Introduction
Recent studies demonstrated that the novel
cyclin-dependent kinase (cdk) inhibitor,
2-[1,1′biphenyl]-4-yl-N-[5-(1,1-dioxo-1λ6-isothiazolidin-2-yl)-1H-indazol-3-yl]acetamide
(BAI) has anticancer effects on various cancer cell lines,
including A549, HCT116, Caki, AMC-HN 4, and AMC-HN-6 (1–3).
Previous studies showed that BAI downregulated Mcl-1(L) at
transcriptional level (2) and B
cell lymphoma-2 (Bcl-2) (4).
However, there are still major gaps in the understanding of BAI,
particularly in terms of its apoptotic mechanisms.
Apoptosis, as an important mechanism of programmed
cell death, is preserved among multi-cellular organisms and
involved in various biological processes including development,
maintenance of tissue homeostasis, and elimination of unwanted or
damaged cells (5,6). There are two major pathways of
apoptosis: the pathway triggered via ligand-binding to the cell
surface death receptors such as Fas (the extrinsic pathway) and the
pathway in which pro-apoptotic Bcl-2 family proteins mediate the
permeabilization of the mitochondrial outer membrane (the intrinsic
pathway) (7). Bcl-2 family members
regulate life/cell death, primarily via interactions between anti-
and pro-apoptotic members (8). For
example, PUMA, unique among BH3-only proteins, functions as a major
mediator of pro-apoptotic p53 function by disrupting the
interaction of p53/Bcl-xL, resulting in apoptosis (9,10).
Therefore, modulating these proteins, such as Bcl-2 family
proteins, PUMA, and p53, is a very promising therapeutic strategy
in the development of compounds for anticancer effects.
In this study, we investigated the underlying
mechanisms of Bcl-2 family proteins involved in BAI-induced
apoptosis in human cancer cells. Our results revealed that the
downregulation of Bcl-xL and the modulations of interactions among
p53 and Bcl-2 family proteins may be involved in BAI-induced
apoptosis in human cancer cells.
Materials and methods
Cell lines and culture
A549 human non-small cell lung cancer cells and
HCT116 human colorectal carcinoma cells were obtained from the
American Type Culture Collection (ATCC, Rockville, MD, USA) and
grown in RPMI-1640 medium (WelGENE Inc., Daegu, Korea) supplemented
with 10% heat-inactivated fetal bovine serum (FBS), 2 mM
L-glutamine, 100 μg/ml streptomycin and 100 μg/ml penicillin. Caki
human renal clear cell carcinoma cells were obtained from the ATCC
and grown in Dulbecco’s modified Eagle’s medium (DMEM), containing
10% heat-inactivated FBS, 20 mM HEPES buffer and 100 μg/ml
streptomycin and 100 μg/ml penicillin.
Drugs and materials
2-[1,1′-biphenyl]-4-yl-N-[5-(1,1-dioxo-1λ6-isothiazolidin-2-yl)-1H-indazol-3-yl]acetamide
(BAI) was kindly supplied by Dr J.H. Lee (Keimyung University,
Daegu, Korea). Anti-Bcl-xL (sc-634, 1:700), anti-AIF (sc-5586,
1:700), anti-p53 (sc-126, 1:1,000), anti-PUMA (sc-19187, 1:700),
anti-cytochrome c oxidase subunit II (sc-23983, 1:700), and
anti-Bcl-2 (sc-783, 1:700) antibodies were purchased from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). Anti-β-actin (A5441,
1:2,000) antibody was purchased from Sigma Chemical Co. (St. Louis,
MO, USA). Anti-poly(ADP-ribose) polymerase (PARP) (#9542, 1:1,000)
antibody was purchased from Cell Signaling Technology (Danvers, MA,
USA). Anti-caspase-3 (610322, 1:1,000), anticytochrome c
(556433, 1:700), and anti-Bax (554104, 1:700) antibodies were
purchased from BD Biosciences (Bedford, MA, USA). Benzyloxy
carbony-Val-Ala-Asp-fluoromethyl ketone (z-VAD-fmk) was purchased
from R&D Systems (Minneapolis, MN, USA). PD-98059 (MEK
inhibitor, PD), SP600125 (JNK inhibitor, SP), and SB-203580 (p38
MAP kinase inhibitor, SB) were purchased from Enzo Life Sciences
(Farmingdale, NY, USA).
Western blot analysis
Cellular lysates were prepared by suspending
0.3×106 cells in 80 μl of lysis buffer (137 mM NaCl, 15
mM EGTA, 0.1 mM sodium orthovanadate, 15 mM MgCl2, 0.1%
Triton X-100, 25 mM MOPS, 100 μM phenylmethylsulfonyl fluoride and
20 μM leupeptin, adjusted to pH 7.2). The cells were disrupted by
vortexing and extracted at 4°C for 30 min. The proteins were
electrotransferred to Immobilon-P membranes (Millipore Corp.,
Bedford, MA, USA). Detection of specific proteins was carried out
with an ECL Western blotting kit according to the manufacturer’s
instructions (Millipore Corp.).
Cell viability assay
The anti-proliferative effect of the BAI on cancer
cells was investigated using a live cell movie analyzer, JuLI™ Br
(NanoEnTek Inc., Seoul, Korea). Briefly, the cells were plated in
6-well culture plates at a density of 0.3×106 cells/well
in medium and allowed to attach for 10 h. The cells treated with
BAI for 24 h. During this study, JuLi Br recorded images of the
cells at 5 min intervals, and confluences were also measured.
Flow cytometric analysis
Approximately 0.5×106 cells were
suspended in 100 μl PBS, and 200 μl of 95% ethanol was added while
vortexing. The cells were incubated at 4°C for 1 h, washed with
PBS, and resuspended in 250 μl of 1.12% sodium citrate buffer (pH
8.4) together with 12.5 μg RNase. Incubation was continued at 37°C
for 30 min. The cellular DNA was then stained by applying 250 μl
propidium iodide (50 μg/ml) for 30 min at room temperature. The
stained cells were analyzed by a FACScan flow cytometer for
relative DNA content based on red fluorescence.
DEVDase activity assay
To evaluate caspase-3 activity, cell lysates were
prepared after their respective treatment with various drugs.
Assays were performed in 96-well microtiter plates by incubating 20
μg cell lysates in 100 μl reaction buffer [1% NP-40, 20 mM Tris-HCl
(pH 7.5), 137 mM NaCl, and 10% glycerol)] containing the caspase 3
substrate (DEVD-pNA) at 5 μM. Lysates were incubated at 37°C for 2
h. Thereafter, the absorbance at 405 nm was measured with a
spectrophotometer.
