Introduction
Interferon-alpha (IFN-α) has a variety of biological
properties, including antiviral effects, antiproliferation and
immune response modulation. Besides, IFN-α also exerts antitumor
activities in a range of haematological and non-haematological
malignancies (1,2). Following IFN-α bond to its receptor,
it affects Janus kinases JAK1 and TYK2 on the phospho-tyrosine
residue and sites at the intracellular domain of each receptor
chain. The signal transducer and transactivator (STAT) proteins are
phosphorylated by JAK1 and TYK2. Moreover, they dissociate from the
receptor, dimerize via SH2 domain, and form the mature ISGF3
complex associated with the IFN regulatory factor family. This
complex further translocates to the nucleus and binds to
interferon-stimulated response elements (ISRE) that initiate gene
transcription contributing to the activation of the cytoplasmic
targets of IFN-α. Of note, the interaction of ISGF3 with ISRE
induces several transcriptional genes such as protein kinase
dependent on dsRNA (PKR) that modulates cancer cell growth. Through
the translational and transcriptional pathways, PKR activates
protein expressions of Fas, p53 and Bax to trigger apoptosis.
IFN-α-induced apoptosis possibly activates the caspase cascade
mediated by mitogen-activated protein kinases (1,3,4).
Alternatively, IFN-α activates its receptors and induces
anti-proliferative signaling via the STAT by cross-talking with the
extracellular signal-regulated kinase (ERK) pathway; it further
leads to the slowing down of G1/S transition without apoptosis in
human hepatocellular carcinoma cells (HCC) (5). Conversely, IFN-α reduces activation
of ERK in haematological malignancies (6). IFN-α also exerts growth inhibition of
human T-cell leukaemia line Jurkat through p38a and p38b (7). These discrepancies depend on the cell
types, time of treatment and dosage used (1). Although the cell growth inhibition
and apoptosis of IFN-α are thought to be a possible explanation for
its antitumor action, the precise mechanisms of this issue are
restricted.
Besides antitumor activity with IFN-α monotherapy,
several studies suggest a combinatorial strategy with IFN-α in
cancer therapies. For instance, the synergic cell growth inhibition
and apoptosis of IFN-α are observed in human T-cell lymphotropic
virus type I-transformed cells combined with arsenic trioxide
(8), in transformed T- and
monocytoid cell lines combined with IL-21 (9), in HCC combined with 5-fluorouracil
(10), or in bladder cancer cells
combined with proanthocyanidin (11). These results indicate that a
combinatorial strategy is more effective to antitumor action.
Nevertheless, serious adverse effects still exist, and they are
limited due to their tolerability and efficacy.
Fluoxetine is widely used in treatment of depression
in patients with cancer or infection of hepatitis C virus (12,13).
The potential of antitumor action of fluoxetine is still
inconclusive due to the dependence on the dosage used and the cell
types (14,15). Besides, fluoxetine is kept to the
range between 5–20 μM as a multidrug resistance reversal agent and
it has been proposed to be considered a fourth-generation
chemo-sensitizer in clinic (15).
However, how fluoxetine regulates cellular signaling to enhance
cellular responses in chemotherapy is still unclear. In the present
study, we used human bladder superficial carcinoma cells, T24, to
investigate the possible mechanisms through which fluoxetine
promotes the antitumor activity of IFN-α. Recent evidence suggests
that peroxisome proliferator-activated receptor alpha (PPAR-α), a
member of the ligand-activated nuclear receptor superfamily, may
regulate cell survival and apoptosis (16). Interaction between PPAR-α and STAT
transcription factors contributes to PPAR-α-mediated
transcriptional repression (17);
however, whether PPAR-α regulates the growth inhibition of IFN-α
associated with the regulation of STAT-1 remains unclear. Thus,
after pretreatments with PPAR-α and STAT-1 inhibitors, we have
examined the IFN-α-mediated anti-proliferation and apoptosis in the
presence of fluoxetine, including cell growth, cell cycle, cyclins,
and signal molecules as well as the levels and co-localization of
activations of STAT-1 and PPAR-α.
Materials and methods
Chemicals
IFN-α-2b (Intron A) was purchased from
Schering-Plough Brinny Co. (Cork, Ireland). Fluoxetine, fludarabine
and GW6471 were purchased from Tocris Bioscience (Ellisville, MO,
USA).
Antibodies
Antibodies against β-actin, phospho-STAT1 (Tyr701
and Ser727), STAT1, cyclin A, cyclin B1, cyclin D1, p27 and p53
were purchased from Cell Signaling Technology, Inc. (Beverly, MA,
USA). PPAR-α was purchased from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). Lamin A was purchased from BD Biosciences (San
Jose, CA, USA).
Cell cultures
The human bladder carcinoma cell line T24 and normal
uroepithelial cell line SV-HUC-1 (Bioresource Collection and
Research Center, Food Industry Research and Development Institute,
Hsinchu, Taiwan) were grown in Dulbecco’s modified Eagle’s medium
and Ham’s F12 medium, respectively, with 10% fetal calf serum (FCS)
and 100 μg/ml gentamicin.
Cell proliferation assay
Cell growth of cultured cells was studied by
colorimetry with a tetrazolium compound
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
MTS] assay kit (Promega Corp., Madison, WI, USA). Briefly, cells
(8×103 cells/well) were seeded on 96-well plates for 24
h and the culture medium was later changed to a new medium
containing IFN-α, fluoxetine, or in combination for 3 days. The
number of viable cells was measured with SpectraMax 340PC
(Molecular Devices, Inc., Sunnyvale, CA, USA) at a wavelength of
490 nm after reacting with the tetrazolium reagent for 1.5 h.
Flow cytometry
Cells (2×106) were harvested from 10-cm
culture dishes, washed with phosphate-buffered saline (PBS),
suspended in 200 μl of ice-cold 70% ethanol and incubated on ice
for a least 1 h. After cells were washed and exposed to RNase A at
37°C for 30 min, these cells were then suspended in propidium
iodide (PI) (Sigma-Aldrich, St. Louis, MO, USA) in PBS. DNA was
analyzed via flow cytometry (FACSCalibur; BD Biosciences) to
evaluate the cell cycle by measuring the percentage of subG1,
G0/G1-, S- and G2/M-phase after treatment with IFN-α and/or
fluoxetine after 24 h.
Caspase-3 activity
Cells (7×105) were treated with GW6471 or
fludarabine for 1 h before IFN-α and fluoxetine, and then harvested
at day 1 for performing ICE-family proteases/caspases activation to
initiate apoptosis. The caspase-3/CPP32 colorimetric assay kit
(BioVision, Inc., Milpitas, CA, USA) was used according to the
manufacturer’s protocols and measured with a spectrophotometer at
405 nm.
Western blot analysis
Cells (7×105) were harvested at indicated
times and lysed with lysis buffer containing 1% Triton X-100, 50 mM
Tris (pH 7.5), 10 mM EDTA, 0.02% NaN3, and protease
inhibitor cocktail (Sigma-Aldrich). The membrane (Millipore,
Billerica, MA, USA) was blocked with 5% skim milk in TBS-T [50 mM
Tris, 150 mM NaCl and 0.05% Tween-20, (pH 7.6)] at room temperature
for 1 h and probed with primary antibodies at 4°C overnight. After
being washed with TBS-T, the blots were incubated with horseradish
peroxidase-conjugated secondary antibodies (1:2,500) at room
temperature for 1 h. The protein expression was visualized via
enhanced chemiluminescence reagent (Perkin-Elmer, Boston, MA, USA)
and analyzed using VisionWorks LS software (Upland, CA, USA) for
the optical densities of phospho-protein/total protein when using
β-actin as the internal control.
