Introduction
Approximately 600,000 new cases of head and neck
squamous cell carcinoma (HNSCC) are diagnosed worldwide annually,
making this cancer the sixth most common human malignancy (1). Systemic therapies have been employed
for treating this cancer with significant progress; however, the
5-year overall survival remains low at ~50% (2), due to subsequent metastasis developed
in such patients which accounts for the poor clinical outcome of
HNSCC (2). Epithelial mesenchymal
transition (EMT), initially described in embryonic development, has
been reported to be one of the most important mechanisms of cell
migration and invasion in physiological and pathological processes,
such as tissue repair, fibrosis and cancer progression (3). When cells undergo EMT, they may
change their morphology and lose cell-cell junctions, while
retaining expression of molecules that promote migration and
invasion (3). Of note, EMT leads
to the poor prognosis of HNSCC (4,5).
Enhancer of zeste homolog 2 (EZH2), a catalytic
subunit of the polycomb repressive complex 2, serves an important
role in the modification of chromatin structure primarily by
trimethylating the lysine 27 residue of histone H3 (H3K27me3)
(6,7). EZH2 is upregulated in several cancer
types, including HNSCC and serves as a negative prognostic factor
(8-11). Reports showed that EZH2 could
regulate tumor angiogenesis (12),
apoptosis (11), EMT (13), and cell migration and invasion
(14,15). Our previous research revealed that
inhibition of EZH2 with 3-deazaneplanocin A (DZNep), a novel
inhibitor of EZH2, induced the apoptosis of HNSCC cells via
mitochondria-dependent cell death (11). EZH2 is therefore a promising
therapeutic target for treating HNSCC.
Signal transducer and activator of transcription
factors 3 (STAT3) is aberrantly activated in 70% of all cancer
types (16,17), and is involved in cancer cell
proliferation, angiogenesis, invasion, and the survival and
maintenance of cancer stem cells (18). STAT3 is constitutively activated
during tumor progression and metastasis, and the level of
tyrosine-phosphorylated STAT3 (Try705) is associated with the poor
prognosis of HNSCC (19). Thus,
STAT3 is a potential therapeutic target for treating HNSCC. A
previous study reported that phosphorylation of EZH2 at serine 21
may enhance STAT3 activity by direct binding to and methylating
STAT3 (20). Furthermore, EZH2
could mediate the oncogenic activity of STAT3 through demethylation
of K49 (21); however, whether and
how EZH2 interacts with STAT3 in HNSCC remains unclear.
Vascular endothelial growth factor receptor 2
[VEGFR2, kinase insert domain receptor (KDR)] is the major receptor
for vascular endothelial growth factor (VEGF) and is upregulated in
HNSCC (22). Emerging evidence has
suggested that STAT3 may participate in the VEGF/VEGFR2 signaling
cascade in human cancers (23).
For example, STAT3 activation has been reported to promote
angiogenesis through upregulation of VEGF expression in lung cancer
(24). Notably, a previous study
demonstrated that VEGF upregulated is induced by STAT3 activation
and thus promotes brain metastasis in human melanoma (25). Based on these studies, we
investigated whether EZH2 mediates metastasis and the EMT process
in HNSCC via regulating the STAT3/VEGFR2 axis. We aimed to
characterize the EZH2-mediated EMT pathway to provide a rationale
for developing novel therapeutic strategies to suppress HNSCC
metastasis.
Materials and methods
Cell culture and reagents
The human HNSCC cell line CAL27 was purchased from
the American Type Culture Collection and the UM1 cell line was a
gift from Professor Jinsong Hou of Guanghua School of Stomatology,
Hospital of Stomatology, Sun Yatsen University. Cells were
maintained in Dulbecco's Modified Eagle's medium (DMEM)/Ham's F-12
and DMEM supplemented with 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.) at 37°C in a 5% CO2
humidified incubator. UM1 and CAL27 cells were treated with DZNep
(2, 5 or 10 µmol/l, Selleck Chemicals) for 48 h at 37°C to
inhibit EZH2 expression. DZNep was dissolved in dimethyl sulfoxide
(DMSO; Sigma-Aldrich, Merck KGaA). UM1 and CAL27 cells were treated
with DMSO as control.
Small interfering RNA (siRNA) and plasmid
transfection
UM1 and CAL27 cell lines were transfected with
siRNA-negative control (siNC) or siRNAs (20 nmol/l, Guangzhou
Ribobio Co., Ltd.) against STAT3 or VEGFR2, labeled as siNC (5′-UUC
UCC GAA CGU GUC ACG U-3′), siSTAT3 (5′-AGT CAG GTT GCT GGT CAA
A-3′) or siKDR (5′-GGT AAA GAT TGA TGA AGA A-3′), respectively,
using Lipofectamine® 2000 reagent according to the
manufacturer's instructions (Invitrogen; Thermo Fisher Scientific,
Inc.). For the transient overexpression of EZH2, the pLVX-IRES
lentiviral vector system was used to deliver EZH2 gene into UM1 and
CAL27 cells, the empty pLVX plasmid (Shanghai GeneChem Co., Ltd.)
was used as a control. FBS-free Opti-MEM medium (Gibco; Thermo
Fisher Scientific, Inc.) was employed for transfection, after 6 h,
the medium was replaced by DMEM/Ham's F-12 or DMEM. Subsequent
analysis were carried out after transfection for 48 h.
Western blotting
Cell lysates were prepared in
radioimmunoprecipitation assay buffer with protease and phosphatase
inhibitors (Beijing Solarbio Science & Technology Co., Ltd.) 48
h after the treatments of cells. Protein concentrations were
determined by using a BCA protein assay kit (Micro BCA Protein
Assay kit; Thermo Fisher Scientific, Inc.). A total of 30 µg
of each protein samples was separated in an analytical 10 or 15%
SDS-PAGE and then transferred onto polyvinylidene difluoride
membranes (Merck KGaA). The membranes were blocked in blocking
buffer (5% non-fat milk, 0.1% Tween 20 in PBS) for 2 h at room
temperature and then incubated overnight at 4°C with primary
antibodies (1:1,000) against human EZH2, STAT3, phosphorylated
(p)-STAT3 (Tyr705), E-cadherin, N-cadherin, Vimentin, Twist-related
protein 1 (Twist-1) obtained from Cell Signaling Technology, Inc.;
primary antibodies against VEGF, VEGFR2 (Abcam), and GAPDH (OriGene
Technologies, Inc.) were employed (Table I). Mouse (sc-2005) or rabbit
(sc-2004) IgG antibodies coupled with horseradish peroxidase (Santa
Cruz Biotechnology, Inc.) were used as secondary antibodies
(1:5,000), which were applied at room temperature for 2 h.