RNA isolation and quantitative real-time
PCR
Total cellular RNA was extracted from tissues using
the TRIzol reagent (Molecular Research Center, Inc., Cincinnati,
OH, USA). RNA was quantified using Nanodrop 1000 (Thermo
Scientific, Wilmington, DE, USA). Each cDNA was synthesized from 2
μg of total RNA using M-MLV reverse transcriptase (Promega,
Madison, WI, USA) according to the manufacturer’s protocol. By
using the specific primer pairs described in Table I and SYBR Green Premix (Toyobo,
Japan). Quantitative real-time PCR (qPCR) was performed on the
LightCycler® 480 real-time PCR system (Roche
Diagnostics, Mannheim, Germany). β-actin was used as a housekeeping
gene for normalization, and no-template sample was used as a
negative control. Then, the qPCR data were analyzed by the
2−ΔΔct method (11).
 | Table IPrimer sequences of miRNA machinery
components used in quantitative PCR. |
Table I
Primer sequences of miRNA machinery
components used in quantitative PCR.
| Components | Position | Sequences |
|---|
| Bcl-2 | Forward |
5′-GCCTTCTTTGAGTTCGGTGG-3′ |
| Reverse |
5′-ATCTCCCGGTTGACGCTCT-3′ |
| Bcl-xL | Forward |
5′-GGTCGCATTGTGGCCTTT-3′ |
| Reverse |
5′-TCCTTGTCTACGCTTTCCACG-3′ |
| β-actin | Forward |
5′-CAGCCATGTACGTTGCTATCCAGG-3′ |
| Reverse |
5′-AGGTCCAGACGCAGGATGGCATG-3′ |
Determination of the mitochondrial
membrane potential by rhodamine 123
Rhodamine 123 (Invitrogen, Molecular Probes, Inc.,
Eugene, OR, USA) uptake by mitochondria is directly proportional to
its membrane potential. Caki cells subjected to 2 h after treatment
were incubated with rhodamine 123 (20 μM) for 10 min in the dark at
37°C. The cells were harvested and suspended in PBS. The
mitochondrial membrane potential was subsequently analyzed using a
flow cytometer (BD Bioscience).
Analysis of mitochondrial cytochrome c
release
Approximately 0.3×106 Caki cells were
harvested, washed once with ice-cold PBS and gently lysed for 2 min
in 80 μl ice-cold lysis buffer (250 mM sucrose, 1 mM EDTA, 20 mM
Tris-HCl pH 7.2, 1 mM DTT, 10 mM KCl, 1.5 mM MgCl2, 5
μg/ml pepstatin A, 10 μg/ml leupeptin, 2 μg/ml aprotinin). Lysates
were centrifuged at 12,000 g at 4°C for 10 min to obtain the
supernatants (cytosolic extracts free of mitochondria) and the
pellets (fraction that contains mitochondria). Cytosolic protein
(30 μg) was resolved on 12% SDS-PAGE and then transferred to
nitrocellulose, and probed with specific anti-cytochrome c
antibody.
Assay for Bax oligomerization
The cells were suspended by conjugation buffer (PBS
