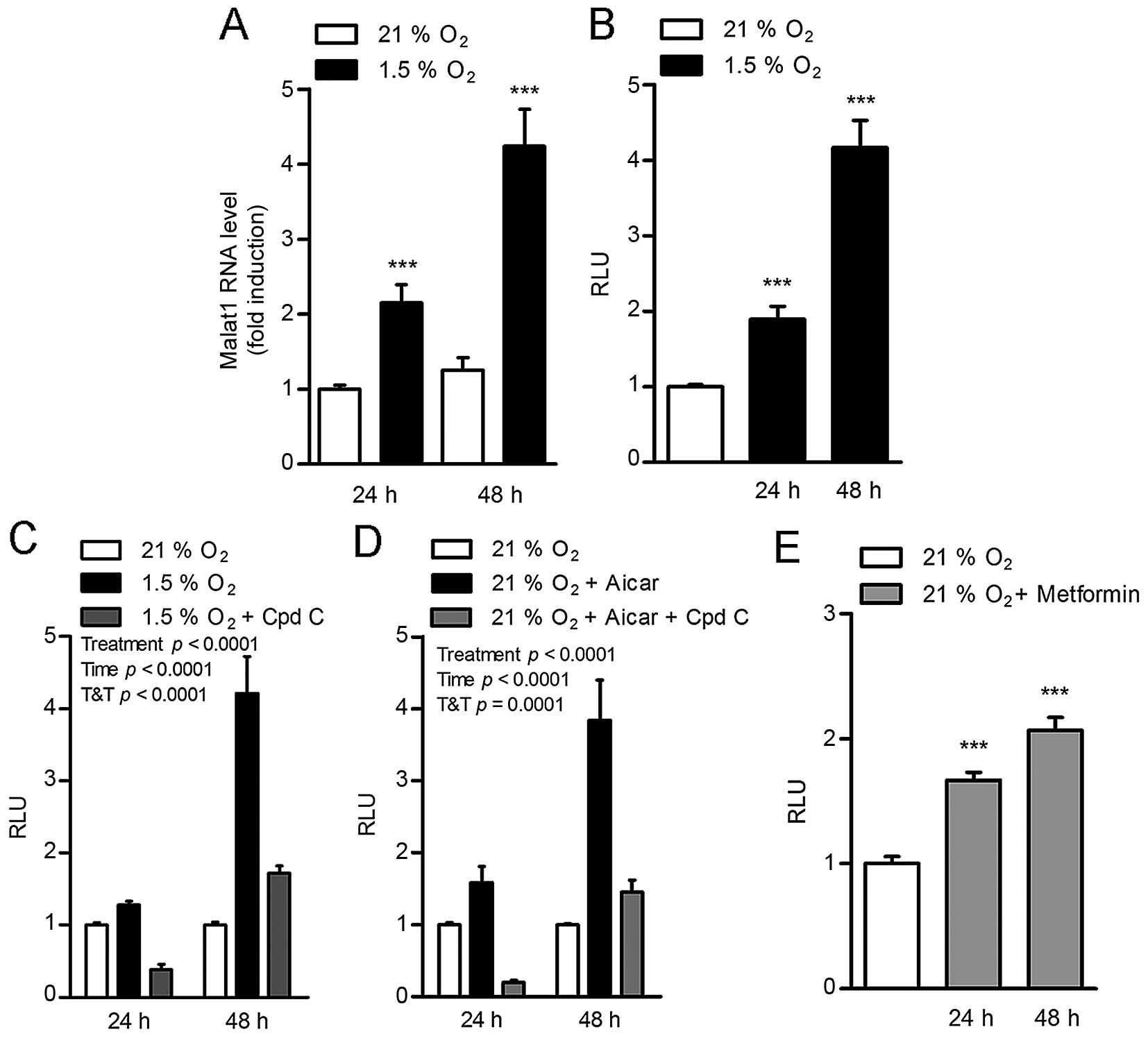
In adenocarcinoma HeLa cells, hypoxia (1.5%
O2) induced a time-dependent increase in Malat1 RNA
levels (Fig. 1A). To determine
that this effect was due to an impact on Malat1 gene transcription,
this experiment was repeated in HEK293T cells transfected with a
construct containing 5.6 kb of the human Malat1 promoter cloned
upstream of the luciferase gene. In this setting, hypoxia
stimulated Malat1 promoter transactivation to a similar
time-dependent extent, resulting in 2- and 4-fold increases after
24 and 48 h of incubation in low oxygen conditions, respectively
(Fig. 1B). This suggested a direct
impact of hypoxia on the Malat1 promoter.
