Introduction
Lung cancer is the leading cause of cancer-related
death worldwide (1). It is the
deadliest malignancy, regardless of the incidence rate (11.4%) or
the fatality rate (18.0%), according to the 2020 Global Cancer
Annual Report (2). Non-small cell
lung carcinoma (NSCLC) accounts for 85% of all lung cancer cases
(3). Numerous recurrent genetic
alterations cause NSCLC, of which KRAS is a classic example. KRAS
mutations are identified in 32% of patients with NSCLC (4) and are associated with a poor
prognosis and a high likelihood of disease recurrence (5). KRAS is an oncogenic driver and its
mutation can directly accelerate lung tumor growth. Therefore, it
is considered a prospective therapeutic target and a possible
diagnostic marker or biomarker for NSCLC-targeted therapy (6). Accordingly, the development of
therapeutic drugs that target KRAS mutations is one of the most
successful lung cancer treatments currently available. AMG-510
(Sotorasib) is a potent, orally bioavailable, and selective
KRASG12C covalent inhibitor with anti-tumor activity. In
a single-group, phase 2 trial, treatment-related adverse events
occurred in 88 of 126 patients (69.8%), including grade 3 events in
25 patients (19.8%) and a grade 4 event in 1 (0.8%) (7). In addition, the response rate of
patients with NSCLC to AMG-510 is only 37% (8). It appears that the identification of
effective and safe treatments for NSCLC caused by KRAS mutations
remains an ongoing process.
Realgar, also known as tetra-arsenic tetra-sulfide
(As4S4 or As2S2), is a
traditional Chinese medicine mainly used in ancient China for
deworming. However, currently it is extensively used as an
anticancer treatment, particularly for malignant tumors (9) and hematological malignancies
(10). The 2020 edition of Chinese
Pharmacopoeia stipulates that the daily dosage of realgar is
between 50 mg/70 kg to 100 mg/70 kg per day. Then main content of
realgar is arsenic, and the therapeutic and toxic concentrations of
arsenic in human blood were 2-70 and 50-250 ng/ml, respectively.
Pharmacokinetic variables were analyzed in 7 volunteers with APL
and HCR by using a single dose of As4S4. The
single dose of up to 60 mg/kg As4S4 was well
tolerated. Arsenic could be detected in the blood 30 min after oral
administration of As4S4. The time peak was
3.4±1.4 h and the Cmax was 24.9±8.0 µg/l (11). Clinicians demonstrated in 1995 that
realgar-indigo naturalis formula-treated patients with acute
promyelocytic leukemia had complete remission rates between 96.7
and 98% and a 5-year overall survival rate of 86.88% (12). Recent studies have demonstrated
that realgar formulations can also be used to treat other
malignancies, including NSCLC. Shi et al (13) demonstrated that realgar burning
could improve the immunological function and hypercoagulability of
patients with lung cancer. The total effective rate was 81.25%. An
additional study indicated that Nano-realgar suppressed lung tumor
growth in vitro and in vivo by inhibiting metabolic
reprogramming (14). Moreover, in
our previous study, it was shown that realgar could reduce Ras
expression via the Ras/MAPK signaling pathway in Caenorhabditis
elegans (C. elegans) (15), suggesting that realgar is a
prospective anticancer medication that prevents Ras mutations.
However, the beneficial effect of realgar in KRAS-driven lung
cancer remains unknown. As a result, the emphasis of the present
study was on the effects of realgar on lung cancer cells that
possess KRAS mutations.
The vital KRAS-controlled pathways are important
players in the growth of certain malignancies (16,17).
Activation of the Ras/Raf/ERK signaling pathway is crucial for the
development and spread of cancer (18). KRAS mediates mitogenic signal
transduction from cell surface receptors to intracellular effectors
and pathways such as Raf-MAPK (19). C-raf is the first known KRAS
effector. KRAS recruits Raf proteins (A-raf, B-raf, and C-Raf) to
the cell membrane, resulting in Raf activation (20,21).