Chemiluminescent horseradish peroxidase substrate (Merck KGaA) was
used as the visualization reagent following the instructions. The
densitometry of the immunoblots was determined using ImageJ
software (version 5.2.5; National Institutes of Health).
 | Table IPrimary antibodies used in the
present study. |
Table I
Primary antibodies used in the
present study.
| Primary
antibody | Cat. no. | Supplier | Application |
|---|
| EZH2 | 5246 | Cell Signaling
Technology, Inc. | WB/IHC |
| STAT3 | 30835 | Cell Signaling
Technology, Inc. | WB/IHC |
| p-STAT3
(Tyr705) | 9145 | Cell Signaling
Technology, Inc. | WB/IHC |
| E-cadherin | 14472 | Cell Signaling
Technology, Inc. | WB/IF/IHC |
| N-cadherin | 13116 | Cell Signaling
Technology, Inc. | WB/IF/IHC |
| Vimentin | 5741 | Cell Signaling
Technology, Inc. | WB/IF/IHC |
| Twist-1 | 46702 | Cell Signaling
Technology, Inc. | WB |
| VEGF | ab155944 | Abcam | WB/IHC |
| VEGFR2 | ab11939 | Abcam | WB/IHC |
| GAPDH | TA-08 | OriGene
Technologies, Inc. | WB |
Wound-healing and Transwell assay
Approximately 2×105 UM1 cells or
5×105 CAL27 cells per well were added into 6-well plate.
A linear scratch was made by 10 µl pipette tips. The plates
were incubated in serum-free medium. Images were taken at 0 and 24
h from several randomly selected wound locations. Cell invasion and
migration assays were performed using Transwell membranes coated
with Matrigel (BD Biosciences) or left uncoated, respectively. The
lower chamber was filled with medium containing with 20% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and cultured at 37°C for 24
h. The non-invading cells were removed with cotton swabs and
invading cells were fixed with 4% paraformaldehyde for 15 min at
room temperature and then stained with 0.1% crystal violet (both
from Beijing Solarbio Science & Technology Co., Ltd.) for 30
min at room temperature. Each image was observed using an inverted
microscope (DMI6000B; Leica Microsystems, Inc.) at ×40
magnification, and cells were counted in five separate fields in
three independent experiments.
Immunofluorescence staining
For immunofluorescence staining, 5×104
cells/well UM1 or CAL27 were seeded on the coverslips and fixed
with 4% paraformaldehyde (Beijing Solarbio Science & Technology
Co., Ltd.) for 15 min at room temperature. Then, the cells were
permeabilized with 0.2% Triton-X 100 for 10 min and treated with 1%
bovine serum albumin (Beijing Solarbio Science & Technology
Co., Ltd.) for 1 h. Immunofluorescence staining was conducted with
antibodies (Table I) against
E-cadherin, N-cadherin, Vimentin (1:100; Cell Signaling Technology,
Inc.) and F-actin was detected by phalloidin (cat no. 40734ES75;
1:200; Yeasen Biotech Co., Ltd.) overnight at 4°C. The cells were
washed with PBS and incubated with Alexa Fluor® 488
(anti-rabbit, cat. no. 4412 or anti-mouse: cat. no. 4408) or Alexa
Fluor 594 (anti-rabbit, cat. no. 8889 or anti-mouse, cat. no. 8890)
secondary antibodies (1:500 dilutions; Cell Signaling Technology,
Inc.) for 1 h at room temperature. Nuclei were stained using DAPI
reagent (Thermo Fisher Scientific, Inc.) for 10 min at room
temperature. Cells were visualized using FV-1000 laser scanning
confocal microscope (Olympus Corporation) in five separate fields;
a ×100 objective lens was used in this experiment.
CAL27 xenograft tumor model and in vivo
treatment
All animal protocols were approved by the Tianjin
Medical University Animal Care and Use Committee and also conformed
to the guidelines published by the National Institutes of Health
for the care and use of laboratory animals (26). A total of 2×106 CAL27
cells were implanted subcutaneously on the right groin region of
4-week-old male nude BALB/c mice (n=25; weight, ~15 g). The housing
conditions for mice were as following: Temperature, 18-22°C;
humidity, 50-60%; ventilation, 15 times/h; 12/12 h light/dark
cycle; free access to food/water. After 30 days following tumor
establishment, the mice were randomly divided into five groups (n=5
per group). Mice in individual groups received an intra-tumoral
injection of designated treatment every three days with 0.07 mg/kg
DZNep (DZNep group), 10 mg/kg Stattic (Stattic group), 0.07 mg/kg
DZNep + 10 mg/kg Stattic (DZNep + Stattic group), 10 mg/kg Apatinib
(Apatinib group; Stattic and Apatinib were purchased from Selleck
Chemicals), or DMSO (DMSO group). Tumor volume (volume=long
diameter × short diameter2/2) and mice weights were
determined by calipers and electronic weighing meter. After 3 weeks
of observation, the mice were sacrificed and the xenograft tumors
were removed for further pathological examination.
Immunohistochemistry (IHC), and
hematoxylin and eosin (H&E) staining
For IHC staining, xenograft tumor samples were fixed
with 4% paraformaldehyde for 24 h at room temperature.
Paraffin-embedded samples were sliced into 4 µm thickness,
and then deparaffinized, rehydrated, and incubated with primary
antibodies (see Table I) against
EZH2, STAT3, p-STAT3 (Tyr705), E-cadherin, N-cadherin, Vimentin,
VEGF or VEGFR2 (1:100) overnight at 4°C. Then, the tumor slices
were incubated with a biotin-labeled secondary antibody (ready to
use; PV-9000; OriGene Technologies, Inc.) for 1 h at 37°C. The IHC
(11) and H&E (27) protocols were performed as
previously described. Images were captured under an inverted
microscope (DMI6000B; Leica Microsystems, Inc.) at ×200
magnification in three separate fields.
Statistical analysis
SPSS software 18.0 (SPSS, Inc.) was used to analyze
all data. The results were presented as the mean ± standard
deviation. Statistical evaluation between two groups was performed
using a Student's t-test. Differences among multiple groups were
determined by two-way ANOVA followed by a Dunnett's test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Inhibition of the EZH2/STAT3 axis
attenuates VEGFR2 expression in HNSCC cell lines in vitro
Our previous research showed that HNSCC cell lines
UM1 and CAL27 possess high expression of EZH2, STAT3 and p-STAT3
(Tyr705) (11,28), we therefore selected UM1 and CAL27
cells for further investigation. We used DZNep, a specific EZH2
small-molecule inhibitor, to suppress EZH2 expression in UM1 and
CAL27 cells. The optimal dose of DZNep treatment in HNSCC cells was
demonstrated in our previous report (11). In DZNep-treated UM1 and CAL27
cells, the expression levels of EZH2, p-STAT3 (Tyr705), VEGF, and
VEGFR2 were markedly decreased than in untreated control cells in a
dose-dependent manner; however, the expression of total STAT3 was
notably unaffected (Fig. 1A).
 | Figure 1EZH2/STAT3 axis regulates VEGFR2
expression in HNSCC cells. (A) UM1 and CAL27 cell lines were
treated with DZNep at designated concentrations (2, 5 or 10
µmol/l) for 48 h. Western blot analyses were performed to
investigate the expression of EZH2, STAT3, p-STAT3 (Tyr705), VEGF,
and VEGFR2. GAPDH was used as a loading control. (B) HNSCC cells
were transfected with pLVX-EZH2 expression plasmids, western blot
analyses were performed to investigate the effects of EZH2 on the
expression of p-STAT3 (Tyr705), VEGF, and VEGFR2. (C) siSTAT3
transfection was performed to determine the impact of STAT3 on
p-STAT3 (Tyr705), VEGF, and VEGFR2 expression in UM1 and CAL27
cells. EZH2, enhancer of zeste homolog 2; HNSCC, head and neck
squamous cell carcinoma; NC, negative control; p, phosphorylated;
si, small interfering RNA; STAT3, signal transducer and activator
of transcription factor 3; VEGF, vascular endothelial growth
factor; VEGFR2, VEGF receptor 2. |
To further investigate the importance of increased
EZH2 expression in HNSCC cells, we transfected the pLVX-EZH2
expression plasmid into UM1 and CAL27 cells. As shown in Fig. 1B, induced EZH2 expression resulted
in the increased expression of p-STAT3 (Tyr705), VEGF, and VEGFR2,
with no notable differences in total STAT3 expression.