Remarkably, the induction of the Malat1 promoter by
hypoxia was completely blocked by compound C (Fig. 1C), an ATP-competitive inhibitor of
AMPK (21), suggesting that AMPK
mediates the effect of 1.5% O2 conditions on Malat1
expression. Consistent with this concept, pharmacological
activation of AMPK with the 5-aminoimidazole-4-carboxamide
ribonucleotide (AICAR) was sufficient to stimulate Malat1 promoter
transactivation in normal oxygen conditions (21% O2)
(Fig. 1D). This effect was
preventable by co-treatment with compound C (Fig. 1D). Interestingly, the anti-diabetic
drug metformin, an indirect AMPK activator shown to lower
carcinogenesis (22), also induced
the activation of the Malat1 promoter, albeit to a lower extent
(Fig. 1E). Taken together, these
findings indicate that hypoxia stimulates Malat1 expression through
the activation of AMPK.
Inhibition of CaMKK blocks
hypoxia-induced Malat1 promoter transactivation
The activity of AMPK is mainly regulated by two
upstream kinases, namely the calcium-dependent kinase CaMKK and
LKB1, itself activated by EPAC (23,24).
In 21% O2 conditions, treatment of HEK293T cells with
the specific EPAC activator 8-CTP-2me-cAMP (25) did not result in the expected
increase in Malat1 promoter transactivation (Fig. 2A). In contrast, incubation with the
pharmacological CaMKK inhibitor STO-609 (26) completely prevented the induction of
the Malat1 promoter under hypoxia (Fig. 2B). These findings suggest that
hypoxia induces the CaMKK/AMPK cascade to stimulate Malat1
expression.
Hypoxia induces Malat1 via the induction
of HIF-1α
Analysis of protein extracts from cells incubated
under 21% and 1.5% O2 conditions indicated that hypoxia
induced the phosphorylation of AMPK at its Thr-172 residue within
60 min (Fig. 3A). In the same
conditions, an increase in HIF-1α was observed within 120 min under
hypoxia (Fig. 3A). This suggests
that the stimulation of HIF-1α protein levels occurs after
AMPK-activating events. Supporting this hypothesis, the
hypoxia-induced upregulation of HIF-1α levels was blocked in cells
incubated with the CaMKK inhibitor STO-609 (Fig. 3B). This further indicates that the
increase in HIF-1α levels is downstream of the CaMKK/AMPK
complex.
To investigate the possibility of a direct impact of
HIF-1α in the hypoxia-induced stimulation of Malat1 expression,
HEK293T cells containing the 5.6-kb human Malat1 promoter reporter
construct were treated with YC-1, a pharmacological inhibitor of
HIF-1α activity (27). Whereas
hypoxia increased Malat1 promoter transactivation in control cells
as expected, this effect was dose-dependently attenuated in cells
treated with YC-1 (Fig. 3C).
Consistent with this finding, transient overexpression of HIF-1α
under 21% O2 conditions was sufficient to transactivate
the Malat1 promoter (Fig. 3D).
This effect was completely abolished by co-treatment with YC-1
(Fig. 3D).
Discussion
The lncRNA Malat1 appears ubiquitously present in
cells during nonpathogenic conditions (8), however, several studies have reported
its high levels in many cancer types. Yet, the metabolic pathways
regulating its transcription in these conditions remain elusive.
This study focused on hypoxia, a powerful physiologic input in
solid tumors. This study strongly suggests that hypoxia triggers a
robust increase in Malat1 expression through the enhanced activity
of the CaMKK/AMPK/HIF-1α axis.
Although most studies on the stimulating impact of
Malat1 on cancer cell proliferation and migration have been
obtained in normoxic conditions (18), upregulation of Malat1 expression
during hypoxia has been recently observed in vitro and in
vivo (19). Our study
confirmed that hypoxia is an initial signal leading to increased
Malat1 expression level (Fig. 1).
Oxygen deprivation triggers several cellular processes required for
survival, including modulation of energy sensors such as AMPK
(3). Indeed, in hypoxic
environments, AMPK is phosphorylated (28), triggering the activation of its
downstream effector acetyl coenzyme A carboxylases 1 and 2
(ACC1/2). Such adaptive phenomenon is not observed in AMPK-null
mouse embryo fibroblasts (MEFs) (28), highlighting the importance of AMPK
in the regulation of energy upon hypoxia. This study also found
that AMPK phosphorylation occurs early after oxygen deprivation,
and that this event is necessary for a full augmentation in Malat1
expression (Fig. 1). Thus, it is
likely that Malat1 overexpression is part of the global adaptive
response to low oxygen conditions.