Nuclear extraction
The commercially available CHEMICON®
nuclear extraction kit (Millipore) was used according to the
manufacturer’s protocols.
Indirect immunofluorescence
The cells (2×104) were fixed with 4%
paraformaldehyde in PBS for 10 min and permeabilized with 0.1%
Triton X-100 for another 10 min at room temperature. These cells
were later stained with primary and secondary antibodies after
being washed with PBS twice. Primary antibodies used were
anti-phospho-STAT1 (Tyr701) or anti-PPAR-α, and Alexa Fluor
568-conjugated goat anti-rabbit IgG and 488-conjuated goat
anti-mouse IgG (Invitrogen, Carlsbad, CA, USA) were used as
secondary antibodies. Stained cells were later washed with PBS and
counterstained with 4,6-diamidino-2-phenylindole (DAPI;
Sigma-Aldrich) at room temperature for 1 h. After staining, these
cells were mounted on glass slides and observed under confocal
laser scanning microscope (Olympus FluoView™ FV1000; Olympus).
Statistical analysis
One-way ANOVA test was used to examine various
experiments among the IFN-α-, fluoxetine-, IFN-α plus
fluoxetine-treated, and the medium control groups. Statistical
significance was set at P<0.05.
Results
Fluoxetine sensitizes cell growth
inhibition to IFN-a
Normal human uroepithelial SV-HUC-1 and bladder
carcinoma T24 cells were treated with IFN-α (500 and 1000 U/ml),
fluoxetine (5 and 10 μM), or in combination, and then harvested for
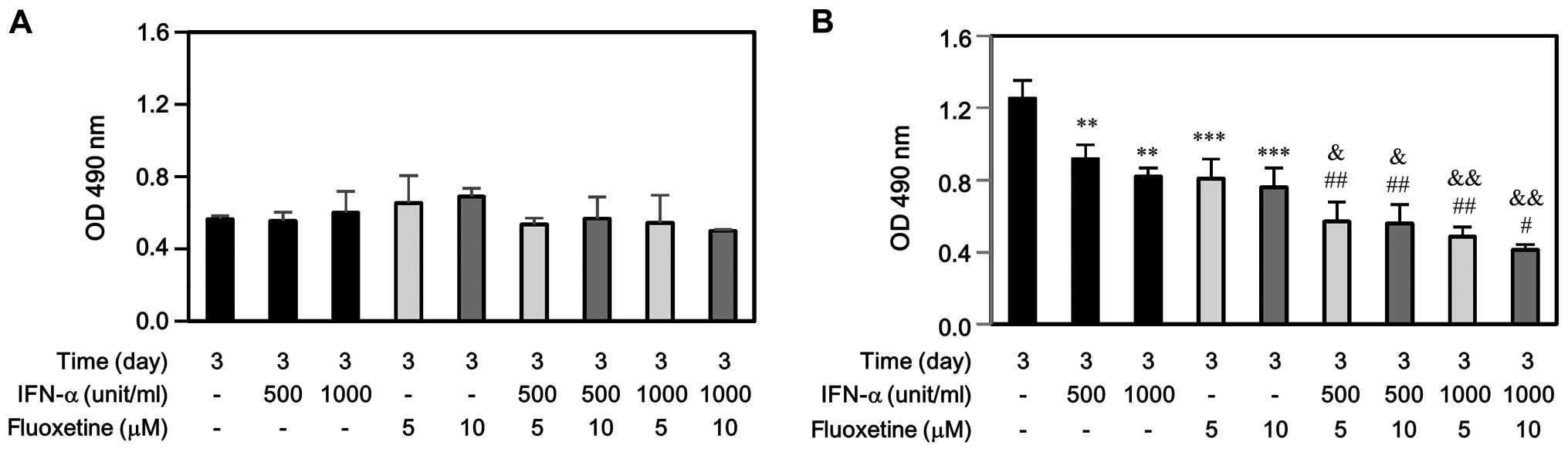
performing cell proliferation by MTS assay. As shown in Fig. 1A, IFN-α, fluoxetine, or in
combination did not affect SV-HUC-1 cell growth at day 3. However,
either IFN-α or fluoxetine only impeded significantly T24 cell
growth at day 3 (Fig. 1B).
Moreover, the decreased levels of T24 proliferation induced by
IFN-α combined with fluoxetine-treated group were much more than
those induced by IFN-α-treated or fluoxetine-treated group.
Fluoxetine sensitized cell growth
inhibition to IFN-α via the STAT-1- and PPAR-α-dependent
pathways
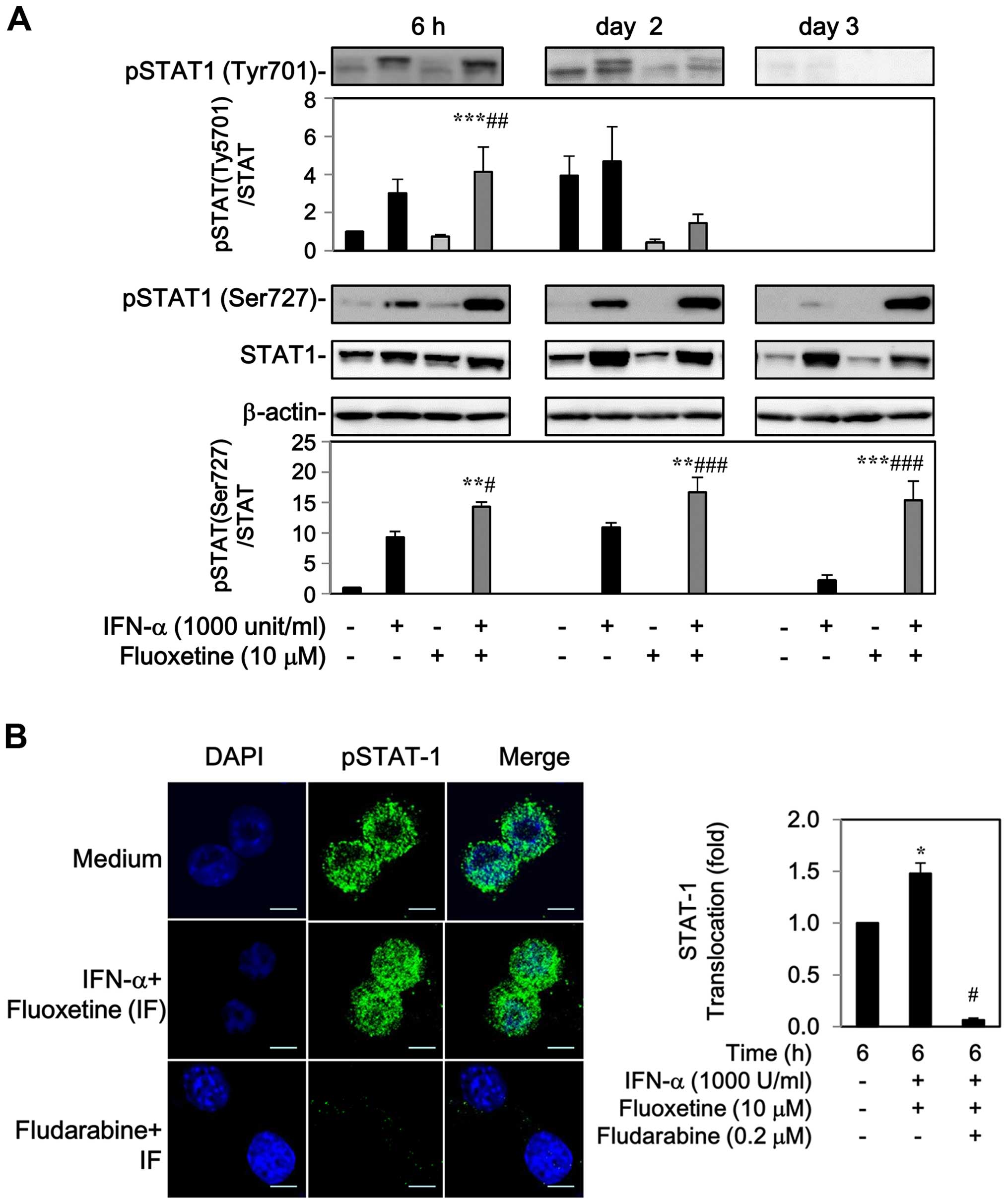
The levels of phospho-STAT-1 at Tyr701 and Ser727
residue were markedly increased by IFN-α after 6 h to 2-day
post-treatment, whereas fluoxetine alone had no effect on the
phosphorylation of STAT-1 (Fig.