Additionally, we used STAT3 siRNA to block STAT3 and p-STAT3
(Tyr705) expression. After transfection for 48 h, the expression
levels of STAT3 and p-STAT3 (Tyr705) were markedly decreased in
both cell lines. Similarly, lower expression of VEGF and VEGFR2 was
observed after STAT3 knockdown (Fig.
1C). These results suggest that the EZH2/STAT3 axis could
regulate VEGFR2 expression in HNSCC cells.
Inhibition of EZH2 attenuates the
migration, invasion, and EMT of HNSCC cells in vitro
We next used DZNep to antagonize EZH2 in UM1 and
CAL27 cells. Treatment with DZNep for 24 h resulted in
significantly delayed wound healing in both cell lines as
determined via scratch tests, compared with the control
(P<0.001; Fig. 2A). The
Transwell assay showed that the number of migrating and invading
cells in the DZNep-treated group was significantly reduced compared
with the control (P<0.001; Fig.
2B). These results suggest that knockdown of EZH2 inhibits the
invasion and migration of HNSCC cells. EMT involves a complex
change in the cell phenotype, and is regarded as one of the most
important mechanisms for cell migration and cancer metastasis
(3,29). Cells undergoing EMT often display
cadherin switching, for example, loss of epithelial cell adhesion
and tight regulation of cadherin adhesion (3). We hypothesized that EZH2 inhibition
could reduce the invasion and migration of HNSCC cells via EMT
interruption. Western blotting revealed that in DZNep-treated group
the EMT marker, E-cadherin, was markedly upregulated, while
N-cadherin, Vimentin and Twist-1 were notably downregulated in a
dose-dependent manner compared with the control (Fig. 2E). Furthermore, immunofluorescence
staining showed similar findings to the western blot assay; in
DZNep-treated group, E-cadherin expression had increased, while
N-cadherin and Vimentin were decreased (Fig. 2C). In the DZNep-treated group,
disrupted F-actin stress fiber networks were observed with notable
phenotypic changes in UM1 and CAL27 cells (Fig. 2D). Taken together, these results
demonstrated that EZH2 inhibition attenuates the aggressive
behavior of HNSCC cells by interrupting the EMT process.
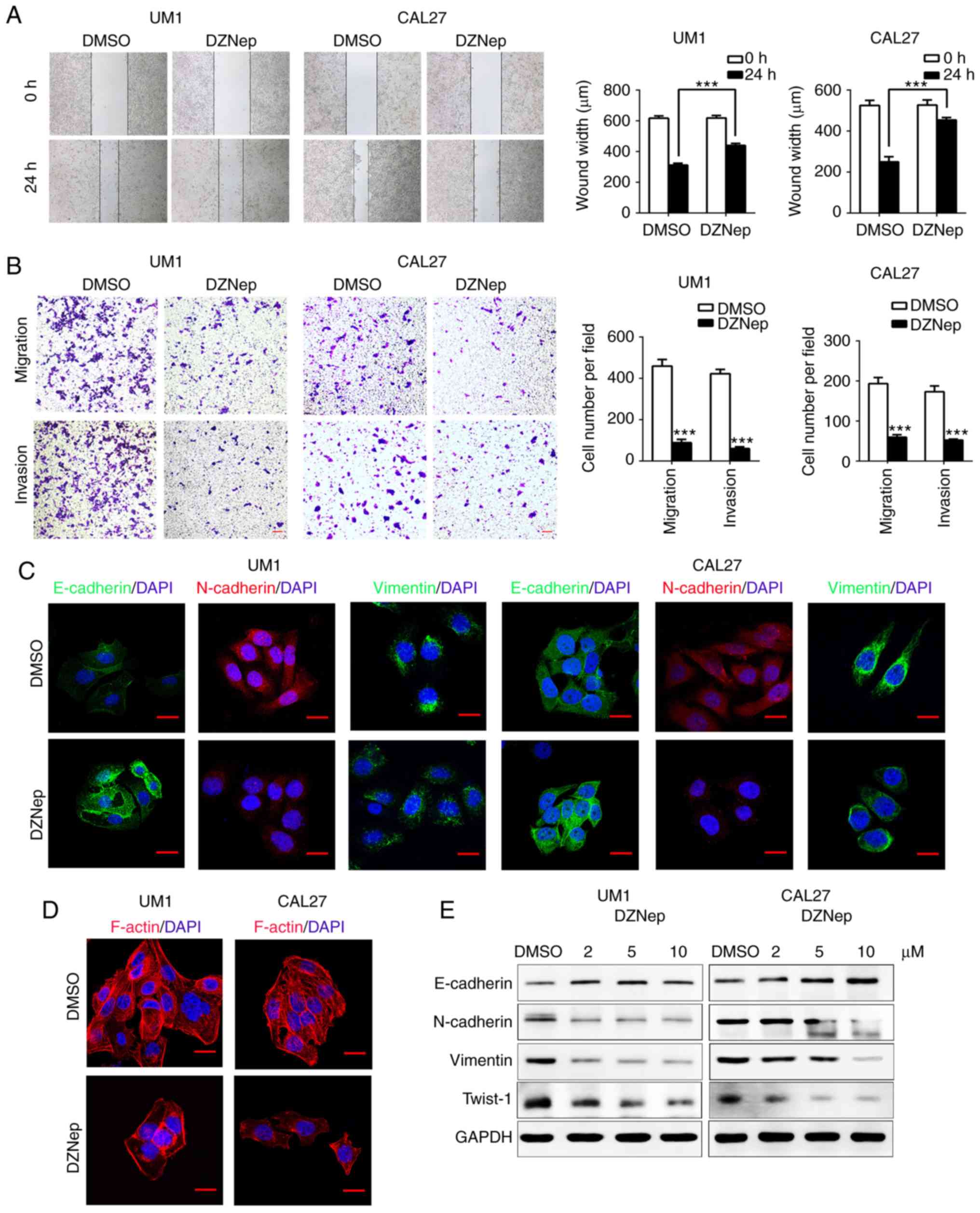 | Figure 2Inhibition of enhancer of zeste
homolog 2 suppresses EMT of head and neck squamous cell carcinoma
cells in vitro. (A) Wound-healing assays were performed to
analyze the migration in DZNep-treated UM1 and CAL27 cells.