with 10 mM EDTA). The cell lysates were incubated with 0.2 mM
bismaleimide (Thermo Scientific, Hudson, NH, USA) at room
temperature for 1 h and then extracted by lysis buffer for western
blot analysis.
Co-immunoprecipitation assays
Caki cells were exposed to 60 nM BAI for the
indicated time periods and cell lysates were prepared in 1× RIPA
buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM
Na2EDTA, 1% NP-40, 1% deoxycholate, 2.5 mM sodium
pyrophosphate, 1 mM β-glycerophosphate, 1 mM
Na3VO4, 1 mg/ml leupeptin, #9806, Cell
Signaling Technology]. The cells were disrupted by sonication and
centrifuged (13,000 rpm) at 4°C for 15 min. Cell lysates were then
subjected to immunoprecipitation with an anti-Bcl-xL antibody.
Protein G PLUS-agarose were added and then the cell lysates were
rotated at 4°C for 2 h. The cell lysates were centrifuged (13,000
rpm) at 4°C for 10 min. The presence of p53 and PUMA in the
anti-Bcl-xL immunoprecipitate (IPs) and lysates was then evaluated
by immunoblot analysis using the specific antibodies.
Statistical analysis
The data were analyzed using a one-way ANOVA
followed by post-hoc comparisons (Student-Newman-Keuls) using the
Statistical Package for Social Sciences version 22.0 (SPSS Inc.,
Chicago, IL, USA).
Results
BAI has anti-proliferative effects on
various human cancer cells
Previous reports demonstrated that BAI induces
apoptosis of various human cancer cell lines. To investigate the
anticancer effects of BAI in detail, we first analyzed the growth
inhibitory effect of BAI in the same human cancer cell lines using
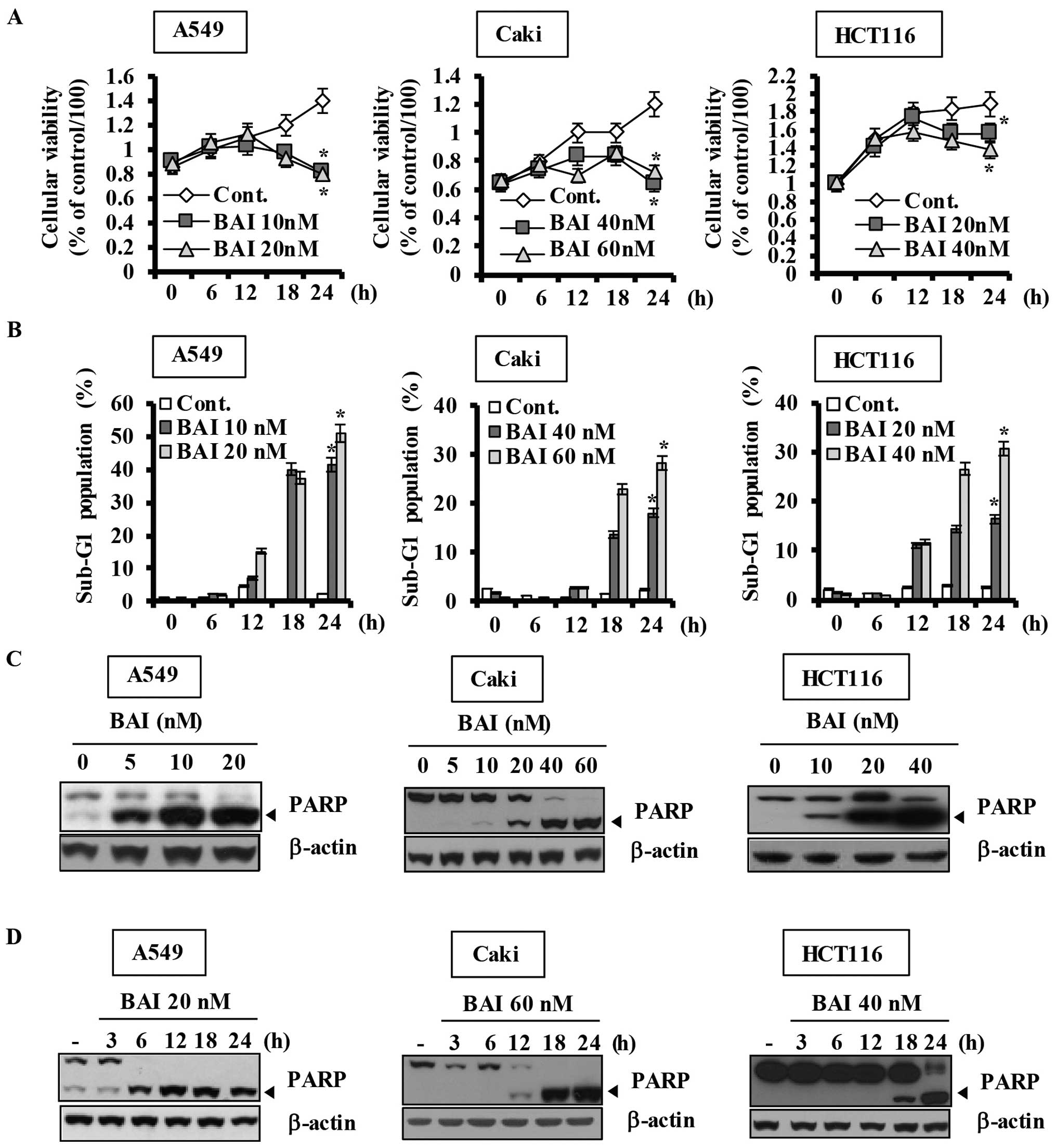
an automated cell counter. As shown in Fig. 1A, BAI markedly inhibited
proliferation of various human cancer cell lines dose- and
time-dependently. To examine the apoptotic effects of BAI, the
cells were next treated with various concentrations of BAI for the
indicated times and then apoptosis was assessed using flow
cytometry to detect hypodiploid cell populations. Treatment of the
cells with BAI resulted in a remarkably increased accumulation of
cells in the sub-G1 population and an increase in PARP cleavage in
a dose-dependent manner (Fig. 1B and
C) and a time-dependent manner (Fig. 1B and D). Furthermore, BAI induced
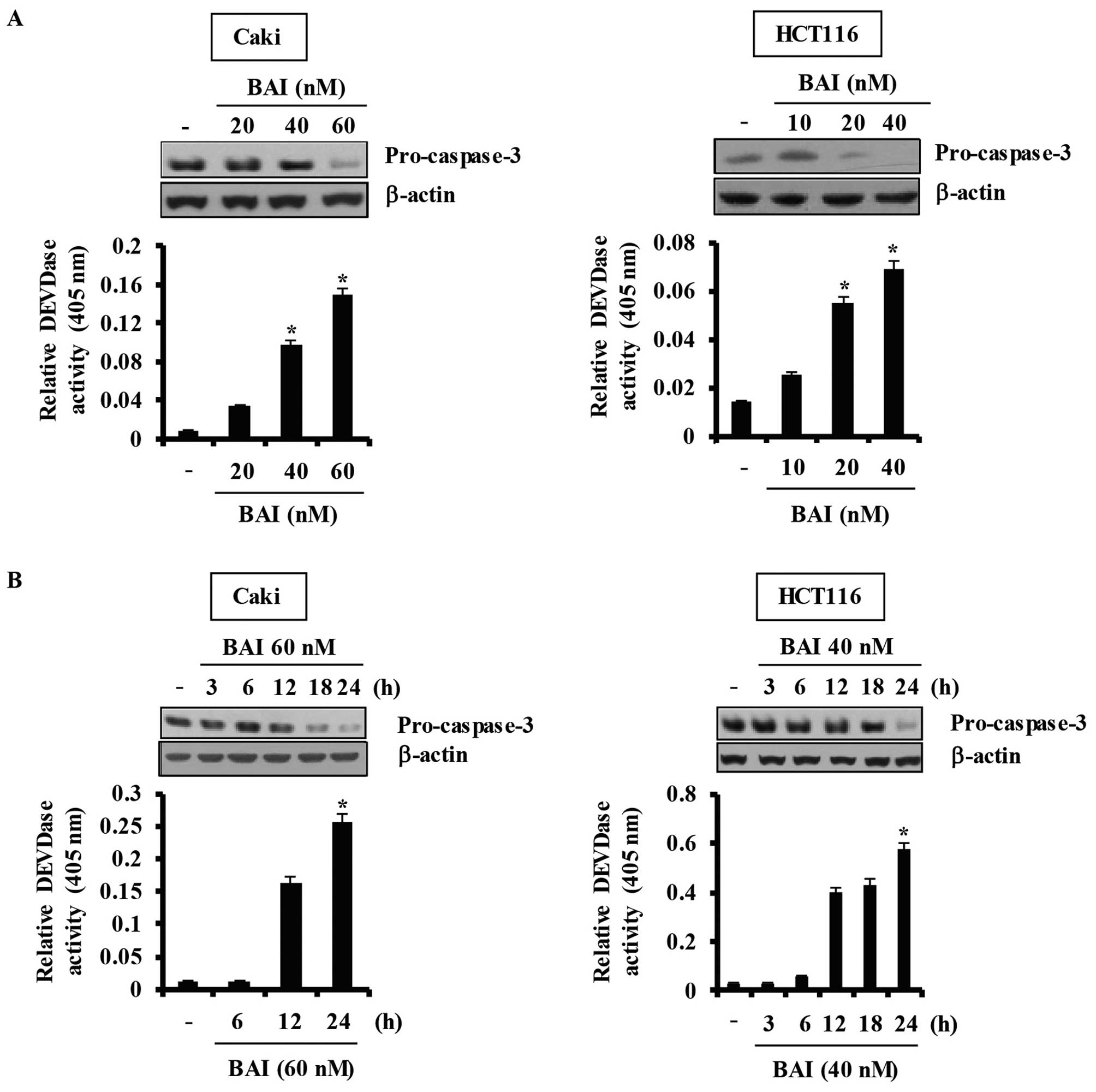
caspase-dependent apoptosis in various cancer cell lines, including
A549, HCT116, and Caki, in a dose- and time-dependent manner
(Fig. 2). Taken together, these
data demonstrate that BAI induces caspase-3-dependent
apoptosis.