2A). Addition of fluoxetine facilitated the IFN-α-mediated
phosphorylation of STAT1 at Tyr701 after 6 h, but declined after 2
days. Moreover, an elevated level of phospho-STAT1 at Ser727 was
also detected after 6 h and maintained continuously to day 3
(Fig. 2A).
The phospho-STAT1 localization was also determined
by indirect immunofluorescence. In the medium of control cells,
phospho-STAT1 was found around the cell surface with slight
fluorescence intensity. Following treatment with IFN-α and
fluoxetine for 6 h, a marked increase in phospho-STAT1 fluorescence
intensity and nuclear translocation was observed, which was
repressed by 0.2 μM of fludarabine, a specific STAT-1 blocker
(18–20) (Fig.
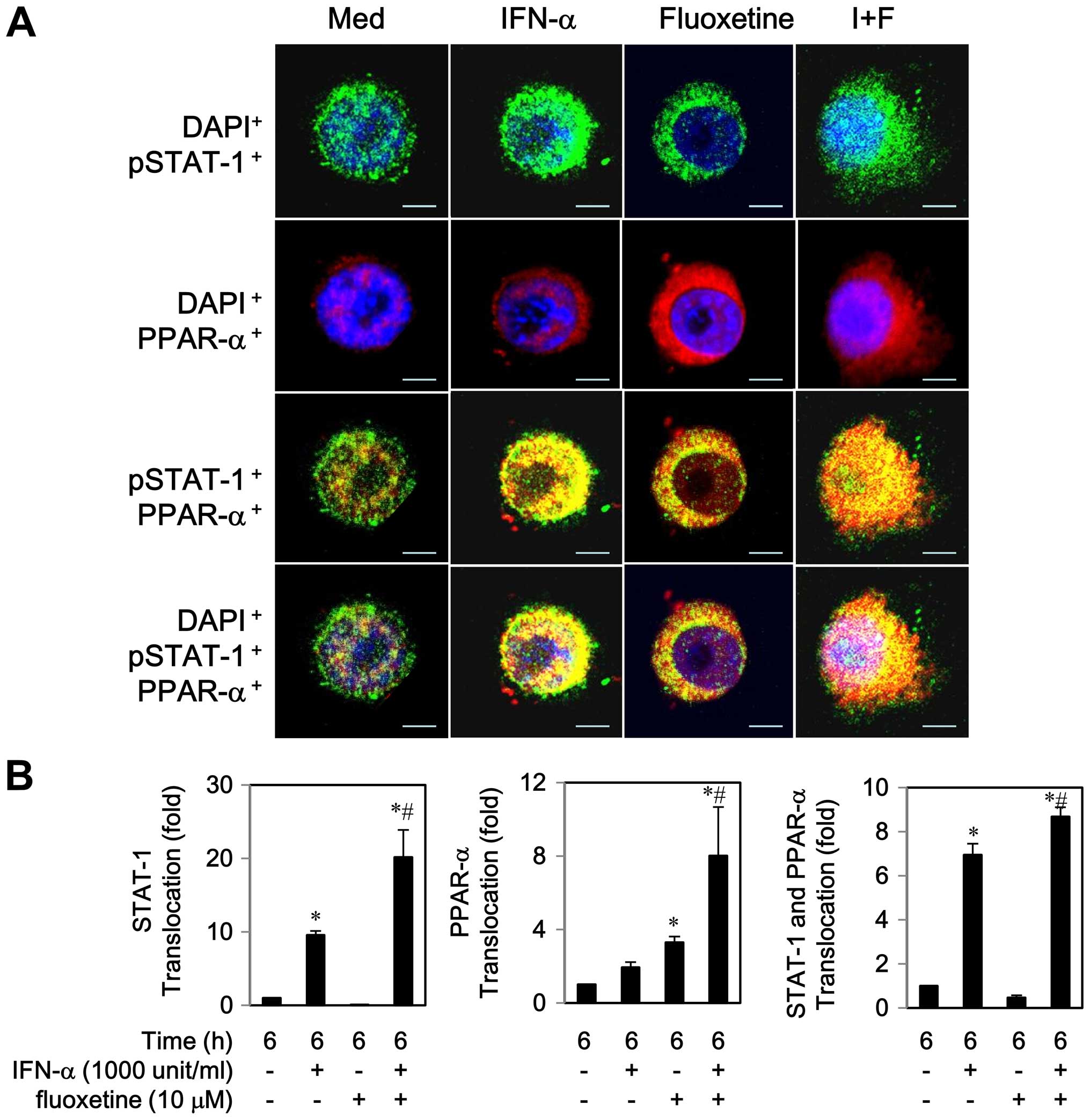
2B). Similarly, an increase in PPAR-α protein expression was
observed during 6–12 h (data not shown). An addition of fluoxetine
to IFN-α caused a translocation of PPAR-α to the nucleus, which was
inhibited by 5 μM of GW6471, a PPAR-α antagonist (Fig. 3).
Notably, co-localization of STAT-1 and PPAR-α was
observed to the some extent in the cytoplasm by IFN-α or fluoxetine
alone after a 6 h post-treatment (Fig.
4A). We also found that IFN-α-treated cells partially activated
and translocated both proteins to the nucleus, whereas
fluoxetine-treated cells predominately triggered PPAR-α
translocation to the nucleus (Fig.
4). As compared with IFN-α-treated cells, co-localization of
STAT-1 and PPAR-α was clearly seen in the nucleus while cells were
exposed to IFN-α in combination with of fluoxetine. These results
indicated that fluoxetine promoted the IFN-α-induced activation and
translocation of STAT1 and PPAR-α.