It has been reported that Raf inhibitors have been shown to have
antitumor activity in KRAS or BRAF mutant cancers (22). Recent studies have shown that the
clinical resistance to the KRAS inhibitor AMG-510 in NSCLC is
characterized by various pathways, the majority of which converge
on the Ras/MAPK pathway (23,24).
Inhibition of Ras/MAPK is consequently a big hurdle for enhancing
the efficacy in KRASG12C patients (8). The process of developing KRAS
inhibitors requires not only examination of the mutational status
of KRAS, but also regulation of the downstream Ras signaling
pathway, which can boost the effectiveness of KRAS inhibitors.
Previous studies have revealed that ferroptosis is
preferentially triggered in cells overexpressing mutant Ras
oncoproteins (25-27). Specific connections have been
reported between ferroptosis and the KRAS mutant NSCLC. Since
ferroptosis is strongly associated with cancer progression
(28), patients with lung cancer
have shown elevated ferritin concentrations in their serum,
bronchoalveolar lavage fluid, and exhaled air condensate samples
(29,30). To form erythroid cells and other
cell types, transferrin receptor 1 (Tfr1) controls the intake of
transferrin-bound circulating iron levels (31). In 88% of NSCLCs, Tfr1 is
significantly expressed implying that lung cancer cells may be able
to boost iron intake by enhancing the effects of the transferrin
protein and the transferrin protein receptor. Increased Tfr1
expression is hypothesized to be a mechanism by which Ras enriches
cellular iron pools, enhancing ferroptosis sensitivity (25). In addition, Ras-selective
inhibitors, including NSCLC cells, can trigger non-apoptotic,
iron-dependent cell death in Ras-mutant cancer cells (32-34).
Cancer cells therefore store more iron than healthy cells (35), and KRAS mutant cells are more
vulnerable to ferroptosis. In the present study, the data indicated
that H23, a KRAS mutant cell line, was more sensitive to realgar
treatment than the non-KRAS mutant cell line H1650.
Therefore, the objective of the present study was to
determine whether realgar has an impact on KRAS-mutant lung cancer
cells and the potential correlation between ferroptosis and the
Ras/Raf pathway. The current study aimed to provide a new clinical
alternative for the treatment of KRAS-mutant NSCLC tumors.
Materials and methods
Preparation of realgar solution
According to the Chinese Pharmacopoeia, realgar from
Shimen (Hunan, China) was refined and identified. Briefly, 100 ml
purified water and 0.1% NaOH were added to 1 g realgar powder. The
solution was stirred overnight to spread evenly. The supernatant
was subsequently washed with 0.1 mol/l HCl until the pH reached
7.38. The solution was filtered through a 0.22-µm
microporous filter to be sterilized, sealed and refrigerated at
4°C. By using inductively coupled plasma mass spectrometry, the
concentration of total arsenic in solution was determined.
Following assessment, the realgar solution had a total arsenic
concentration of 2,592.067 µg/ml.
Cell culture and experimental groups
KRAS mutant H23, A549 and H460 cells, as well as
non-KRAS mutant H1650 cells, were cultured in RPMI-1640 (cat. no.
SH30809.01; Hyclone; Cytiva;) with 10% fetal bovine serum (cat. no.
11011-8611; Evergreen; www.hzsjq.com). The cells were incubated at 37°C in a
5% CO2 incubator, digested with trypsin (0.25%; Servier)
and seeded into plates once they reached the logarithmic growth
phase. Control, AMG-510 (10 µg/ml), realgar (1, 2 and 4
µg/ml), ferrostatin-1 (Fer-1, 1 µM), and sorafenib
(8, 16 and 32 nM) were used for the experimental groups. During the
logarithmic growth phase, all cells used in the experiments were
harvested.
MTT assay
Cell viability was determined using the MTT assay.