Student's t-test ***P<0.001. (B) Transwell assays
were performed to investigate migration (without Matrigel) and
invasion (with Matrigel) in the DZNep-treated group of both cell
lines. Student's t-test. ***P<0.001 vs. DMSO. Scale
bar, 50 µm. (C) EMT-related proteins were nalyzed by
immunofluorescence staining in UM1 and CAL27 cells after DZNep or
DMSO treatment. Scale bar, 20 µm. (D) F-actin was detected
by staining with phalloidin. In DZNep-treated group, F-actin was
remodeled and polarized, whereas in the DMSO group F-actin
presented a stress pattern. Scale bar, 20 µm. (E) Western
blot assays were performed to show the expression of EMT-related
proteins after treatment with DZNep (2, 5 or 10 µmol/).
DMSO, dimethyl sulfoxide; DZNep, 3-deazaneplanocin A; EMT,
epithelial-mesenchymal transition; NC, negative control; si, small
interfering RNA; STAT3, signal transducer and activator of
transcription factor 3; STAT3, signal transducer and activator of
transcription factor 3; Twist-1, Twist-related protein 1. |
Inhibition of STAT3 attenuates HNSCC cell
migration, invasion and EMT in vitro
Since EZH2 mediates the EMT process of HNSCC cells
as suggested by our results (Fig.
2), we aimed to determine whether downstream effector molecules
of EZH2 participate in EMT. As shown in Fig. 1A and B, STAT3 acted as a downstream
target of EZH2, we investigated whether EZH2 mediates EMT in HNSCC
cells through regulating STAT3. To inhibit STAT3 activity, we
transfected UM1 and CAL27 cells with siSTAT3. After 48 h of
transfection, the expression of STAT3 and p-STAT3 (Tyr705) were
significantly decreased in both UM1 and CAL27 cell lines compared
with the control (Fig. 1C).
Notably, STAT3 knockdown significantly inhibited HNSCC cells
migration compared with the scramble transfected cells as suggested
by a scratch-wound assay (P<0.001; Fig. 3A). Additionally, a Transwell
chamber assay confirmed that siSTAT3 significantly inhibited the
invasion and migration of HNSCC cells in vitro compared with
the control (P<0.001; Fig. 3B).
We next determined whether STAT3 participates in the EMT process of
UM1 and CAL27 cells by western blotting. After STAT3 knockdown, the
expression of the EMT marker, E-cadherin, was markedly increased,
while that of N-cadherin, Vimentin and Twist-1 was decreased,
compared with the control (Fig.
3E). Immunofluorescence staining for the EMT-related markers
E-cadherin, N-cadherin and Vimentin, further validated the results
of western blot assays (Fig. 3C).
Furthermore, F-actin presented a notable stress fiber pattern in
HNSCC cells treated with siSTAT3 compared with the control cells
(Fig. 3D). Overall, our results
indicated that knockdown of STAT3 efficiently suppressed the EMT
process and rearranged the cytoskeletal proteins in HNSCC
cells.
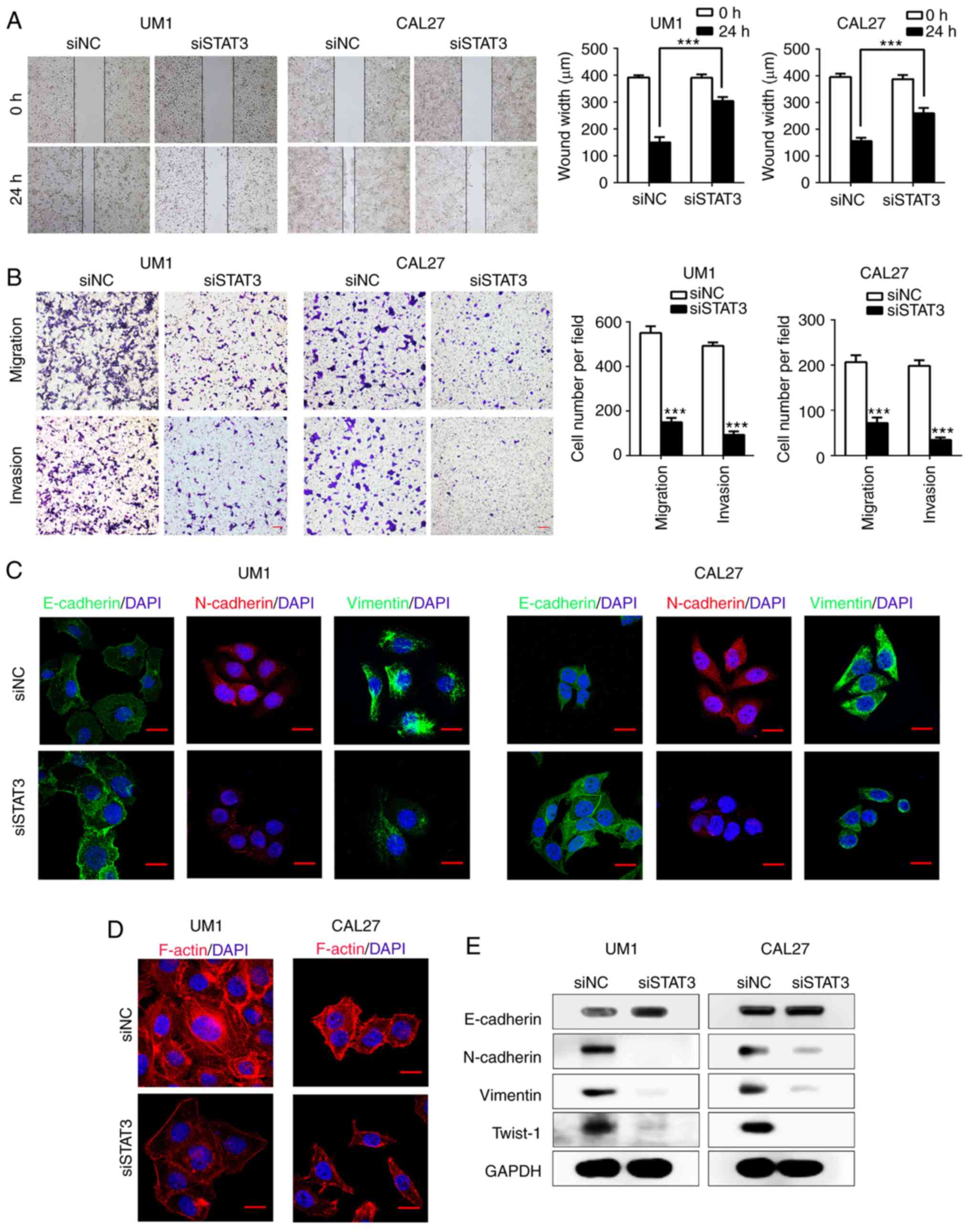 | Figure 3STAT3 inhibition suppresses the EMT
of HNSCC cells in vitro. (A) Wound-healing assays performed
with siSTAT3-treated UM1 and CAL27 cells, compared with scramble
HNSCC cells. ***P<0.001. (B) Transwell assays
performed to show migration (without Matrigel) and invasion (with
Matrigel) in siSTAT3-treated group. ***P<0.001 vs.
siNC. Scale bar, 50 µm. (C) Confocal immunofluorescence
analysis performed to show the change of EMT-related proteins in
siSTAT3-treated cells compared with the scramble cells. Scale bar,
20 µm. (D) Confocal immunofluorescence analysis performed to
show F-actin rearrangement in STAT3 siRNA-treated HNSCC cells.