The roles of specific apoptosis-related
pathways in BAI-induced apoptosis: MAPKase pathways or ROS
generation
Mitogen-activated protein kinases (MAPKs) are key
participants in cell proliferation, survival, and differentiation
(12,13). To explore the signaling events
regulated during BAI-induced apoptosis, we used specific
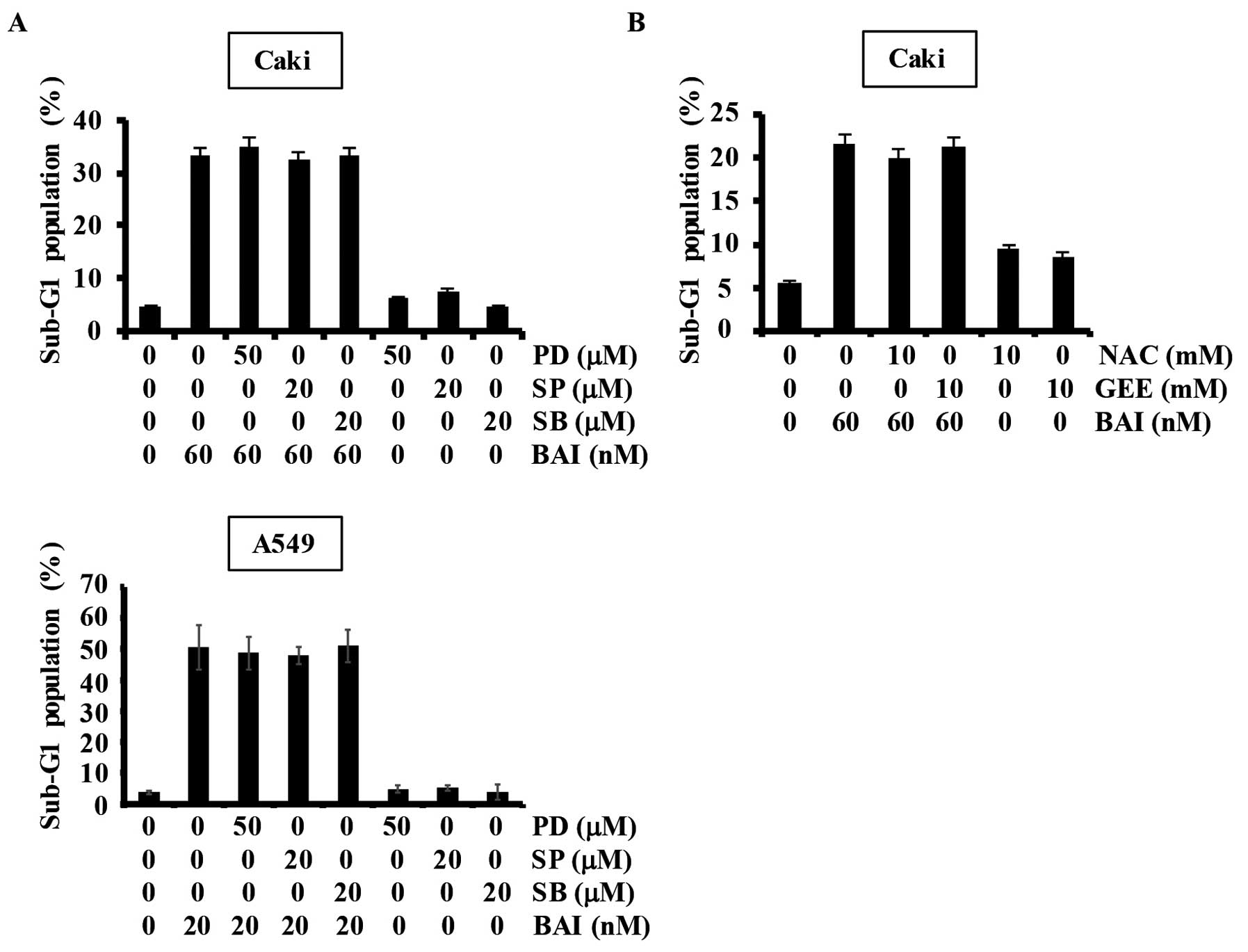
inhibitors. Our results showed that specific MAPK inhibitors (PD,
MEK inhibitor; SP, JNK inhibitor; SB, p38 MAPK inhibitor) did not
affect BAI-induced apoptosis in Caki and A549 cells (Fig. 3A). Reactive oxygen species (ROS),
natural byproducts of the normal metabolism of oxygen, play a
crucial role in apoptosis under both physiologic and pathologic
processes (14). Therefore, we
investigated whether ROS generation is involved in BAI-induced
apoptosis in Caki cells. As shown in Fig. 3B, BAI-induced apoptosis was not
attenuated by pretreatment with N-acetylcysteine (NAC) or
glutathione (GEE). These data indicate that BAI-induced apoptosis
is not associated with MAPK pathways or ROS generation.
BAI reduces mitochondrial membrane
potential (MMP) and induces Bax activation
In general, apoptosis induction is correlated with,
and probably mediated by, perturbations of mitochondrial function,
a manifestation of which is the dissipation of the transmembrane
potential (ΔΨm). Therefore, we evaluated ΔΨm
during apoptosis induction in BAI-treated human cancer cells. As
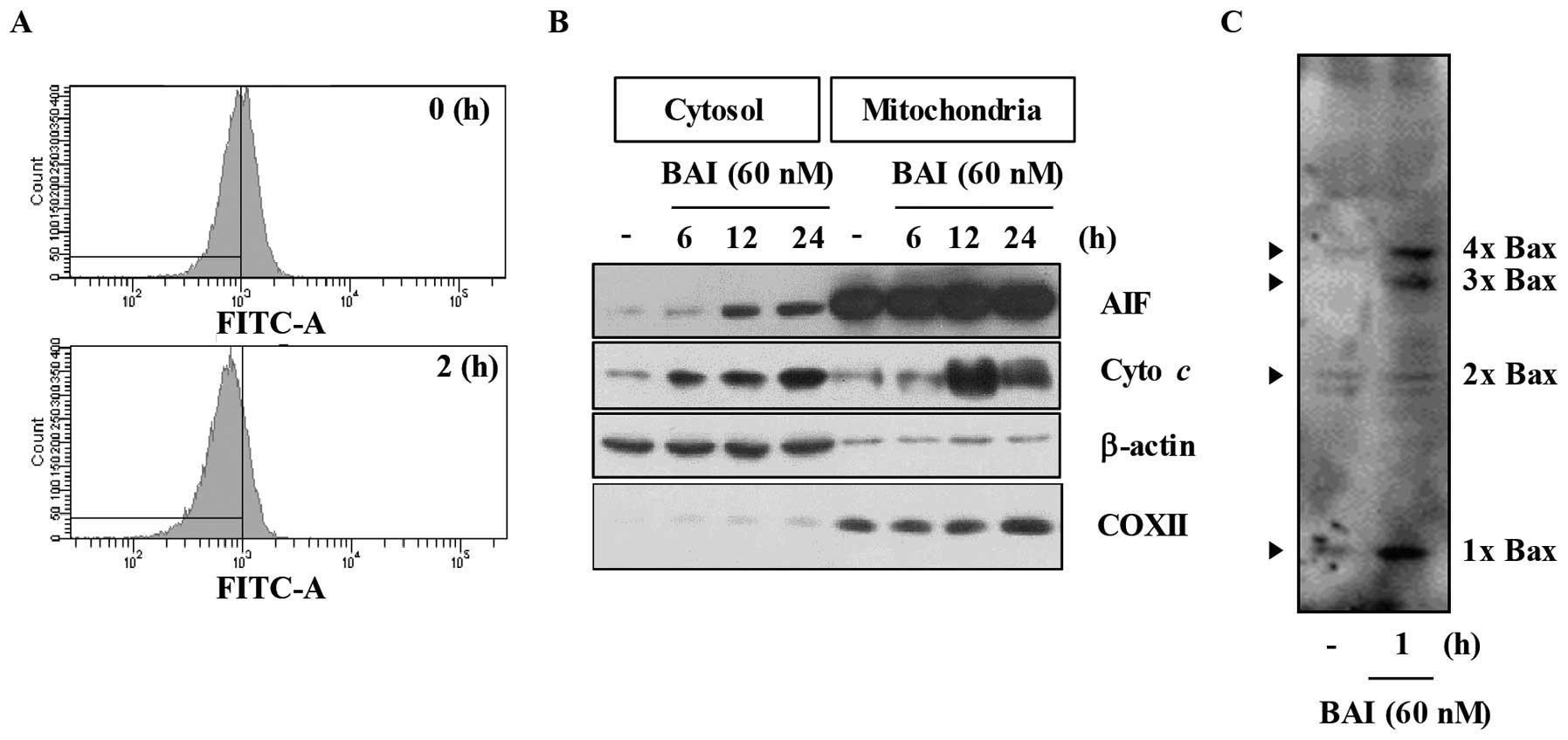
shown in Fig. 4A, treatment with
BAI markedly decreased ΔΨm in Caki cells. Mitochondria
mediates apoptosis by releasing apoptogenic effectors such as
cytochrome c and apoptosis-inducing factor (AIF) (15,16).