PPAR-a and STAT-1 were partially involved
in the growth inhibition of IFN-α in the presence of fluoxetine via
the alterations of cell cycle subpopulations and cell cycle
regulatory proteins
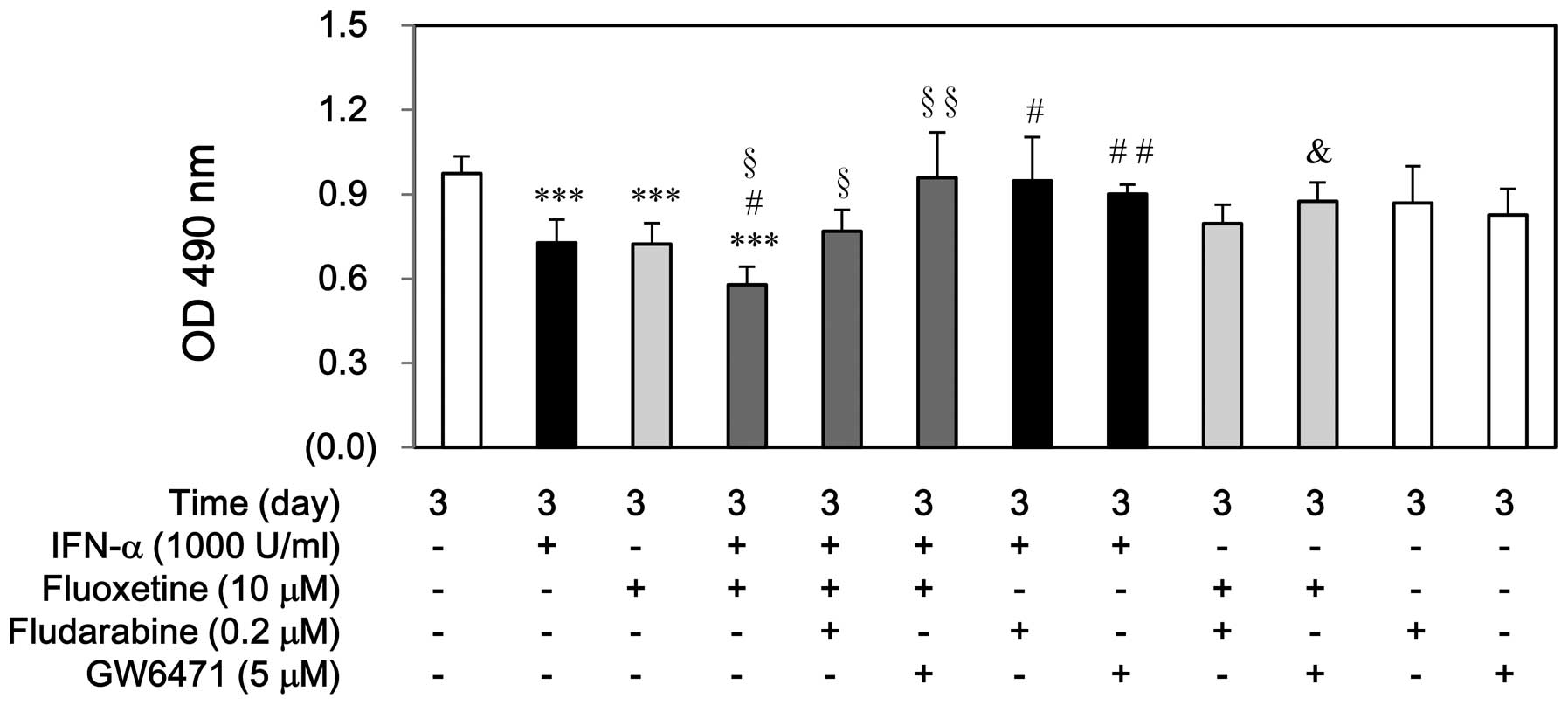
First, in order to investigate the roles of STAT-1
and PPAR-α proteins in cell growth inhibition of IFN-α or
fluoxetine, fludarabine and GW6471 were used before treatment with
IFN-α, fluoxetine, or in combination. Fludarabine or GW6471
partially prevented the cell growth inhibition induced by IFN-α or
in combination with fluoxetine, whereas pretreatment of fludarabine
could not reverse this inhibition induced by fluoxetine. Otherwise,
either fludarabine or GW6471 alone did not have this result
(Fig. 5).
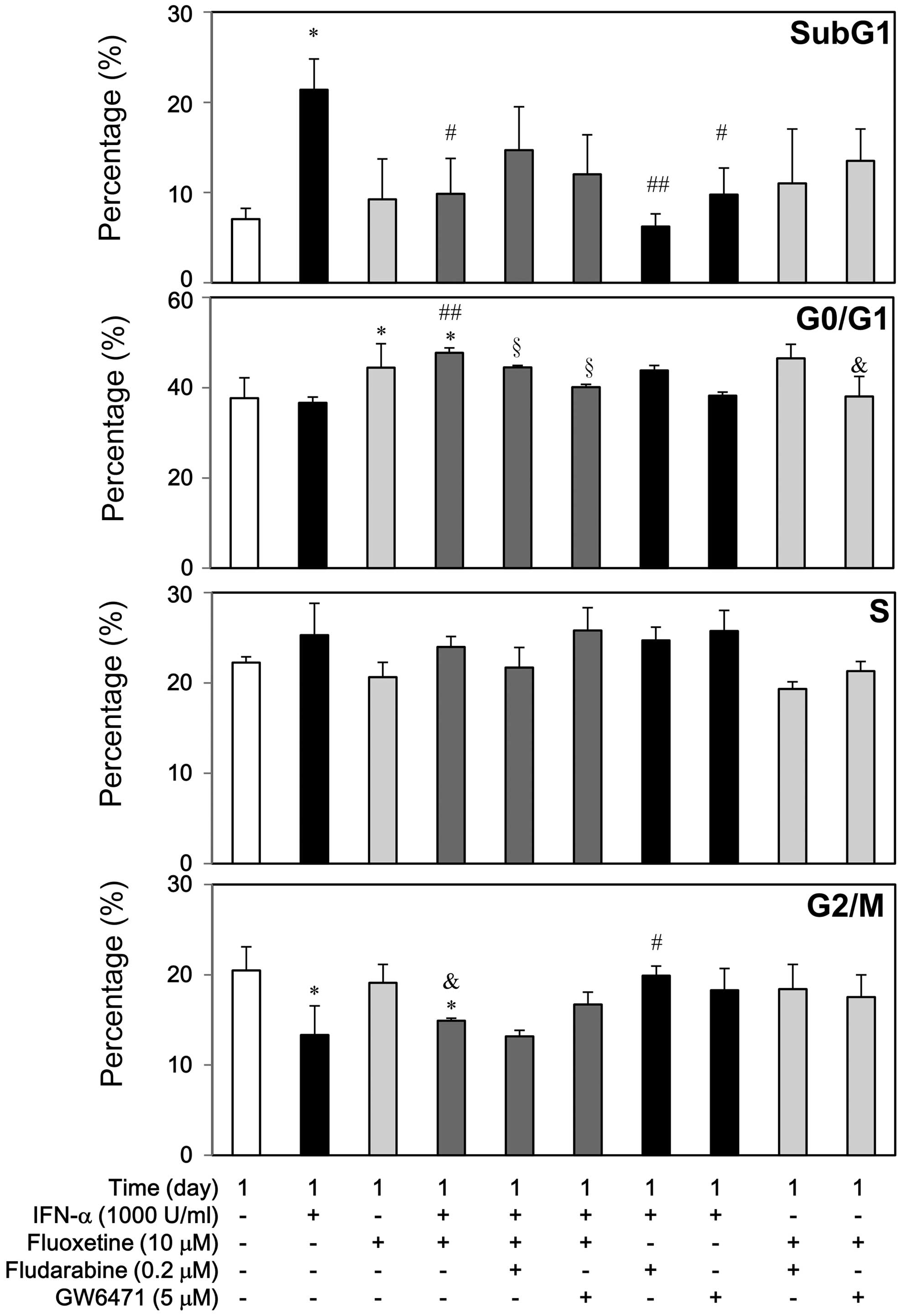
Second, we stained the cells with PI to carry out
flow cytometry and detect changes of cell cycle subpopulations to
examine the cellular mechanisms of IFN-α growth inhibition in the
presence of fluoxetine (Fig. 6).