Scale bar, 20 µm. (E) Western blot analysis was performed to
show the effects of STAT3 knockdown on the expression of
E-cadherin, N-cadherin, Vimentin and Twist-1 in UM1 and CAL27
cells. EMT, epithelial-mesenchymal transition; HNSCC, head and neck
squamous cell carcinoma; siSTAT3, STAT3, small interfering RNA
signal transducer and activator of transcription factor 3; Twist-1,
Twist-related protein 1. |
Inhibition of VEGFR2 attenuates the
migration, invasion and EMT of HNSCC cells in vitro
Previous studies have demonstrated that
constitutively activated STAT3 plays a pivotal role in tumor
angiogenesis through upregulating VEGF expression (30,31).
Recent reports showed that inhibition of STAT3 activity could
downregulate VEGF and VEGFR2 expression (32), while VEGFR2 mediates cancer
metastasis (33). We therefore
hypothesized that VEGFR2 may act as a downstream factor of STAT3
and mediate the EMT process in HNSCC. We transfected UM1 and CAL27
cells with siKDR. As presented in Fig.
4E, the expression of VEGFR2 was notably lower after siKDR
transfection in UM1 and CAL27 cells. Similarly, VEGF expression was
also reduced. Intriguingly, the Transwell assay showed that siKDR
treatment significantly inhibited UM1 and CAL27 cells migration and
invasion compared with the control (P<0.001; Fig. 4B). The results of the scratch-wound
assays showed a significant reduction in the migration in both UM1
and CAL27 cell lines compared with the control (P<0.001 and
P<0.01; Fig. 4A). To further
demonstrate that VEGFR2 mediates the invasion and migration of
HNSCC cells, we knocked down VEGFR2 expression in UM1 and CAL27
cells with the EMT-phenotype. Western blotting showed that the EMT
marker, E-cadherin, was notably upregulated, while the expression
of N-cadherin, Vimentin and Twist-1 was markedly downregulated in
the siKDR-treated group (Fig. 4E).
Immunofluorescence staining demonstrated that the expression of
E-cadherin was increased, and that of N-cadherin and Vimentin was
decreased (Fig. 4C). In addition,
the levels of F-actin in siKDR-treated group were markedly lowered
and caused the changes in the cellular morphology compared with the
control group (Fig. 4D).
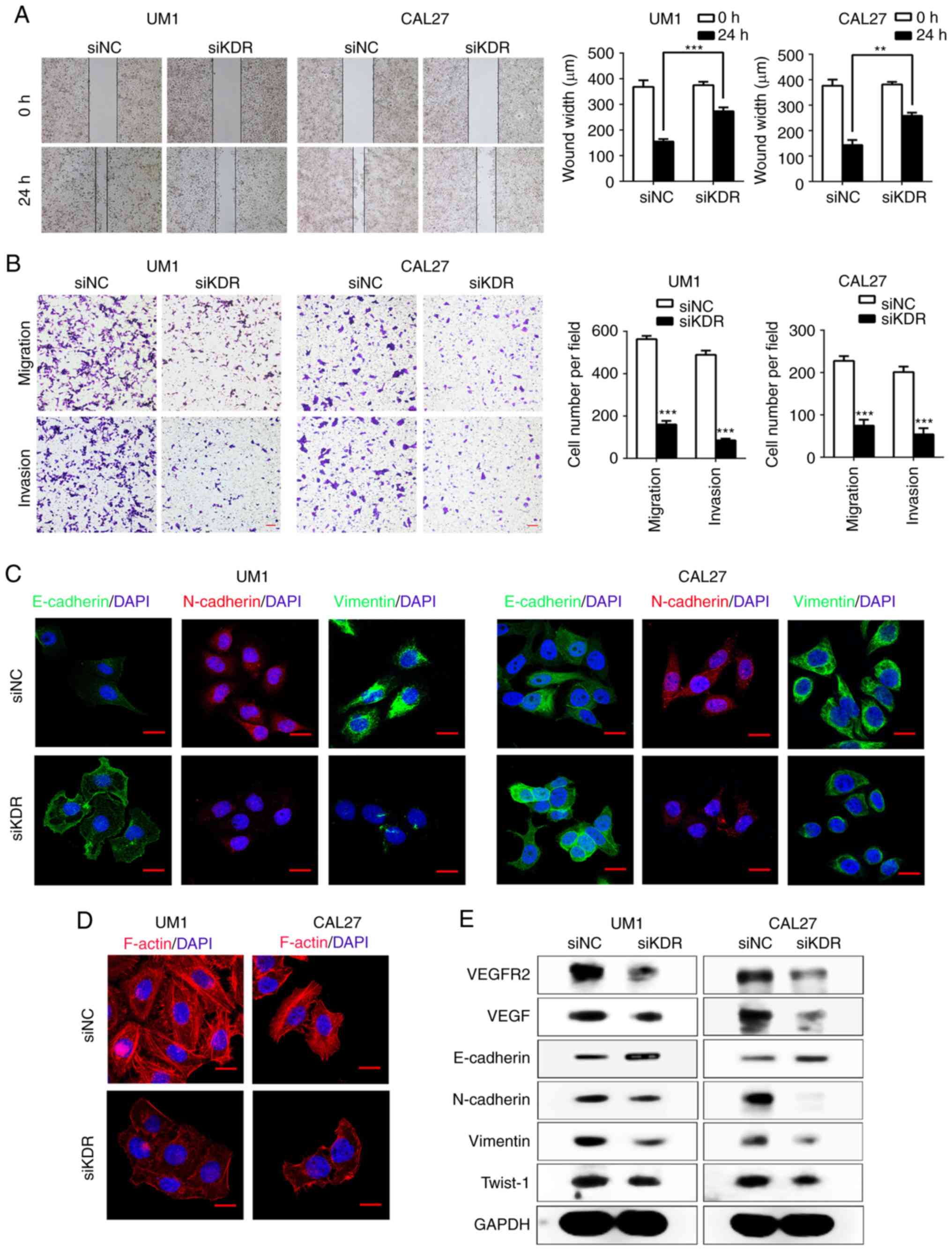 | Figure 4VEGFR2 regulates EMT in HNSCC cells
in vitro. (A) Wound-healing assays performed to investigate
the migration in siKDR-treated UM1 and CAL27 cells
(**P<0.01, ***P<0.001). (B) Transwell
assays performed to show migration (without Matrigel) and invasion
(with Matrigel) in the siKDR-treated group.