In 96-well plates, the cells were seeded at a density of
5×104 cells/ml. The cells were subsequently treated for
24 or 48 h with a specific concentration of realgar. MTT assay was
performed by adding 20 µl of MTT reagent (cat. no. M8180;
Beijing Solarbio Science & Technology Co., Ltd.) into each well
and continued with incubation for 4 h. After removing the
supernatant, 150 µl of DMSO (cat. no. G0004; Beijing
Solarbio Science & Technology Co., Ltd.) was mixed and then
placed on a plate shaker to dissolve formazan crystals. A
microplate reader was used to measure the absorbance at 570 nm
(Bio-Rad Laboratories, Inc.). The following formula was used to
calculate cell viability: Cell viability=(OD experimental group-OD
blank control group)/(OD normal group-OD blank control) ×100%.
Hoechst 33258 staining assay
Nuclear morphology was examined with Hoechst 33258
staining to assess cell apoptosis. A six-well plate was used and
the cells were plated at a density of 1×106 cells/ml.
The cells were fixed in 4% paraformaldehyde at room temperature for
15 min following 24 or 48 h of specific intervention. Subsequently,
the cells were gently rinsed three times with PBS prior to
incubation in the dark for 30 min at 37°C with 1 ml (5
µg/ml) Hoechst 33258 solution in each well. The images were
captured and analyzed using an Olympus IX71 fluorescence microscope
(Olympus Corporation) with an excitation wavelength range of
330-380 nm.
Apoptosis measurement
Annexin V/propidium iodide (PI) double labeling was
used to assess the induction of apoptosis in H23 and H1650 cells.
Annexin V/PI staining was carried out according to the
manufacturer's instructions using an apoptosis detection kit [cat.
no. MA0220-Jun-25G; Multi Sciences (lianke) Biotech, Co., Ltd.].
Following harvesting and staining with Annexin V/PI, the treated
cells were analyzed using flow cytometry (FACS CelestaTM; BD
Biosciences). FlowJo was used to analyze the data (version 10.6.2;
FlowJo LLC).
Transcriptomic analyses performed through
RNA sequencing
Briefly, mRNA was isolated using the Isolate RNA
mini kit (Bioline) and converted into cDNA. Subsequently, cDNA was
sequenced in a 150 bp paired-ended fashion on the Illumina
NovaSeq6000 to a depth of 40 million reads at the Amsterdam UMC
Genomics Core Facility. The quality control of the reads was
performed with FastQC (v0.11.2) and summarization through MultiQC
(v1.0). Differential expression (DE) analysis was performed using
the limma (v3.32.10) package DESeq2 (v1.28.0) in the R statistical
environment (v3.46.0). Differentially expressed genes (DEGs) were
defined as genes whose difference presented a limma |log2FC|>=1
and P-Value <=0.05. Functional analysis for genes in the key
module was performed using Metascape (https://metascape.org/gp/index.html#/main/step1) and
Gene Ontology (GO) was performed using the DAVID 6.8 database
(https://david.ncifcrf.gov) to elucidate
the mechanism of realgar in the treatment of H23. Subsequently,
building protein-protein interaction (PPI) Network with Cytoscape
(V3.8.2; http://www.cytoscape.org/).
Reactive oxygen species (ROS)
detection
To detect the intracellular ROS levels in H23 cells,
a ROS Assay kit (cat. no. S0033S; Beyotime Institute of
Biotechnology) was used. Realgar-treated cells were incubated for
20 min with 10 µM dichlorofluorescin diacetate. Prior to
counting the cells with a flow cytometer (BD Biosciences), they
were washed in PBS. FlowJo was used to analyze the average
fluorescence intensity (version 10.6.2; FlowJo LLC).
Measurement of Fe2+,
glutathione (GSH), and malondialdehyde (MDA) levels
A reduced GSH assay kit (Nanjing Jiancheng
Bioengineering Institute), Ferro Orange (Dojindo Molecular
Technologies, Inc.), and an MDA assay kit (Nanjing Jiancheng
Bioengineering Institute) were used to measure GSH, Fe2+
and MDA levels, respectively.