As shown in Fig. 4B, BAI
remarkably induced time-dependent release of cytochrome c
and AIF into the cytoplasm in Caki cells. Several lines of evidence
strongly support the notion that activation of the pro-apoptotic
Bcl-2 protein, Bax, plays a critical role in apoptosis by changes
of MMP levels and release of cytochrome c (17). Therefore, we next evaluated the
effect of BAI on Bax activation. As shown in Fig. 4C, BAI markedly promoted Bax
oligomerization. Taken together, these results suggest that BAI
induces loss of MMP levels and release of cytochrome c
through activation of Bax.
Downregulation of Bcl-2 is not associated
with BAI-induced apoptosis in Caki cells
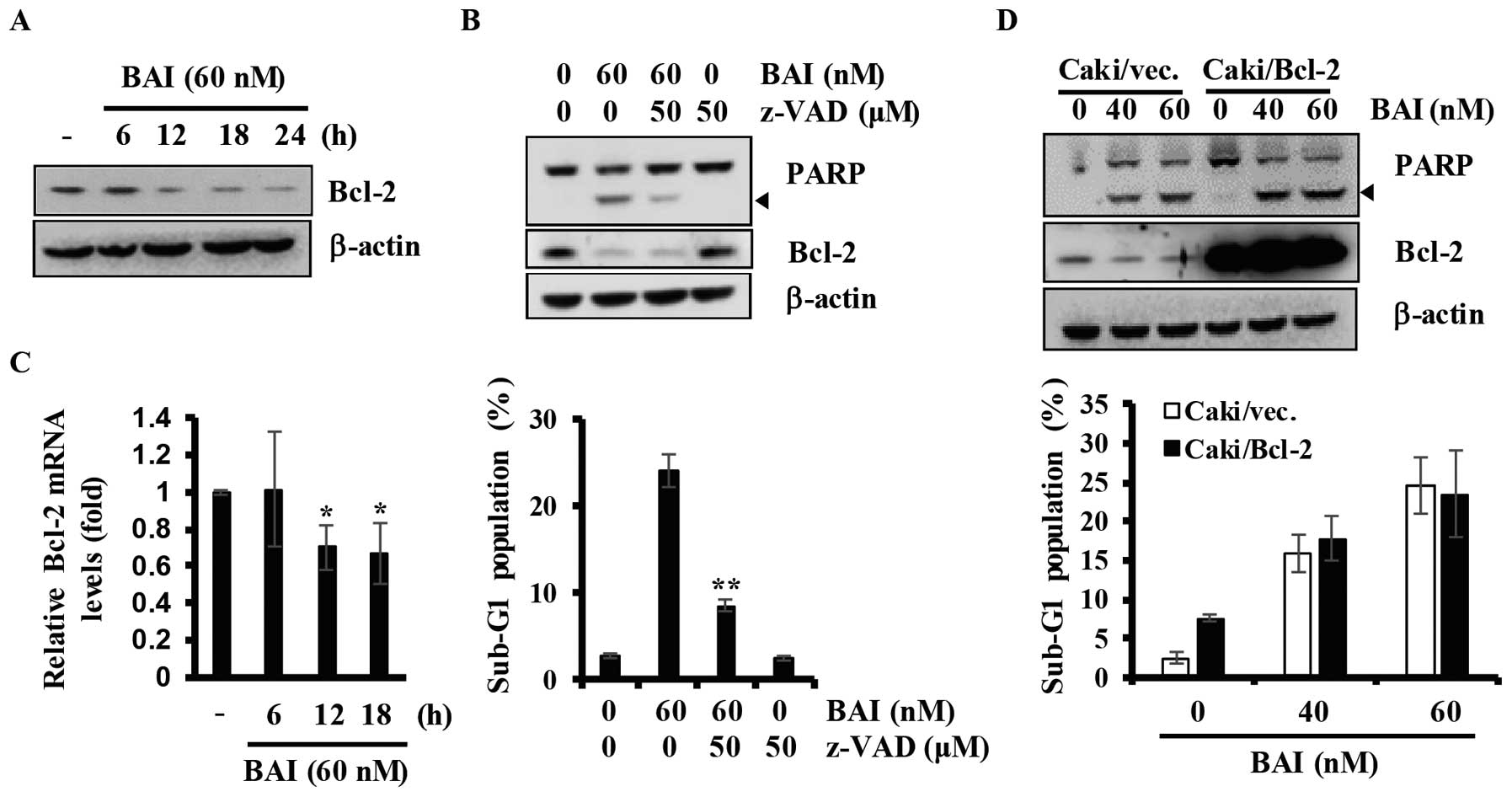
We next determined the effect of BAI on Bcl-2
regulation in Caki cells. As shown in Fig. 5A, data from kinetic analysis showed
that treatments with BAI for various time-points (6–24 h) led to a
marked downregulation of Bcl-2. To identify the Bcl-2 regulating
mechanisms by BAI, we treated Caki cells with or without BAI in the
presence or absence of z-VAD-fmk, a pan-caspase inhibitor, for 24
h, and then measured sub-G1 populations and the cellular levels of
PARP, Bcl-2, and β-actin by FACS and western blot analysis,
respectively. BAI induced cleavage of PARP and increased the
population of Caki cells in the sub-G1 phase, which were largely
suppressed by pre-treatment with z-VAD-fmk (Fig. 5B). However, BAI-induced
downregulation of Bcl-2 was not blocked by pre-treatment with
z-VAD-fmk, suggesting that the downregulation of Bcl-2 protein is
not involved in caspase activity (Fig.