***P<0.001 vs. siNC. Scale bar, 50 µm. (C)
Confocal immunofluorescence analysis performed to show the change
in the expression of EMT-related markers in siKDR-treated cells
compared with the scramble cells. Scale bar, 20 µm. (D)
Confocal immunofluorescence analysis to show F-actin rearrangement
in siKDR-treated HNSCC cells. Scale bar, 20 µm. (E) Western
blot analysis was performed to show the effects of VEGFR2 knockdown
on the expression of E-cadherin, VEGF, N-cadherin, Vimentin and
Twist-1 in UM1 and CAL27 cell lines. EMT, epithelial-mesenchymal
transition; HNSCC, head and neck squamous cell carcinoma; NC,
negative control; si, small interfering RNA; Twist-1, Twist-related
protein 1; VEGF, vascular endothelial growth factor; VEGFR2, VEGF
receptor 2; KDR, kinase insert domain receptor. |
Inhibition of the EZH2/STAT3/VEGFR2 axis
suppresses the growth of HNSCC xenograft tumors and EMT in
vivo
To examine whether inhibition of EZH2/STAT3/VEGFR2
axis reverses EMT in vivo, we established HNSCC xenograft
tumors in immunocompromised mice using the CAL27 cell line.
Compared with DMSO-treated tumors, the DZNep-treated group
exhibited significantly lower tumor volume and tumor weight
compared with the control (P<0.05; Fig. 5A-C). In addition, the volume and
weight of tumors of the Stattic group [a small molecule inhibitor
selectively inhibits the expression levels of p-STAT3 (Try705),
without any effect on total STAT3], those of the Stattic + DZNep,
and Apatinib groups (a highly potent tyrosine-kinase inhibitor
targeting VEGFR2) were also significantly reduced compared with
DMSO group (P<0.01, P<0.01, P<0.05; Fig. 5A-C). Interestingly, H&E
staining in DMSO-treated CAL27 tumors had crowded nuclei with
hyperchromatism and pleomorphism, while in the DZNep-treated group
the nucleus of tumor cells exhibited a reduced degree of atypia.
Moreover, IHC staining showed decreased expression of EZH2, p-STAT3
(Tyr705), VEGF, VEGFR2, N-cadherin and Vimentin, and increased
expression of E-cadherin, without notable alterations in total
STAT3 levels in DZNep-treated CAL27 tumors (Fig. 5D and E). These results further
validated that EZH2, STAT3 and VEGFR2 were closely linked and
inhibition of this axis could profoundly impact the EMT of HNSCC
cells.
 | Figure 5Targeting the EZH2/STAT3/VEGFR2 axis
inhibits the growth of HNSCC xenograft tumors and reverses EMT in
HNSCC tumors in vivo. (A-C) The volume and weight of CAL27
xenograft tumors treated with DMSO, DZNep, Stattic, DZNep +
Stattic, and Apatinib (two-way ANOVA; *P<0.05,
**P<0.01 vs. DMSO). (D) Immunohistochemical staining
was performed to show EMT-related markers N-cadherin, Vimentin, and
E-cadherin expression in the DZNep-treated group (magnification,
×400). (E) Immunohistochemical staining performed to determine the
expression of EZH2, STAT3, p-STAT3 (Tyr705), VEGF, and VEGFR2 in
the DZNep-treated group (magnification, 400). DMSO, dimethyl
sulfoxide; DZNep, 3-deazaneplanocin A; EMT, epithelial-mesen-chymal
transition; EZH2, enhancer of zeste homolog 2; HNSCC, head and neck
squamous cell carcinoma; p, phosphorylated; STAT3, signal
transducer and activator of transcription factor 3; VEGF, vascular
endothelial growth factor; VEGFR2, VEGF receptor 2. |
Discussion
The development of novel targets for the treatment
of metastatic HNSCC is necessary. Accumulating evidence suggests
that EMT and cancer metastatic ability are inextricably associated
during cancer development (4,5);
however, the molecular mechanisms underlying EMT in HNSCC remain
unclear. In this study, we reported that an EZH2-centered axis
regulates HNSCC progression, as manifested by our observations that
targeting the EZH2/STAT3/VEGFR2 pathway suppresses HNSCC metastatic
progression both in vitro and in vivo.
Tumor metastasis is the major cause of
cancer-associated mortality and the process has been divided into
at least four steps: Local invasion, intravasation, extravasation
and metastatic colonization (3,4). EMT
is considered to play a critical role in initiating cancer
metastasis, in association with loss of E-cadherin expression in
HNSCC, particularly in patients with lymph node metastasis and with
poor prognosis (4,34). As of its role in epigenetic gene
silencing and chromatin remodeling, EZH2 is crucial in
tumorigenesis and cancer progression (13,14).
Accumulating evidence suggests that EZH2 is closely related to EMT
in several cancer types (35), and
our previous study demonstrated that EZH2 is upregulated in HNSCC
and acts as a negative prognostic factor for patients with HNSCC
(11). The present study, to the
best of our knowledge, we are the first to demonstrate that
inhibition of EZH2 reverses EMT in HNSCC cells and xenograft tumors
by regulating the STAT3/VEGFR2 axis in vitro and in
vivo.
Transcription factor STAT3 was shown to be
abnormally activated in various cancer types. With a well-known
role in promoting tumor cell proliferation, survival, tumor
invasion, angiogenesis and immunosuppression, STAT3 also promotes
cancer development through inflammation, stem cells, obesity, and
the pre-metastatic niche formation (16-19,36-38).
Our previous study demonstrated that targeting STAT3 by using
HJC0152 and other inhibitors significantly suppressed the growth
and invasion of HNSCC via regulating the microRNA-21/β-catenin axis
(28). Recent research has shown
that EZH2 could enhance cancer cell invasion and metastasis through
regulating the activation of STAT3 (39). One study reported that targeting
EZH2/STAT3 signals with EZH2 inhibitor GSK-126 or
ASC-J9® (a novel androgen receptor degradation enhancer)
decreases the invasion of prostate cancer stem/progenitor cells
(40). Another study validated
that phosphorylation of EZH2 at serine 21 via AKT promotes STAT3
methylation and enhances the activation of STAT3 (20). STAT3 is closely associated with
VEGF (24,25) or cyclin dependent kinase 2
(41) in cancer progression. Lee
et al (42) demonstrated
that arsenic herbal mixture PROS exerts antitumor effects by
targeting STAT3/VEGF/CDK2 axis in non-small lung cancer cells.
In our in vivo experiments, we found that
inhibition of EZH2 in HNSCC cells resulted in the suppression of
p-STAT3 (Tyr705) expression, along with reduced expression of VEGF,
VEGFR2, and EMT-related markers but E-cadherin was elevated. In
contrast, overexpression of EZH2 resulted in increased the
expression p-STAT3 (Tyr705), VEGF, and VEGFR2. Additionally, after
targeting STAT3 by siRNA, the expression of p-STAT3 (Tyr705), VEGF,
VEGFR2, and EMT-related markers except E-cadherin were similarly
attenuated. Consistently, EZH2 inhibition decreased the migration
and invasion of HNSCC cells, similar results were found in the
HNSCC cells treated with siSTAT3. Angiogenesis plays an important
role in tumor growth and metastasis (24). VEGF and its receptors, mainly
VEGFR2, have a crucial role in promoting angiogenesis (43). Niu et al (30) reported that constitutively
activated STAT3 upregulates VEGF expression and thereby induces
tumor angiogenesis. Xue et al (32) demonstrated that STAT3 inhibition
decreases the expression of VEGF and VEGFR2. In current study, we
found that targeting STAT3 by siRNA in HNSCC cells attenuates the
expression of VEGF and VEGFR2, suggesting that the VEGF/VEGFR2 axis
served as the downstream effectors of STAT3 in HNSCC cells.