The upstream mechanisms leading to hypoxia-driven
AMPK activation are unclear. In response to low energy status, AMPK
activation has been linked to phosphorylation of LKB1, a
serine/threonine kinase also associated with tumor suppression
(29). However, incubation of
cells with the LKB1 upstream kinase activator 8-CTP-2me-cAMP
suggests that LKB1 does not robustly modify Malat1 expression in
our model (Fig. 2A), which is
consistent with the absence of impact of PKA activators on the same
system (data not shown).
More recently, other studies indicated that CaMKK
may play an important role in hypoxia-induced AMPK stimulation
(30). Indeed, knockdown of LKB1
in MEFs cultured in hypoxic conditions had no impact on ACC1/2
phosphorylation, suggesting that LKB1 is not the upstream kinase
leading to AMPK activation under hypoxia (30). In contrast, knockdown of CaMKK in
the same system clearly diminished AMPK and ACC1/2 phosphorylation
status (30). This is in agreement
with the fact that hypoxia increases intracellular calcium
concentrations (31), which has
been recently shown to be regulated by the effects of
STIM1-mediated store-operated calcium entry (32). Our study is consistent with these
studies, since the specific CaMKK inhibitor STO-609 abolished the
activation of Malat1 under hypoxia (Fig. 2). The possible control of Malat1 by
STIM1 remains to be investigated.
Calcium plays major functions in the regulation of
gene expression. Notably, calcium chelation modulates HIF-1α
activity (33–35). Moreover, calcium entry rapidly
stimulates CaMKK-induced p300 phosphorylation, which stabilizes
HIF-1α (32,36). Our study also corroborates that
hypoxia-induced HIF-1α protein stabilization is under CaMKK
regulation, as it occurred after AMPK phosphorylation (Fig. 3), and that cells treated with
STO-609 did not show high levels of HIF-1α under hypoxic conditions
(Fig. 3). More importantly, this
study shows that Malat1 overexpression upon hypoxia is dependent of
HIF-1α, and that an increase in HIF-1α levels is sufficient to
stimulate Malat1 transcription to a similar extent as did hypoxia
(Fig. 3). Further supporting the
large contribution of HIF-1α in the increase in Malat1 in low
oxygen conditions, loss of HIF-1α activity by YC-1 treatment
completely blocked hypoxia-induced Malat1 transcription.
Interestingly, bioinformatics analysis indicated four putative
HIF-1α binding sites, corresponding to the consensus hypoxia
response element ([A/G]CGTG), within 5.6 kb of the human Malat1
promoter, at positions −2246, −1687, −1317, and −259 from the
initiation start of the Malat1 coding sequence. The relative
importance of each of these binding sites in the transactivation of
the Malat1 promoter by HIF-1α remains to be tested.
Interestingly, a recent study reported a possible
involvement of p53 as a transcriptional repressor of Malat1
expression in early stage of hematopoietic cell proliferation
(37). It is established that
oxygen deprivation modulates the levels and activity of p53, a
major transcription regulator of cellular fate under intensive
stress (38). Depending on oxygen
availability (39), it is thus
possible that p53 influenced the action of HIF-1α in this study,
since reciprocal transcriptional effects by HIF-1α and p53 have
been reported (1,40). At the molecular level, this is a
likely event as they share and compete for the nuclear cofactor
p300 (41).
In conclusion, this study was conducted to
understand the mechanisms regulating Malat1 expression levels upon
hypoxia, an important characteristic of cancer tissues. This study
indicates that the enhanced transcription of the Malat1 gene upon
low oxygen conditions is under the control of HIF-1α, itself
regulated by the activation of the CaMKK/AMPK complex, a master
regulator of cellular energy. Our results also support and extend
published literature that calcium influx is an early signal in the
adaptive response to hypoxic stress. Finally, since Malat1 induces
angiogenesis in cancer (18,42),
it would be of interest to determine its role in physiological
processes in which increased tissue mass is associated with high
demand for oxygen and energy substrates, such as exercise-induced
myogenesis, or cold-induced brown adipose tissue hyperplasia.
References
|
1
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hardie DG and Carling D: The AMP-activated
protein kinase - fuel gauge of the mammalian cell? Eur J Biochem.
246:259–273. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Imamura K, Ogura T, Kishimoto A, Kaminishi
M and Esumi H: Cell cycle regulation via p53 phosphorylation by a
5′-AMP activated protein kinase activator,
5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, in a human
hepatocellular carcinoma cell line. Biochem Biophys Res Commun.
287:562–567. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Inoki K, Zhu T and Guan KL: TSC2 mediates
cellular energy response to control cell growth and survival. Cell.
115:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maxwell PH, Dachs GU, Gleadle JM, Nicholls
LG, Harris AL, Stratford IJ, Hankinson O, Pugh CW and Ratcliffe PJ:
Hypoxia-inducible factor-1 modulates gene expression in solid
tumors and influences both angiogenesis and tumor growth. Proc Natl
Acad Sci USA. 94:8104–8109. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Salceda S and Caro J: Hypoxia-inducible
factor 1alpha (HIF-1alpha) protein is rapidly degraded by the
ubiquitin-proteasome system under normoxic conditions. Its
stabilization by hypoxia depends on redox-induced changes. J Biol
Chem. 272:22642–22647. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ji P, Diederichs S, Wang W, Böing S,
Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, et
al: MALAT-1, a novel noncoding RNA, and thymosin beta4 predict
metastasis and survival in early-stage non-small cell lung cancer.
Oncogene. 22:8031–8041. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin R, Maeda S, Liu C, Karin M and
Edgington TS: A large noncoding RNA is a marker for murine
hepatocellular carcinomas and a spectrum of human carcinomas.
Oncogene. 26:851–858. 2007. View Article : Google Scholar
|
|
10
|
Gutschner T, Hämmerle M, Eissmann M, Hsu
J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Gross M, et al:
The noncoding RNA MALAT1 is a critical regulator of the metastasis
phenotype of lung cancer cells. Cancer Res. 73:1180–1189. 2013.
View Article : Google Scholar
|
|
11
|
Lai MC, Yang Z, Zhou L, Zhu QQ, Xie HY,
Zhang F, Wu LM, Chen LM and Zheng SS: Long non-coding RNA MALAT-1
overexpression predicts tumor recurrence of hepatocellular
carcinoma after liver transplantation. Med Oncol. 29:1810–1816.
2012. View Article : Google Scholar
|
|
12
|
Xu C, Yang M, Tian J, Wang X and Li Z:
MALAT-1: A long non-coding RNA and its important 3′ end functional
motif in colorectal cancer metastasis. Int J Oncol. 39:169–175.
2011.PubMed/NCBI
|
|
13
|
Ying L, Chen Q, Wang Y, Zhou Z, Huang Y
and Qiu F: Upregulated MALAT-1 contributes to bladder cancer cell
migration by inducing epithelial-to-mesenchymal transition. Mol
Biosyst. 8:2289–2294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tano K, Mizuno R, Okada T, Rakwal R,
Shibato J, Masuo Y, Ijiri K and Akimitsu N: MALAT-1 enhances cell
motility of lung adenocarcinoma cells by influencing the expression
of motility-related genes. FEBS Lett. 584:4575–4580. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tripathi V, Shen Z, Chakraborty A, Giri S,
Freier SM, Wu X, Zhang Y, Gorospe M, Prasanth SG, Lal A, et al:
Long noncoding RNA MALAT1 controls cell cycle progression by
regulating the expression of oncogenic transcription factor B-MYB.
PLoS Genet. 9:e10033682013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tripathi V, Ellis JD, Shen Z, Song DY, Pan
Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, et al: The
nuclear-retained noncoding RNA MALAT1 regulates alternative
splicing by modulating SR splicing factor phosphorylation. Mol
Cell. 39:925–938. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eissmann M, Gutschner T, Hämmerle M,
Günther S, Caudron-Herger M, Gross M, Schirmacher P, Rippe K, Braun
T, Zörnig M, et al: Loss of the abundant nuclear non-coding RNA
MALAT1 is compatible with life and development. RNA Biol.
9:1076–1087. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Michalik KM, You X, Manavski Y,
Doddaballapur A, Zörnig M, Braun T, John D, Ponomareva Y, Chen W,
Uchida S, et al: Long noncoding RNA MALAT1 regulates endothelial
cell function and vessel growth. Circ Res. 114:1389–1397. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lelli A, Nolan KA, Santambrogio S,
Gonçalves AF, Schönenberger MJ, Guinot A, Frew IJ, Marti HH,
Hoogewijs D and Wenger RH: Induction of long non coding RNA MALAT1
in hypoxic mice. Hypoxia (Auckl). 2015:45–52. 2015.
|
|
20
|
Miard S, Dombrowski L, Carter S, Boivin L
and Picard F: Aging alters PPARgamma in rodent and human adipose
tissue by modulating the balance in steroid receptor coactivator-1.
Aging Cell. 8:449–459. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fu D, Wakabayashi Y, Lippincott-Schwartz J
and Arias IM: Bile acid stimulates hepatocyte polarization through
a cAMP-Epac-MEK-LKB1-AMPK pathway. Proc Natl Acad Sci USA.