5B). Therefore, we next investigated the effect of BAI on the
transcriptional regulation of Bcl-2 by RT-qPCR analysis. As shown
in Fig. 5C, BAI reduced levels of
Bcl-2 transcripts in a time-dependent manner. To further
investigate the role of Bcl-2 in BAI-induced apoptosis, we used
Caki renal carcinoma cells engineered for overexpression of Bcl-2.
As shown in Fig. 5D,
overexpression of Bcl-2 could not attenuate the apoptosis induced
by BAI. Collectively, these results indicate that downregulation of
Bcl-2 is not associated with BAI-induced apoptosis in Caki
cells.
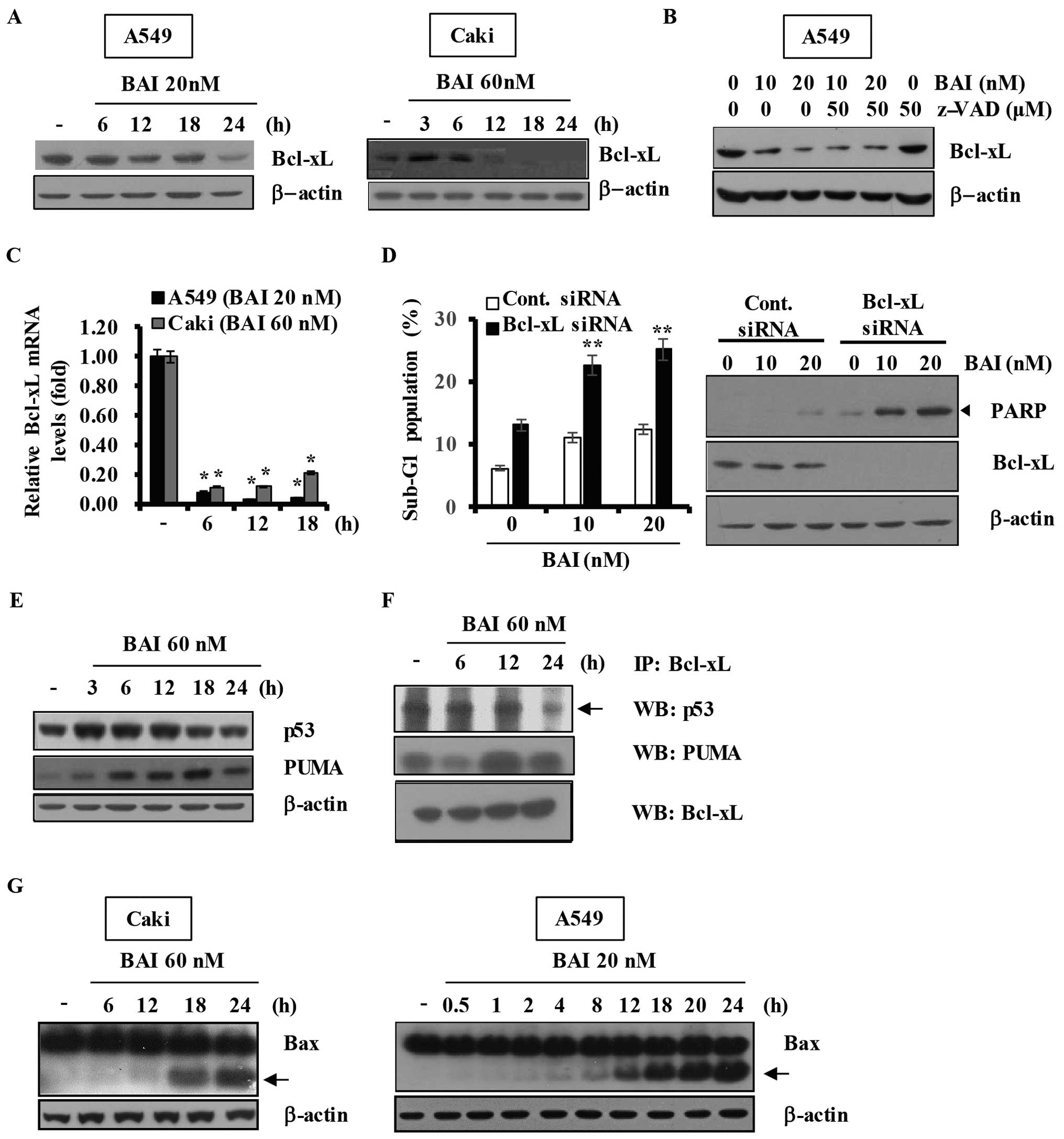
BAI modulates the expression of Bcl-xL
and the interactions among p53 and Bcl-2 family proteins in human
cancer cells
Bcl-xL is a widely studied factor of resistance to
cytotoxic anticancer agents. We first examined whether Bcl-xL is
associated with BAI-induced apoptosis, cancer cells were treated
with BAI at different times. As shown in Fig. 6A, BAI treatment of cancer cells for
various time-points resulted in markedly decreased expression
levels of Bcl-xL in A549 and Caki cells. We explored the possible
link between loss of Bcl-xL protein and activation of caspases in
BAI-treated A549 cells. As shown in Fig. 6B, pretreatment with z-VAD-fmk had
no effect on the reduction of Bcl-xL protein by BAI, implying that
the BAI-induced downregulation of Bcl-xL protein is not associated
with caspase activity. This led us to investigate the effect of BAI
on transcriptional regulation of Bcl-xL. Notably, results of
RT-qPCR analysis, as shown in Fig.
6C, demonstrated a marked reduction of Bcl-xL transcripts in
BAI-treated cells, suggesting that BAI downregulates Bcl-xL at the
transcriptional levels. To evaluate the functional significance of
BAI-induced Bcl-xL downregulation, we transfected A549 cells with
siRNA targeting Bcl-xL mRNA and treated cells with or without BAI
for 24 h. The concentrations of BAI were sub-cytotoxic in
comparison with the results of previous experiments. Immunoblot
analysis confirmed that transfection with Bcl-xL siRNA resulted in
suppression of Bcl-xL expression in A549 cells compared with cells
transfected with control GFP siRNA (Fig. 6D). Notably, the BAI-induced
accumulation of sub-G1 phase was markedly increased in cells
transfected with Bcl-xL siRNA as compared with control
siRNA-transfected cells (Fig. 6D).
In addition, the expression of cleaved PARP was induced only in
cells transfected with Bcl-xL siRNA (Fig. 6D).
Bcl-2 family members regulate survival/death
decisions through a network of interactions among the pro-survival
member Bcl-xL, the pro-apoptotic member PUMA, and p53 (9,10,18).