Finally, we demonstrated that knocking-down of VEGFR2 by siKDR
decreased the expression of EMT-related markers but increased
E-cadherin, meanwhile the invasion and migration abilities of HNSCC
cells were attenuated. These findings suggest that targeting EZH2
reverses EMT in HNSCC cells via regulating STAT3/VEGFR2 axis.
The results of our in vivo study consistently
confirmed the in vitro findings. EZH2 inhibition suppressed
the growth and EMT process of CAL27 cell-derived xenograft tumors.
To the best of our knowledge, our findings indicated that the
EZH2/STAT3/VEGFR2 axis plays a critical role in promoting the
metastasis of HNSCC. One potential application of this study is the
targeting of the EZH2/STAT3/VEGFR2 axis as a novel and effective
approach in treating invasive, progressive, and metastatic
HNSCC.
In summary, we demonstrated that EZH2 promotes the
progression and EMT process of HNSCC via regulating STAT3 and
VEGFR2. This EZH2-centered mechanism in HNSCC metastasis may serve
as a rationale to develop general and/or specific inhibitory
approaches for targeting the EZH2/STAT3/VEGFR2 axis effectively
control HNSCC progression and metastasis.
Funding
This work was supported by grants from the National
Natural Science Foundation of China (grant nos. 81572492, 81872206,
81872495, 81702678 and 81673026), the National Clinical Research
Center for Cancer (NCRCC) and Supported by Special Program of
Talents Development for Excellent Youth Scholars in Tianjin.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MZ, XH, YX, CW, JC and LK performed the experiments
and data analysis, and wrote the manuscript. LK, SS, YR and RJ were
involved in revising the manuscript critically for important
intellectual content. XZ and LZ also gave final approval of the
version to be published. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Animal Care and Use Committee of Tianjin Medical University Cancer
Institute and Hospital (Tianjin, China).
Patient consent for publication
Not applicable.
Competing interests
The authors have declared that no competing interest
exists.
Acknowledgments
Not applicable.
Abbreviations:
|
DZNep
|
3-deazaneplanocin A
|
|
EMT
|
epithelial-mesenchymal transition
|
|
EZH2
|
enhancer of zeste homolog 2
|
|
H3K27me3
|
trimethylation of lysine 27 on histone
H3
|
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
STAT3
|
signal transducer and activator of
transcription factor 3
|
|
VEGF
|
vascular endothelial growth factor
|
|
VEGFR2
|
vascular endothelial growth factor
receptor 2
|
|
KDR
|
kinase insert domain receptor
|
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
2
|
Leemans CR, Braakhuis BJ and Brakenhoff
RH: The molecular biology of head and neck cancer. Nat Rev Cancer.
11:9–22. 2011. View
Article : Google Scholar
|
|
3
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Smith A, Teknos TN and Pan Q: Epithelial
to mesenchymal transition in head and neck squamous cell carcinoma.
Oral Oncol. 49:287–292. 2013. View Article : Google Scholar :
|
|
5
|
Sun SS, Zhou X, Huang YY, Kong LP, Mei M,
Guo WY, Zhao MH, Ren Y, Shen Q and Zhang L: Targeting STAT3/miR-21
axis inhibits epithelial-mesenchymal transition via regulating CDK5
in head and neck squamous cell carcinoma. Mol Cancer. 14:2132015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cao R, Wang L, Wang H, Xia L,
Erdjument-Bromage H, Tempst P, Jones RS and Zhang Y: Role of
histone H3 lysine 27 methylation in polycomb-group silencing.
Science. 298:1039–1043. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kuzmichev A, Nishioka K, Erdjument-Bromage
H, Tempst P and Reinberg D: Histone methyltransferase activity
associated with a human multiprotein complex containing the
enhancer of zeste protein. Genes Dev. 16:2893–2905. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karanikolas BD, Figueiredo ML and Wu L:
Comprehensive evaluation of the role of EZH2 in the growth,
invasion, and aggression of a panel of prostate cancer cell lines.
Prostate. 70:675–688. 2010.PubMed/NCBI
|
|
9
|
Ahani N, Shirkoohi R, Rokouei M, Alipour
Eskandani M and Nikravesh A: Overexpression of enhancer of zeste
human homolog 2 (EZH2) gene in human cytomegalovirus positive
glioblastoma multiforme tissues. Med Oncol. 31:2522014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reijm EA, Timmermans AM, Look MP,
Meijer-van Gelder ME, Stobbe CK, van Deurzen CH, Martens JW,
Sleijfer S, Foekens JA, Berns PM and Jansen MP: High protein
expression of EZH2 is related to unfavorable outcome to tamoxifen
in metastatic breast cancer. Ann Oncol. 25:2185–2190. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou X, Ren Y, Kong LP, Cai GH, Sun SS,
Song WZ, Wang Y, Jin R, Qi L, Mei M, et al: Targeting EZH2
regulates tumor growth and apoptosis through modulating
mitochondria dependent cell-death pathway in HNSCC. Oncotarget.
6:33720–33732. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun J, Zheng G, Gu Z and Guo Z: MiR-137
inhibits proliferation and angiogenesis of human glioblastoma cells
by targeting EZH2. J Neurooncol. 122:481–489. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luo H, Jiang Y, Ma S, Chang H, Yi C, Cao
H, Gao H, Guo H, Hou J, Yan J, et al: EZH2 promotes invasion and
metastasis of laryngeal squamous cells carcinoma via
epithelial-mesenchymal transition through H3K27me3. Biochem Biophys
Res Commun. 479:253–259. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mu Z, Li H, Fernandez SV, Alpaugh KR,
Zhang R and Cristofanilli M: EZH2 knockdown suppresses the growth
and invasion of human inflammatory breast cancer cells. J Exp Clin
Cancer Res. 32:702013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang C, Liu X, Chen Z, Huang H, Jin Y,
Kolokythas A, Wang A, Dai Y, Wong DT and Zhou X: Polycomb group
protein EZH2-mediated E-cadherin repression promotes metastasis of
oral tongue squamous cell carcinoma. Mol Carcinog. 52:229–236.
2013. View
Article : Google Scholar
|
|
16
|
Frank DA: STAT3 as a central mediator of
neoplastic cellular transformation. Cancer Lett. 251:199–210. 2007.
View Article : Google Scholar
|
|
17
|
Bar-Natan M, Nelson EA, Xiang M and Frank
DA: STAT signaling in the pathogenesis and treatment of myeloid
malignancies. JAKSTAT. 1:55–64. 2012.PubMed/NCBI
|
|
18
|
Xiong A, Yang Z, Shen Y, Zhou J and Shen
Q: Transcription factor STAT3 as a novel molecular target for
cancer prevention. Cancers (Basel). 6:926–957. 2014. View Article : Google Scholar
|
|
19
|
Masuda M, Suzui M, Yasumatu R, Nakashima
T, Kuratomi Y, Azuma K, Tomita K, Komiyama S and Weinstein IB:
Constitutive activation of signal transducers and activators of
transcription 3 correlates with cyclin D1 overexpression and may
provide a novel prognostic marker in head and neck squamous cell
carcinoma. Cancer Res. 62:3351–3355. 2002.PubMed/NCBI
|
|
20
|
Kim E, Kim M, Woo DH, Shin Y, Shin J,
Chang N, Oh YT, Kim H, Rheey J, Nakano I, et al: Phosphorylation of
EZH2 activates STAT3 signaling via STAT3 methylation and promotes
tumorigenicity of glioblastoma stem-like cells. Cancer Cell.