IFN-α at 1000 U/ml caused apoptosis, slightly increased S phase,
and decreased G2/M phase, but it did not affect G1 phase markedly.
However, fludarabine and GW6471 could reverse the IFN-α-mediated
apoptosis, whereas fludarabine but not GW6471 could reverse the
decreased G2/M phase. On the contrary, fluoxetine at 10 μM caused
G1 arrest without apoptosis, but it did not affect S and G2/M
phase. GW6471 but not fludarabine attenuated the
fluoxetine-mediated G1 arrest.
Compared with the IFN-α-treated group, addition of
fluoxetine attenuated the IFN-α-induced apoptosis significantly,
which was not significantly reversed by fludarabine and GW6471
(Fig. 6). Similarly, IFN-α
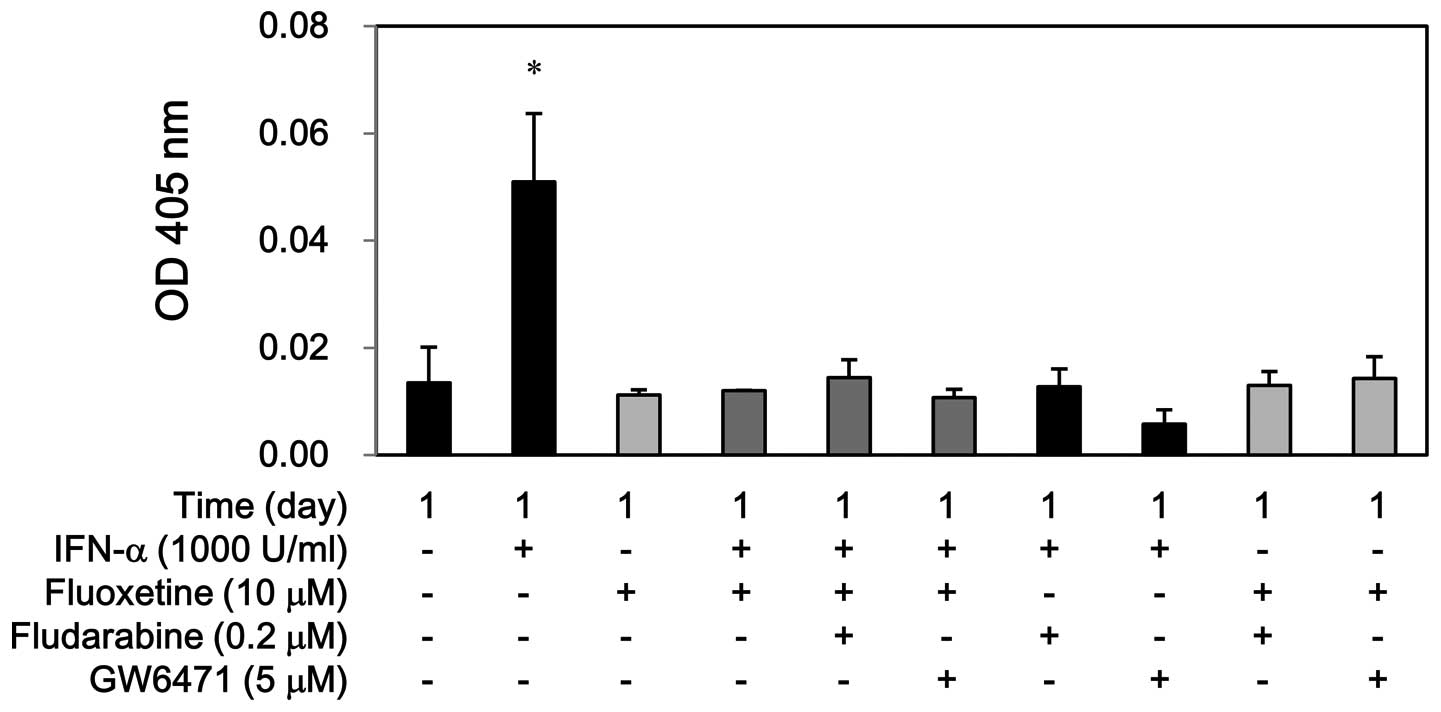
increased the caspase-3 activity at day 1, whereas fluoxetine alone
or in combination with IFN-α did not show this effect. Moreover,
fludarabine and GW6471 hampered the caspase-3 activity by IFN-α but
not by fluoxetine alone or in combination (Fig. 7). Conversely, this co-treatment
also significantly caused G1 arrest, which was partially reversed
by fludarabine and GW6471. Otherwise, fluoxetine did not affect the
S phase and the G2/M reduction of IFN-α (Fig. 6).
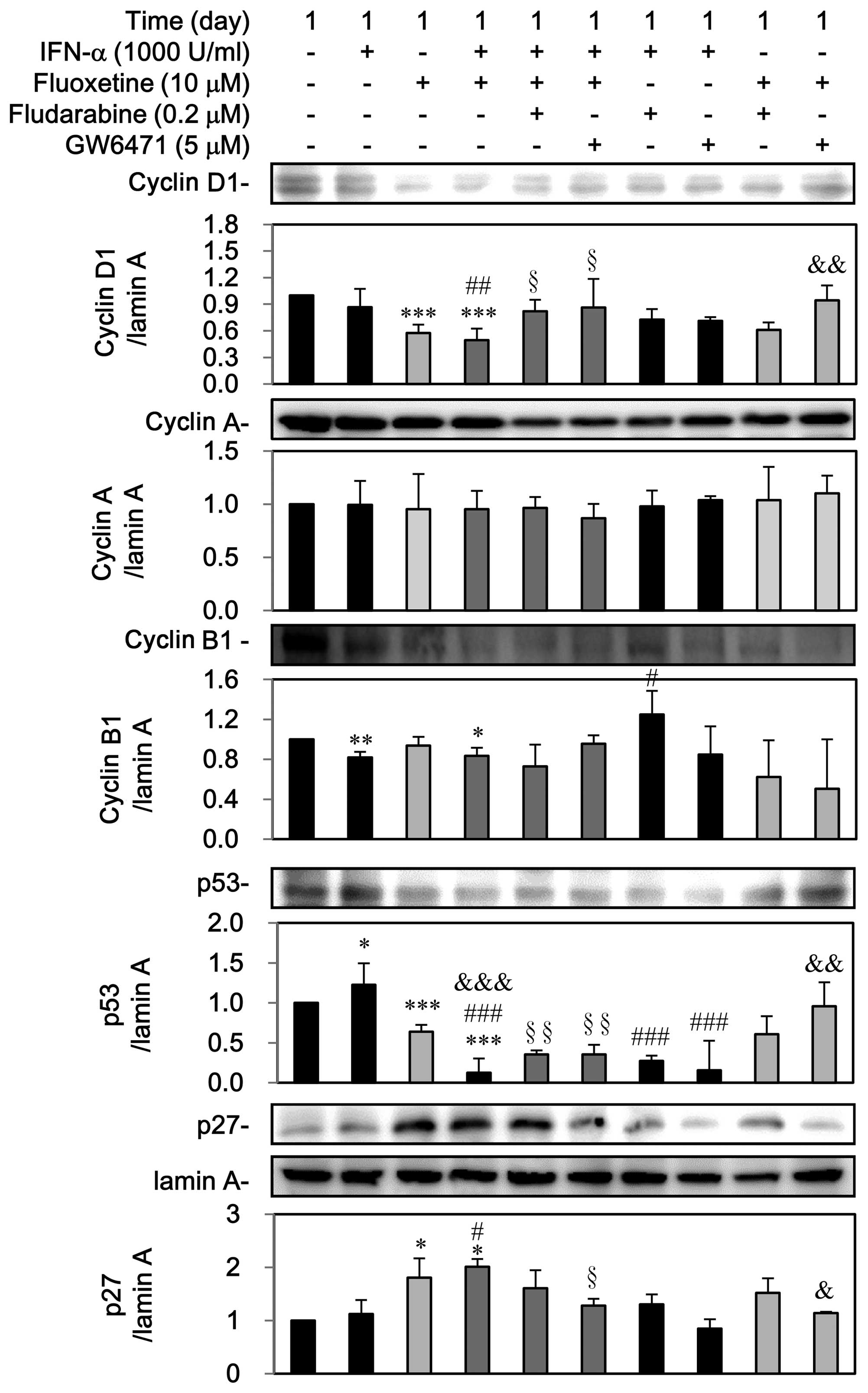
Third, as shown in Fig.