We next investigated whether BAI affects the expression levels of
p53 and PUMA proteins in cancer cells. As shown in Fig. 6E, Caki cells treated with BAI
showed upregulation of p53 and PUMA in a time-dependent manner. We
then determined whether BAI modulates the interactions between
specific Bcl-2 families in Caki cells using co-immunoprecipitation
assays. As shown in Fig. 6F, BAI
not only efficiently disrupted the Bcl-xL/p53 interaction but also
induced the binding between PUMA and Bcl-xL in Caki cells in a
time-dependent manner. Additionally, A549 and Caki cells treated
with BAI showed induction of Bax cleavage in a time dependent
manner (Fig. 6G). Taken together,
these results suggest that downregulation of Bcl-xL protein is
importantly associated with the BAI-induced apoptosis and that BAI
modulates interactions among p53 and Bcl-2 family proteins in human
cancer cells.
Discussion
Until recently, targeted cancer therapy was widely
accepted as an effective means for cancer therapeutic strategies
(19). However, recent reports
have shown that intratumoral heterogeneity plays an important role
in tumor adaptation and therapeutic failure (20). For this reason, appropriate
validation and balanced modulation of multiple targets have been
attractive therapeutic strategies in treating cancer. Recent
studies reported the synthesis and anticancer effects of the novel
cyclin-dependent kinase inhibitor BAI (1–4). BAI
was shown to exhibit various apoptotic effects, including caspase
activation, inactivation of Akt (2), and sensitizing effect on
farnesyltransferase inhibitor, LB42708-mediated apoptosis through
the downregulation of Bcl-2 and c-FLIP (L) (4). In this study, we further investigated
the apoptotic mechanisms of BAI in the human renal cell carcinoma
Caki cell line and human non-small cell lung cancer A549 cell
line.
Among the apoptosis-related pathways, ROS generation
and MAPK pathways have been known to modulate apoptosis in cancer.
It is a promising cancer therapeutic strategy to eliminate cancer
cells by regulating oxidative stress-mediated apoptosis induced by
cytotoxic drugs (21).
Furthermore, MAPK pathways play an important role in modulating
survival and apoptosis of cancer cells (22). Our data using specific inhibitors
on ROS generation or MAPK pathways showed that these inhibitors did
not influence BAI-induced apoptosis in Caki cells, indicating that
ROS generation and MAPK pathways are not involved in BAI-induced
apoptosis.
Anti-apoptotic Bcl-2 family proteins such as Bcl-xL
are frequently overexpressed in cancers (23). Downregulation of cell survival
proteins may render cancer cells sensitive to anticancer agents. A
previous study showed that BAI downregulated the expression levels
of XIAP and Mcl-1 (L) proteins (2). However, downregulations of XIAP and
Mcl-1 (L) proteins were not associated with BAI-induced apoptosis
(2). On the other hand, BAI
inhibited activation of p-Akt, and the inactivation of p-Akt
contributed to BAI-facilitated PI3K/Akt inhibitor LY294002-induced
apoptosis (2). In this study, we
investigated the role of anti-apoptotic Bcl-2 family proteins, such
as Bcl-2 and Bcl-xL in BAI-induced apoptosis. Our data showed that
BAI downregulated Bcl-2 expression at the transcriptional levels,
but overexpression of Bcl-2 could not block BAI-induced apoptosis.
These results suggest that downregulation of Bcl-2 is not involved
in BAI-induced apoptosis in Caki cells. Bcl-2 proteins, such as
Bcl-2, are related to chemoresistance in a variety of human cancers
(24,25). Therefore, targeting Bcl-2 members
represents a promising anticancer strategy (26). Notably, our data showed that BAI
could induce apoptosis in Bcl-2-overexpressing Caki cells. These
results suggest that BAI could overcome the increased activity of
Bcl-2, suggesting that BAI may be a potentially useful anticancer
agent against Bcl-2-overexpressing malignancies. We also found that
BAI downregulated Bcl-xL at the transcriptional level and that
Bcl-xL siRNA increased the sensitivity of BAI in the human cancer
cells, suggesting that downregulation of Bcl-xL plays an important
role in BAI-induced apoptosis.
Following DNA damage, nuclear or cytoplasmic
accumulation of the tumor suppressor p53 is an important mechanism
in apoptosis (27). Cytoplasmic
p53 is sequestered by antiapoptotic Bcl-2 family proteins, such as
Bcl-xL (18,27). The BH3-only protein PUMA, induced
by nuclear p53, mediates cytosolic pro-apoptotic p53 function
(9). When DNA damage induces
apoptosis, cytoplasmic p53 is released from the complex with Bcl-xL
and can directly activate Bax, subsequently promoting apoptosis via
mitochondrial outer membrane permeabilization (28). Based on the DNA
damagep53-PUMA-Bcl-xL-mediated apoptotic signaling pathway, we
hypothesized that BAI-induced apoptosis follows this apoptotic
signaling pathway. We previously reported that BAI induces DNA
fragmentation (2). In this study,
BAI upregulated the expression levels of p53 and PUMA in a
time-dependent manner. Furthermore, we demonstrated that BAI
disrupts the interaction between p53 and Bcl-xL, and induces PUMA
binding to Bcl-xL in Caki cells. Further studies are required to
elucidate the precise regulatory mechanisms underlying the
interactions among p53 and Bcl-2 family proteins (PUMA and Bcl-xL)
in BAI-induced apoptosis, however, our results demonstrate that p53
and Bcl-2 family proteins play important roles in BAI-induced
apoptosis of human cancer cells. Additionally, we found that
upregulation of p53 was followed by that of PUMA in BAI-treated
cancer cells (Fig. 6E). PUMA is a
mediator of p53-induced apoptosis (29,30).
Therefore, it is required to investigate whether p53 or PUMA play
an important role in BAI-induced apoptosis.
Bax cleavage is a well-known and important
phenomenon in caspase-dependent apoptosis (31–33).
Our findings demonstrated that BAI induces Bax cleavage and
promotes Bax oligomerization.
Mitochondria play an essential role in apoptosis by
releasing apoptogenic effectors such as AIF and cytochrome c
(15,34). We found that BAI markedly decreased
MMP in human cancer cells and induced a marked release of
cytochrome c and AIF into the cytoplasm. Given that release
of AIF and cytochrome c from the mitochondria to the
cytoplasm triggers activation of the caspase-3 pathway (35), it is likely that the release of AIF
and cytochrome c induced by BAI is implicated in
caspase-dependent apoptosis in human cancer cells.
Together our data show that BAI induces apoptosis in
various cancer cells through loss of MMP, activation of Bax,
downregulation of Bcl-xL, and regulation of interactions among p53,
PUMA, and Bcl-xL. These findings support the idea that BAI may be
useful for development as an attractive multi-target drug against
cancer.
Acknowledgements
This study was supported by the Bisa Research Grant
of Keimyung University in 2012.