23:839–852. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dasgupta M, Dermawan JK, Willard B and
Stark GR: STAT3-driven transcription depends upon the dimethylation
of K49 by EZH2. Proc Natl Acad Sci USA. 112:3985–3990. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Neuchrist C, Erovic BM, Handisurya A,
Steiner GE, Rockwell P, Gedlicka C and Burian M: Vascular
endothelial growth factor receptor 2 (VEGFR2) expression in
squamous cell carcinomas of the head and neck. Laryngoscope.
111:1834–1841. 2001. View Article : Google Scholar
|
|
23
|
Lu W, Chen H, Ye F, Wang F and Xie X: VEGF
induces phosphorylation of STAT3 through binding VEGFR2 in ovarian
carcinoma cells in vitro. Eur J Gynaecol Oncol. 27:363–369.
2006.PubMed/NCBI
|
|
24
|
Zhao M, Gao FH, Wang JY, Liu F, Yuan HH,
Zhang WY and Jiang B: JAK2/STAT3 signaling pathway activation
mediates tumor angiogenesis by upregulation of VEGF and bFGF in
non-small-cell lung cancer. Lung Cancer. 73:366–374. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xie TX, Huang FJ, Aldape KD, Kang SH, Liu
M, Gershenwald JE, Xie K, Sawaya R and Huang S: Activation of stat3
in human melanoma promotes brain metastasis. Cancer Res.
66:3188–3196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
U.S. National Institutes of Health:
Laboratory animal welfare: Public Health Service policy on humane
care and use of laboratory animals by awardee institutions; notice.
Fed Regist. 50:19584–19585. 1985.PubMed/NCBI
|
|
27
|
Feldman AT and Wolfe D: Tissue processing
and hematoxylin and eosin staining. Methods Mol Biol. 1180:31–43.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Y, Wang S, Wu Y, Ren Y, Li Z, Yao X,
Zhang C, Ye N, Jing C, Dong J, et al: Suppression of the growth and
invasion of human head and neck squamous cell carcinomas via
regulating STAT3 signaling and the miR-21/β-catenin axis with
HJC0152. Mol Cancer Ther. 16:578–590. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nieto MA: The ins and outs of the
epithelial to mesenchymal transition in health and disease. Annu
Rev Cell Dev Biol. 27:347–376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Niu G, Wright KL, Huang M, Song L, Haura
E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, et al:
Constitutive Stat3 activity up-regulates VEGF expression and tumor
angio-genesis. Oncogene. 21:2000–2008. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wei D, Le X, Zheng L, Wang L, Frey JA, Gao
AC, Peng Z, Huang S, Xiong HQ, Abbruzzese JL and Xie K: Stat3
activation regulates the expression of vascular endothelial growth
factor and human pancreatic cancer angiogenesis and metastasis.
Oncogene. 22:319–329. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xue C, Xie J, Zhao D, Lin S, Zhou T, Shi
S, Shao X, Lin Y, Zhu B and Cai X: The JAK/STAT3 signalling pathway
regulated angiogenesis in an endothelial cell/adipose-derived
stromal cell co-culture, 3D gel model. Cell Prolif. 50:2017.
View Article : Google Scholar
|
|
33
|
Balakrishnan S, Bhat FA, Raja Singh P,
Mukherjee S, Elumalai P, Das S, Patra CR and Arunakaran J: Gold
nanoparticle-conjugated quercetin inhibits epithelial-mesenchymal
transition, angiogenesis and invasiveness via EGFR/VEGFR-2-mediated
pathway in breast cancer. Cell Prolif. 49:678–697. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Diniz-Freitas M, Garcia-Caballero T,
Antúnez-López J, Gándara-Rey JM and Garcia-Garcia A: Reduced
E-cadherin expression is an indicator of unfavourable prognosis in
oral squamous cell carcinoma. Oral Oncol. 42:190–200. 2006.
View Article : Google Scholar
|
|
35
|
Chang JW, Gwak SY, Shim GA, Liu L, Lim YC,
Kim JM, Jung MG and Koo BS: EZH2 is associated with poor prognosis
in head-and-neck squamous cell carcinoma via regulating the
epithelial-to-mesenchymal transition and chemosensitivity. Oral
Oncol. 52:66–74. 2016. View Article : Google Scholar
|
|
36
|
Priceman SJ, Kujawski M, Shen SD,
Cherryholmes GA, Lee H, Zhang C, Kruper L, Mortimer J, Jove R,
Riggs AD and Yu H: Regulation of adipose tissue T cell subsets by
Stat3 is crucial for diet-induced obesity and insulin resistance.
Proc Natl Acad Sci USA. 110:13079–13084. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schroeder A, Herrmann A, Cherryholmes G,
Kowolik C, Buettner R, Pal S, Yu H, Müller-Newen G and Jove R: Loss
of androgen receptor expression promotes a stem-like cell phenotype
in prostate cancer through STAT3 signaling. Cancer Res.
74:1227–1237. 2014. View Article : Google Scholar
|
|
38
|
Yu H, Lee H, Herrmann A, Buettner R and
Jove R: Revisiting STAT3 signalling in cancer: New and unexpected
biological functions. Nat Rev Cancer. 14:736–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu Z, Sun Y, Guo Y, Qin G, Mu S, Fan R,
Wang B, Gao W, Wu H, Wang G and Zhang Z: NF-YA promotes invasion
and angiogenesis by upregulating EZH2-STAT3 signaling in human
melanoma cells. Oncol Rep. 35:3630–3638. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wen S, Tian J, Niu Y, Li L, Yeh S and
Chang C: ASC-J9(®), and not casodex or enzalutamide, suppresses
prostate cancer stem/progenitor cell invasion via altering the
EZH2-STAT3 signals. Cancer Lett. 376:377–386. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Steinman RA, Wentzel A, Lu Y, Stehle C and
Grandis JR: Activation of Stat3 by cell confluence reveals negative
regulation of Stat3 by cdk2. Oncogene. 22:3608–3615. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee H, Lee HJ, Bae IJ, Kim JJ and Kim SH:
Inhibition of STAT3/VEGF/CDK2 axis signaling is critically involved
in the antiangiogenic and apoptotic effects of arsenic herbal
mixture PROS in non-small lung cancer cells. Oncotarget.
8:101771–101783. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hoeben A, Landuyt B, Highley MS, Wildiers
H, Van Oosterom AT and De Bruijn EA: Vascular endothelial growth
factor and angio-genesis. Pharmacol Rev. 56:549–580. 2004.
View Article : Google Scholar : PubMed/NCBI
|