8, IFN-α can decline cyclin B1 but induced p53, which was
reversed by fludarabine. Otherwise, GW6471 can block the
IFN-α-mediated upregulation of p53, however, IFN-α did not notably
affect the expressions of cyclin D1, cyclin A and p27. On the
contrary, fluoxetine itself decreased the expression of cyclin D1
and p53 as well as induced the nuclear p27 expression, which was
reversed by GW6471 but not by fludarabine. However, fluoxetine did
not affect cyclin A and cyclin B1.
A reduction in the expression of cyclin D1, cyclin
B1, and p53 as well as a marked increase of p27, but not cyclin A,
was observed in the group of IFN-α in combination of fluoxetine.
Pretreatment with fludarabine and GW6471 could reverse
downregulations of cyclin D1 and p53 but not cyclin B1, whereas
GW6471 could block the upregulation of p27 by IFN-α combined with
fluoxetine (Fig. 8).
Discussion
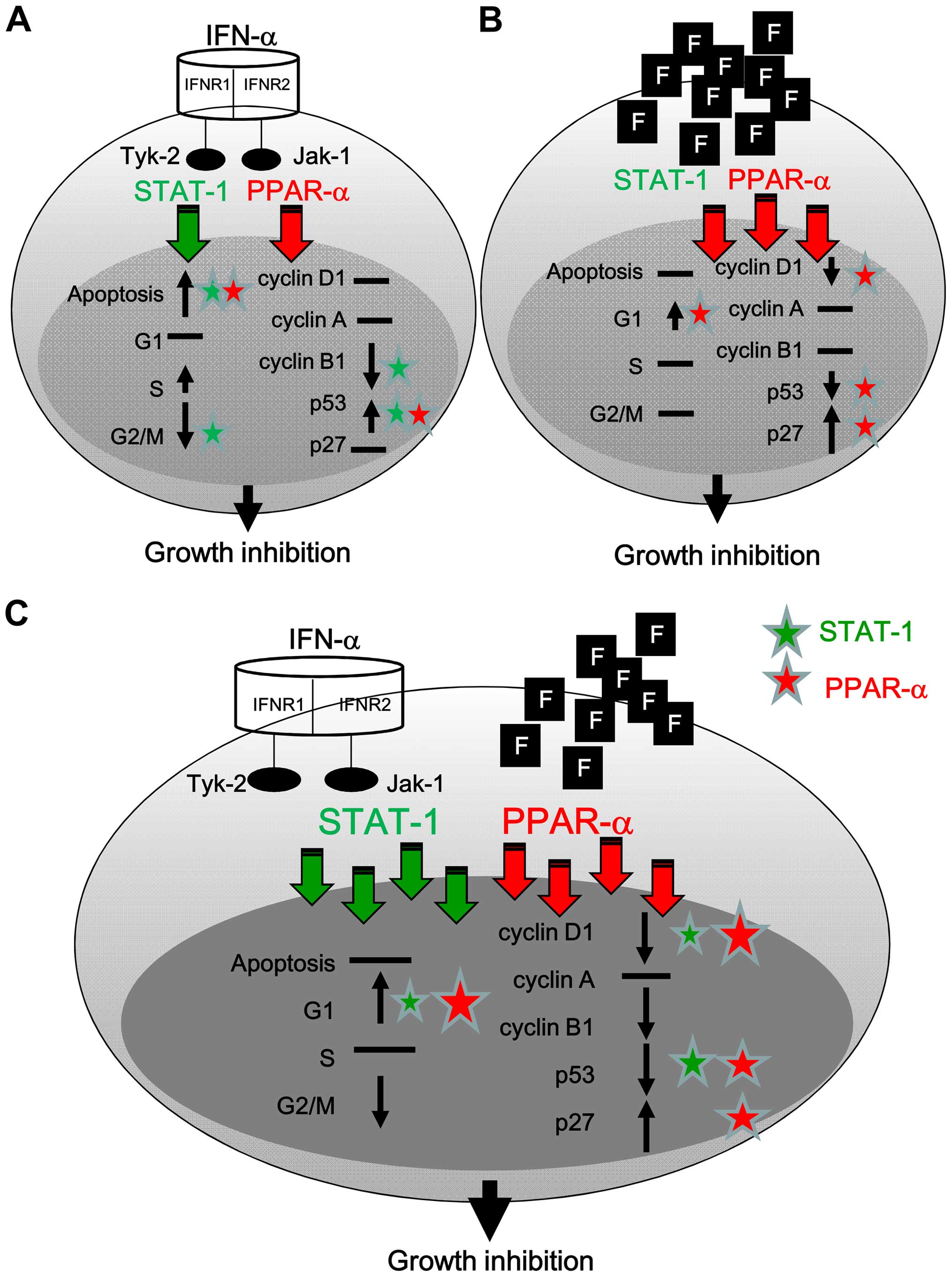
The present study demonstrate that IFN-α can
activate STAT-1 and PPAR-α, translocate to the nucleus, and induce
apoptosis via the induction of p53 (Fig. 9A). Fluoxetine predominately
activated PPAR-α to further cause G1 arrest via the reduction of
cyclin D1 and p53 and induction of p27 (Fig. 9B). The addition of fluoxetine
facilitated the cell growth inhibition of IFN-α and caused cell
arrest via a boosted activation of STAT-1 and PPAR-α accompanied
with the downregulation of cyclin D1 and p53 and the upregulation
of p27 (Fig. 9C).
In the present study, IFN-α caused apoptosis and
blocked the G2/M phase, but did not affect markedly G1 and S phases
of cell cycle accompanied by the induced p53 as well as declined
cyclin B1 and showed no effect on the expression of cyclin D1,
cyclin A and p27. Our results were similar to previous studies with
IFN-α treatment in various cell lines, including hematopoietic (H9)
(21), Ba/F3 pro-B cells (22), and HCC (23); yet, discorded with some reports in
chronic myelogenous leukemia (K562) (24), carcinoid tumor Bon1 (25), and human glioblastoma, U-373MG and
T98G (26). Therefore, the
influence of IFN-α on the cell cycle progression appears to be
variable. These discrepancies depend on the cell types, the length
of time in treatment and the dosage used (1).