108:1403–1408. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lo B, Strasser G, Sagolla M, Austin CD,
Junttila M and Mellman I: Lkb1 regulates organogenesis and early
oncogenesis along AMPK-dependent and -independent pathways. J Cell
Biol. 199:1117–1130. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Enserink JM, Christensen AE, de Rooij J,
van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL and
Bos JL: A novel Epac-specific cAMP analogue demonstrates
independent regulation of Rap1 and ERK. Nat Cell Biol. 4:901–906.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda
M, Saji I and Kobayashi R: STO-609, a specific inhibitor of the
Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem.
277:15813–15818. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yeo EJ, Chun YS, Cho YS, Kim J, Lee JC,
Kim MS and Park JW: YC-1: A potential anticancer drug targeting
hypoxia-inducible factor 1. J Natl Cancer Inst. 95:516–525. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Laderoute KR, Amin K, Calaoagan JM, Knapp
M, Le T, Orduna J, Foretz M and Viollet B: 5′-AMP-activated protein
kinase (AMPK) is induced by low-oxygen and glucose deprivation
conditions found in solid-tumor microenvironments. Mol Cell Biol.
26:5336–5347. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shaw RJ, Kosmatka M, Bardeesy N, Hurley
RL, Witters LA, DePinho RA and Cantley LC: The tumor suppressor
LKB1 kinase directly activates AMP-activated kinase and regulates
apoptosis in response to energy stress. Proc Natl Acad Sci USA.
101:3329–3335. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mungai PT, Waypa GB, Jairaman A, Prakriya
M, Dokic D, Ball MK and Schumacker PT: Hypoxia triggers AMPK
activation through reactive oxygen species-mediated activation of
calcium release-activated calcium channels. Mol Cell Biol.
31:3531–3545. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arnould T, Michiels C, Alexandre I and
Remacle J: Effect of hypoxia upon intracellular calcium
concentration of human endothelial cells. J Cell Physiol.
152:215–221. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Guo B, Xie Q, Ye D, Zhang D, Zhu Y,
Chen H and Zhu B: STIM1 mediates hypoxia-driven
hepatocarcinogenesis via interaction with HIF-1. Cell Rep.
12:388–395. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mottet D, Michel G, Renard P, Ninane N,
Raes M and Michiels C: ERK and calcium in activation of HIF-1. Ann
NY Acad Sci. 973:448–453. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mottet D, Michel G, Renard P, Ninane N,
Raes M and Michiels C: Role of ERK and calcium in the
hypoxia-induced activation of HIF-1. J Cell Physiol. 194:30–44.
2003. View Article : Google Scholar
|
|
35
|
Berchner-Pfannschmidt U, Petrat F, Doege
K, Trinidad B, Freitag P, Metzen E, de Groot H and Fandrey J:
Chelation of cellular calcium modulates hypoxia-inducible gene
expression through activation of hypoxia-inducible factor-1alpha. J
Biol Chem. 279:44976–44986. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hui AS, Bauer AL, Striet JB, Schnell PO
and Czyzyk-Krzeska MF: Calcium signaling stimulates translation of
HIF-alpha during hypoxia. FASEB J. 20:466–475. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ma XY, Wang JH, Wang JL, Ma CX, Wang XC
and Liu FS: Malat1 as an evolutionarily conserved lncRNA, plays a
positive role in regulating proliferation and maintaining
undifferentiated status of early-stage hematopoietic cells. BMC
Genomics. 16:6762015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Koumenis C, Alarcon R, Hammond E, Sutphin
P, Hoffman W, Murphy M, Derr J, Taya Y, Lowe SW, Kastan M, et al:
Regulation of p53 by hypoxia: Dissociation of transcriptional
repression and apoptosis from p53-dependent transactivation. Mol
Cell Biol. 21:1297–1310. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou CH, Zhang XP, Liu F and Wang W:
Modeling the interplay between the HIF-1 and p53 pathways in
hypoxia. Sci Rep. 5:138342015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sermeus A and Michiels C: Reciprocal
influence of the p53 and the hypoxic pathways. Cell Death Dis.
2:e1642011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Schmid T, Zhou J, Köhl R and Brüne B: p300
relieves p53-evoked transcriptional repression of hypoxia-inducible
factor-1 (HIF-1). Biochem J. 380:289–295. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tee AE, Liu B, Song R, Li J, Pasquier E,
Cheung BB, Jiang C, Marshall GM, Haber M, Norris MD, et al: The
long noncoding RNA MALAT1 promotes tumor-driven angiogenesis by
up-regulating pro-angiogenic gene expression. Oncotarget.
7:8663–8675. 2016.PubMed/NCBI
|