Abbreviations:
|
Bcl-2
|
B cell lymphoma-2
|
|
Bcl-xL
|
B-cell lymphomaextra large
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
ROS
|
reactive oxygen species
|
|
MAPK
|
mitogen-activated protein kinases
|
|
MMP
|
mitochondrial membrane potential
|
|
z-VAD-fmk
|
benzyloxy
carbony-Val-Ala-Asp-fluoromethyl ketone
|
|
NAC
|
N-acetylcysteine
|
|
GEE
|
glutathione
|
|
COXII
|
cytochrome c oxidase subunit
II
|
|
IP
|
immunoprecipitate
|
|
AIF
|
apoptosis-inducing factor
|
References
|
1
|
Lee J, Choi H, Kim KH, Jeong S, Park JW,
Baek CS and Lee SH: Synthesis and biological evaluation of
3,5-diaminoindazoles as cyclin-dependent kinase inhibitors. Bioorg
Med Chem Lett. 18:2292–2295. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim S, Lee J, Jang BC, Kwon TK and Park
JW: BAI, a novel cyclin-dependent kinase inhibitor induces
apoptosis in A549 cells through activation of caspases and
inactivation of Akt. J Cell Biochem. 114:282–293. 2013. View Article : Google Scholar
|
|
3
|
Shin HC, Song DW, Baek WK, Lee SR, Kwon
TK, Lee J, Park SH, Jang BC and Park JW: Anticancer activity and
differentially expressed genes in head and neck cancer cells
treated with a novel cyclin-dependent kinase inhibitor.
Chemotherapy. 55:353–362. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jang JH, Cho YC, Kim KH, Lee KS, Lee J,
Kim DE, Park JS, Jang BC, Kim S, Kwon TK, et al: BAI, a novel Cdk
inhibitor, enhances farnesyltransferase inhibitor LB42708-mediated
apoptosis in renal carcinoma cells through the downregulation of
Bcl-2 and c-FLIP (L). Int J Oncol. 45:1680–1690. 2014.PubMed/NCBI
|
|
5
|
Jacobson MD, Weil M and Raff MC:
Programmed cell death in animal development. Cell. 88:347–354.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thompson CB: Apoptosis in the pathogenesis
and treatment of disease. Science. 267:1456–1462. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Green DR and Llambi F: Cell death
signaling. Cold Spring Harb Perspect Biol. 7:72015. View Article : Google Scholar
|
|
8
|
Kroemer G: The proto-oncogene Bcl-2 and
its role in regulating apoptosis. Nat Med. 3:614–620. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Follis AV, Chipuk JE, Fisher JC, Yun MK,
Grace CR, Nourse A, Baran K, Ou L, Min L, White SW, et al: PUMA
binding induces partial unfolding within BCL-xL to disrupt p53
binding and promote apoptosis. Nat Chem Biol. 9:163–168. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu J, Wang Z, Kinzler KW, Vogelstein B and
Zhang L: PUMA mediates the apoptotic response to p53 in colorectal
cancer cells. Proc Natl Acad Sci USA. 100:1931–1936. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Graves JD, Campbell JS and Krebs EG:
Protein serine/threonine kinases of the MAPK cascade. Ann NY Acad
Sci. 766(1 Receptor Acti): 320–343. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Low HB and Zhang Y: Regulatory roles of
MAPK phosphatases in cancer. Immune Netw. 16:85–98. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar
|
|
15
|
Le Bras M, Rouy I and Brenner C: The
modulation of interorganelle cross-talk to control apoptosis. Med
Chem. 2:1–12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wei MC, Zong WX, Cheng EH, Lindsten T,
Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB and
Korsmeyer SJ: Proapoptotic BAX and BAK: A requisite gateway to
mitochondrial dysfunction and death. Science. 292:727–730. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chipuk JE, Bouchier-Hayes L, Kuwana T,
Newmeyer DD and Green DR: PUMA couples the nuclear and cytoplasmic
proapoptotic function of p53. Science. 309:1732–1735. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sawyers C: Targeted cancer therapy.
Nature. 432:294–297. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gerlinger M, Rowan AJ, Horswell S, Larkin
J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A,
Tarpey P, et al: Intratumor heterogeneity and branched evolution
revealed by multiregion sequencing. N Engl J Med. 366:883–892.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ivanova D, Bakalova R, Lazarova D, Gadjeva
V and Zhelev Z: The impact of reactive oxygen species on anticancer
therapeutic strategies. Adv Clin Exp Med. 22:899–908. 2013.
|
|
22
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Walensky LD: BCL-2 in the crosshairs:
Tipping the balance of life and death. Cell Death Differ.
13:1339–1350. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Johnstone RW, Ruefli AA and Lowe SW:
Apoptosis: A link between cancer genetics and chemotherapy. Cell.
108:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fernandez-Luna JL: Regulation of
pro-apoptotic BH3-only proteins and its contribution to cancer
progression and chemoresistance. Cell Signal. 20:1921–1926. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Azmi AS, Wang Z, Philip PA, Mohammad RM
and Sarkar FH: Emerging Bcl-2 inhibitors for the treatment of
cancer. Expert Opin Emerg Drugs. 16:59–70. 2011. View Article : Google Scholar :
|
|
27
|
Green DR and Kroemer G: Cytoplasmic
functions of the tumour suppressor p53. Nature. 458:1127–1130.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chipuk JE, Kuwana T, Bouchier-Hayes L,
Droin NM, Newmeyer DD, Schuler M and Green DR: Direct activation of
Bax by p53 mediates mitochondrial membrane permeabilization and
apoptosis. Science. 303:1010–1014. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hemann MT, Zilfou JT, Zhao Z, Burgess DJ,
Hannon GJ and Lowe SW: Suppression of tumorigenesis by the p53
target PUMA. Proc Natl Acad Sci USA. 101:9333–9338. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jeffers JR, Parganas E, Lee Y, Yang C,
Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al:
Puma is an essential mediator of p53-dependent and -independent
apoptotic pathways. Cancer Cell. 4:321–328. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yanase N, Ohshima K, Ikegami H and
Mizuguchi J: Cytochrome c release, mitochondrial membrane
depolarization, caspase-3 activation, and Bax-alpha cleavage during
IFN-alpha-induced apoptosis in Daudi B lymphoma cells. J Interferon
Cytokine Res. 20:1121–1129. 2000. View Article : Google Scholar
|
|
32
|
Kim YH, Shin HC, Song DW, Lee SH, Furumai
T, Park JW and Kwon TK: Arisostatins A induces apoptosis through
the activation of caspase-3 and reactive oxygen species generation
in AMC-HN-4 cells. Biochem Biophys Res Commun. 309:449–456. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yeo JK, Cha SD, Cho CH, Kim SP, Cho JW,
Baek WK, Suh MH, Kwon TK, Park JW and Suh SI:
Se-methylselenocysteine induces apoptosis through caspase
activation and Bax cleavage mediated by calpain in SKOV-3 ovarian
cancer cells. Cancer Lett. 182:83–92. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Budihardjo I, Oliver H, Lutter M, Luo X
and Wang X: Biochemical pathways of caspase activation during
apoptosis. Annu Rev Cell Dev Biol. 15:269–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
van Gurp M, Festjens N, van Loo G, Saelens
X and Van den Abeele P: Mitochondrial intermembrane proteins in
cell death. Biochem Biophys Res Commun. 304:487–497. 2003.
View Article : Google Scholar : PubMed/NCBI
|