In synergistic and human xenograft mouse tumor
models, fluoxetine impedes multidrug resistance extrusion pumps and
enhances the responses to chemotherapy. Thus, fluoxetine may help a
drug to be sensitive to tumor cells and accumulate to a sufficient
level that culminates and undergoes cell growth inhibition
(15,27). In addition to our observation in
T24 cells, similar results of IFN-α in combination with fluoxetine
were observed in human Jurkat-T, Huh7.5 and U2OS cells (data not
shown). Since fluoxetine belongs to the pump-protein of ATP-binding
cassette sub-family B member 1 (also known as P-glycoprotein 1 or
multidrug resistance protein 1), our preliminary results indicated
that IFN-α and fluoxetine alone could inhibit the P-glycoprotein
expression in early stage cancer. Unfortunately, fluoxetine failed
to facilitate the down-regulation of this protein in the presence
of IFN-α (data not shown). Instead, fluoxetine itself caused a
slight G1 arrest along with a marked induction of p27 as well as
slight reduction of cyclin D1 and p53. Our results were similar to
previous studies related to the effect of fluoxetine on G1 arrest
and upregulations of p27 in human cervical cancer cells (SiHa),
breast cancer cells (MDA-MB-231) and colon cancer cells HT29
(28–30). In addition, the dosage of
fluoxetine as a chemosensitizer is kept under the range of 5–20 μM,
where this agent itself does not affect cell viability (15). In the present study, fluoxetine at
10 μM did not induce apoptosis, whereas higher concentrations of
fluoxetine (>25 μM) could suppress glioblastoma cells by
calcium-dependent apoptosis (31).
Alternatively, the addition of fluoxetine lessened the
IFN-α-induced apoptosis but switched to cause G1 arrest, and
maintained the IFN-α-mediated reduction in G2/M phase (Fig. 6). Accordingly, fluoxetine
sensitized the growth inhibition of IFN-α via the alteration of
cell cycle progression, especially G1 arrest.
Activation of STAT-1, especially on Ser727, might
modulate pro- and anti-apoptotic genes upon stress-induced
responses via the caspase-3 dependent pathway. Moreover, STAT-1, a
novel modulator of p53 function, can interact with p53 and induce
apoptosis (32), whereas the
STAT-1-deficient cells are more resistant to apoptosis-inducing
agents (33). In the present
study, the IFN-α-mediated STAT-1 activation caused apoptosis
related to the induction of p53. Alternatively, through the
enhancement of STAT-1 activation, the addition of fluoxetine
lessened the IFN-α-mediated apoptosis and lowered the p53
expression but enhanced G1 arrest related to subsiding expression
of cyclin D1 and upregulation of p27. These results might possibly
be explained by the fact that STAT-1 serine 727 phosphorylation
site can interact directly with cyclin D1, accelerate cyclin
D1-dependent proteosomal degradation, and downregulate cyclin D1,
which further mediates cell cycle arrest (34). Conversely, inhibiting STAT-1 could
reverse the cell growth delay of IFN-α (Fig. 5), but this blockade did not affect
the increased amounts of cell cycle inhibitors p27 of IFN-α in the
presence of fluoxetine (Fig. 8).
These findings were partially consistent with previous a study that
STAT-1 deficient cells proliferated but reduced p27 in human
fibrosarcoma cell lines, 2fTGH and U3A (34).
Besides controlling lipid metabolism, recent
evidence suggests that PPAR-α suppresses apoptosis and induces
proliferation in hepatocytes, in response to peroxisome
proliferators (16,35). Conversely, loss of PPAR-α inhibits
radiation-induced apoptosis in the mouse kidney through the
activation of NF-κB and the upregulation of anti-apoptosis factors
(36). PPAR-α activation also
causes the release of STAT-1 from gene promoters concomitant with
the downregulation of gene expression in human hepatocellular
carcinoma HepG2 cells (17).
IFN-α2b affects the expression of various drug-metabolizing enzymes
and transporters in human primary hepatocytes related to the
upregulation of STAT-1 and PPAR-α-regulated genes (37). However, cross talk between
PPAR-α-mediated survival signaling and cell cycle associated with
STAT-1 activation remains unclear. In the present study,
co-localization of STAT-1 and PPAR-α protein to some extent were
observed in the cytoplasm by IFN-α or fluoxetine alone. We also
found that IFN-α-treated cells partially activated and translocated
both proteins to the nucleus, whereas fluoxetine-treated cells
predominantly triggered PPAR-α translocation to the nucleus.
Notably, the addition of fluoxetine to IFN-α-treated cells caused
significant co-localization of both proteins in the nucleus
(Fig. 4), which mediated cell
growth inhibition.
In conclusion, STAT-1 and PPAR-α might contribute
distinct functions in cell cycle progression to achieve the growth
inhibition of IFN-α, fluoxetine, or in combination. Moreover,
fluoxetine regulates cell growth inhibition of IFN-α via a boosted
activation of STAT-1 and PPAR-α.
Acknowledgements
The present study was supported by the Shin Kong Wu
Ho-Su Memorial Hospital (SKH-8302-103-DR-09 and
SKH-8302-103-DR-10), the Ministry of Education, and the Chi-Mei
Medical Center, Taiwan. We are also grateful for the support from
the Core Research Laboratory, College of Medicine, National Cheng
Kung University, Tainan, Taiwan, R.O.C.
References
|
1
|
Caraglia M, Marra M, Pelaia G, Maselli R,
Caputi M, Marsico SA and Abbruzzese A: Alpha-interferon and its
effects on signal transduction pathways. J Cell Physiol.
202:323–335. 2005. View Article : Google Scholar
|
|
2
|
Santhanam S, Decatris M and O’Byrne K:
Potential of interferon-alpha in solid tumours: Part 2. BioDrugs.
16:349–372. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Caraglia M, Vitale G, Marra M, Budillon A,
Tagliaferri P and Abbruzzese A: Alpha-interferon and its effects on
signalling pathways within cells. Curr Protein Pept Sci. 5:475–485.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Caraglia M, Vitale G, Marra M, Del Prete
S, Lentini A, Budillon A, Beninati S and Abbruzzese A:
Translational and post-translational modifications of proteins as a
new mechanism of action of alpha-interferon: Review article. Amino
Acids. 26:409–417. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Inamura K, Matsuzaki Y, Uematsu N, Honda
A, Tanaka N and Uchida K: Rapid inhibition of MAPK signaling and
anti-proliferation effect via JAK/STAT signaling by
interferon-alpha in hepatocellular carcinoma cell lines. Biochim
Biophys Acta. 1745:401–410. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Romerio F, Riva A and Zella D:
Interferon-alpha2b reduces phosphorylation and activity of MEK and
ERK through a Ras/Raf-independent mechanism. Br J Cancer.
83:532–538. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee WH, Liu FH, Lee YL and Huang HM:
Interferon-alpha induces the growth inhibition of human T-cell
leukaemia line Jurkat through p38alpha and p38beta. J Biochem.
147:645–650. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bazarbachi A, El-Sabban ME, Nasr R,
Quignon F, Awaraji C, Kersual J, Dianoux L, Zermati Y, Haidar JH,
Hermine O, et al: Arsenic trioxide and interferon-alpha synergize
to induce cell cycle arrest and apoptosis in human T-cell
lymphotropic virus type I-transformed cells. Blood. 93:278–283.
1999.
|
|
9
|
Eriksen KW, Søndergaard H, Woetmann A,
Krejsgaard T, Skak K, Geisler C, Wasik MA and Odum N: The
combination of IL-21 and IFN-alpha boosts STAT3 activation,
cytotoxicity and experimental tumor therapy. Mol Immunol.
46:812–820. 2009. View Article : Google Scholar
|
|
10
|
Kondo M, Nagano H, Wada H, Damdinsuren B,
Yamamoto H, Hiraoka N, Eguchi H, Miyamoto A, Yamamoto T, Ota H, et
al: Combination of IFN-alpha and 5-fluorouracil induces apoptosis
through IFN-alpha/beta receptor in human hepatocellular carcinoma
cells. Clin Cancer Res. 11:1277–1286. 2005.PubMed/NCBI
|
|
11
|
Fishman AI, Johnson B, Alexander B, Won J,
Choudhury M and Konno S: Additively enhanced antiproliferative
effect of interferon combined with proanthocyanidin on bladder
cancer cells. J Cancer. 3:107–112. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rodin G, Lloyd N, Katz M, Green E, Mackay
JA and Wong RK; Supportive Care Guidelines Group of Cancer Care
Ontario Program in Evidence-Based Care. The treatment of depression
in cancer patients: A systematic review. Support Care Cancer.
15:123–136. 2007. View Article : Google Scholar
|
|
13
|
Sockalingam S and Abbey SE: Managing
depression during hepatitis C treatment. Can J Psychiatry.
54:614–625. 2009.PubMed/NCBI
|
|
14
|
Serafeim A, Holder MJ, Grafton G, Chamba
A, Drayson MT, Luong QT, Bunce CM, Gregory CD, Barnes NM and Gordon
J: Selective serotonin reuptake inhibitors directly signal for
apoptosis in biopsy-like Burkitt lymphoma cells. Blood.
101:3212–3219. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Peer D and Margalit R: Fluoxetine and
reversal of multidrug resistance. Cancer Lett. 237:180–187. 2006.
View Article : Google Scholar
|
|
16
|
Roberts RA, Chevalier S, Hasmall SC, James
NH, Cosulich SC and Macdonald N: PPAR alpha and the regulation of
cell division and apoptosis. Toxicology. 181–182:167–170. 2002.
View Article : Google Scholar
|
|
17
|
van der Meer DL, Degenhardt T, Väisänen S,
de Groot PJ, Heinäniemi M, de Vries SC, Müller M, Carlberg C and
Kersten S: Profiling of promoter occupancy by PPARalpha in human
hepatoma cells via ChIP-chip analysis. Nucleic Acids Res.
38:2839–2850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fryknäs M, Dhar S, Oberg F, Rickardson L,
Rydåker M, Göransson H, Gustafsson M, Pettersson U, Nygren P,
Larsson R, et al: STAT1 signaling is associated with acquired
crossresistance to doxorubicin and radiation in myeloma cell lines.
Int J Cancer. 120:189–195. 2007. View Article : Google Scholar
|
|
19
|
Torella D, Curcio A, Gasparri C, Galuppo
V, De Serio D, Surace FC, Cavaliere AL, Leone A, Coppola C, Ellison
GM, et al: Fludarabine prevents smooth muscle proliferation in
vitro and neointimal hyperplasia in vivo through specific
inhibition of STAT-1 activation. Am J Physiol Heart Circ Physiol.
292:H2935–H2943. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao L, Huang Y, Tian C, Taylor L,
Curthoys N, Wang Y, Vernon H and Zheng J: Interferon-αregulates
glutaminase 1 promoter through STAT1 phosphorylation: Relevance to
HIV-1 associated neurocognitive disorders. PLoS One. 7:e329952012.
View Article : Google Scholar
|
|
21
|
Sangfelt O, Erickson S, Castro J, Heiden
T, Gustafsson A, Einhorn S and Grandér D: Molecular mechanisms
underlying interferon-alpha-induced G0/G1 arrest: CKI-mediated
regulation of G1 Cdk-complexes and activation of pocket proteins.
Oncogene. 18:2798–2810. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Prietzsch H, Brock J, Kleine HD, Liebe S
and Jaster R: Interferon-alpha inhibits cell cycle progression by
Ba/F3 cells through the antagonisation of interleukin-3 effects on
key regulators of G1/S transition. Cell Signal.
14:751–759. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maeda S, Wada H, Naito Y, Nagano H,
Simmons S, Kagawa Y, Naito A, Kikuta J, Ishii T, Tomimaru Y, et al:
Interferon-α acts on the S/G2/M phases to induce apoptosis in the
G1 phase of an IFNAR2-expressing hepatocellular carcinoma cell
line. J Biol Chem. 289:23786–23795. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Grebenová D, Kuzelová K, Fuchs O, Halada
P, Havlícek V, Marinov I and Hrkal Z: Interferon-alpha suppresses
proliferation of chronic myelogenous leukemia cells K562 by
extending cell cycle S-phase without inducing apoptosis. Blood
Cells Mol Dis. 32:262–269. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou Y, Wang S, Yue BG, Gobl A and Oberg
K: Effects of interferon alpha on the expression of p21cip1/waf1
and cell cycle distribution in carcinoid tumors. Cancer Invest.
20:348–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tanabe T, Kominsky SL, Subramaniam PS,
Johnson HM and Torres BA: Inhibition of the glioblastoma cell cycle
by type I IFNs occurs at both the G1 and S phases and correlates
with the upregulation of p21WAF1/CIP1. J Neurooncol.
48:225–232. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peer D, Dekel Y, Melikhov D and Margalit
R: Fluoxetine inhibits multidrug resistance extrusion pumps and
enhances responses to chemotherapy in syngeneic and in human
xenograft mouse tumor models. Cancer Res. 64:7562–7569. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kannen V, Hintzsche H, Zanette DL, Silva
WA Jr, Garcia SB, Waaga-Gasser AM and Stopper H: Antiproliferative
effects of fluoxetine on colon cancer cells and in a colonic
carcinogen mouse model. PLoS One. 7:e500432012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stopper H, Garcia SB, Waaga-Gasser AM and
Kannen V: Antidepressant fluoxetine and its potential against colon
tumors. World J Gastrointest Oncol. 6:11–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Krishnan A, Hariharan R, Nair SA and
Pillai MR: Fluoxetine mediates G0/G1 arrest by inducing functional
inhibition of cyclin dependent kinase subunit (CKS)1. Biochem
Pharmacol. 75:1924–1934. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu KH, Yang ST, Lin YK, Lin JW, Lee YH,
Wang JY, Hu CJ, Lin EY, Chen SM, Then CK, et al: Fluoxetine, an
antidepressant, suppresses glioblastoma by evoking AMPAR-mediated
calcium-dependent apoptosis. Oncotarget. 6:5088–5101. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stephanou A and Latchman DS: STAT-1: A
novel regulator of apoptosis. Int J Exp Pathol. 84:239–244. 2003.
View Article : Google Scholar
|
|
33
|
Lee CK, Smith E, Gimeno R, Gertner R and
Levy DE: STAT1 affects lymphocyte survival and proliferation
partially independent of its role downstream of IFN-gamma. J
Immunol. 164:1286–1292. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dimco G, Knight RA, Latchman DS and
Stephanou A: STAT1 interacts directly with cyclin D1/Cdk4 and
mediates cell cycle arrest. Cell Cycle. 9:4638–4649. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Roberts RA, James NH, Woodyatt NJ,
Macdonald N and Tugwood JD: Evidence for the suppression of
apoptosis by the peroxisome proliferator activated receptor alpha
(PPAR alpha). Carcinogenesis. 19:43–48. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao W, Iskandar S, Kooshki M, Sharpe JG,
Payne V and Robbins ME: Knocking out peroxisome
proliferator-activated receptor (PPAR) alpha inhibits
radiation-induced apoptosis in the mouse kidney through activation
of NF-kappaB and increased expression of IAPs. Radiat Res.
167:581–591. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen C, Han YH, Yang Z and Rodrigues AD:
Effect of interferon-α2b on the expression of various
drug-metabolizing enzymes and transporters in co-cultures of
freshly prepared human primary hepatocytes. Xenobiotica.
41:476–485. 2011. View Article : Google Scholar : PubMed/NCBI
|