Measurement of mitochondrial membrane
potential
The membrane potential of H23 cells was determined
using a mitochondrial membrane potential assay kit and JC-1
(Beijing Solarbio Science & Technology Co., Ltd.). H23 cells
were seeded in a six-well plate and stained with JC-1 in the
working solution for 20 min prior to flow cytometry analysis (BD
Biosciences).
Transmission electron microscopy
(TEM)
H23 cells in the logarithmic growth phase were
cultivated in six-well plates with 2 ml cell suspension per well
for 48 h in culture medium containing different concentrations of
realgar. The cells were digested by trypsin without EDTA, prepared
following fixation in 2.5% glutaraldehyde 4 h and stored at 4°C,
and observed using TEM (Hitachi, Ltd.).
Western blotting and antibodies
The following commercially available antibodies were
used: Rabbit anti-c-Raf antibody (1:1,000; cat. no. cst-9422T),
rabbit anti-phosphorylated (p)-c-Raf antibody (1:1,000; cat. no.
cst-9427T), rabbit anti-ERK1/2 antibody (1:1,000; cat. no.
cst-4695T), rabbit anti-p-ERKl/2 antibody (1:2,000; cat. no.
cst-4370T), rabbit anti-JNK anti-body (1:1,000; cat. no.
cst-9252T), rabbit anti-p-JNK antibody (1:1,000; cat. no.
cst-4668T), rabbit anti-p38 antibody (1:1,000; cat. no. cst-8690T)
and rabbit anti-p-p38 antibodies (1:1,000; cat. no. cst-4511T) were
purchased from Cell Signaling Technology, Inc. A rabbit
anti-glutamate-cystine antiporter (Xct/SCL7A11; 1:1,000; cat. no.
ab175186) antibody was obtained from Abcam. The rabbit
anti-acyl-CoA synthetase long chain family member 4 (ACSL4)
antibody (1:1,000; cat. no. abs106075), rabbit anti-glutatione
peroxidase (GPX) 4 antibody (1:1,000; cat. no. abs136221),
anti-GAPDH (cat. no. abs830030ss) and anti-rabbit/mouse IgG,
horseradish peroxidase (HRP)-linked antibody (1:10,000; cat. no.
abs20040ss) were obtained from Absin (www.absin.cn).
Western blotting was used to assess the changes in
protein expression. Protein extracts were isolated from each group
cells using RIPA protein lysis buffer containing 1 mM PMSF (cat.
no. R0010; Beijing Solarbio Science & Technology Co., Ltd.).
They were centrifuged at 10,000 × g for 15 min at 4°C. A
bicinchoninic acid protein test kit was used to determine the
protein concentrations (Beijing Solarbio Science & Technology
Co., Ltd.). The protein lysates (30 µg) were separated by
10% SDS-PAGE and transferred to polyvinylidene fluoride membranes
(EMD Millipore, 0.25 µm). The membranes were subsequently
blocked in TBST buffer (TBS buffer with 0.1% Tween 20) containing
5% BSA (cat. no. PH0420; Phygene) for 2 h at room temperature. The
membrane was washed three times with TBST for 10 min each prior to
incubation with primary antibodies overnight at 4°C. The membrane
was subsequently incubated for 1 to 2 h at room temperature in
HRP-conjugated secondary antibodies. Finally, an enhanced
chemiluminescence detection kit was used with a chemiluminescence
detector (C300; Azure Biosystems, Inc.) to detect HRP luminescence
(Absin). Densitometric analysis was conducted to compare the
expression level of proteins using ImageJ (version 1.8.0; National
Institutes of Health).
Statistical analysis
The SPSS Statistics 23 software (IBM Corp.) was used
for statistical analysis. GraphPad Prism (version 8.0.2; GraphPad
Software, Inc.) was used for statistical analysis of the results.
One-way ANOVA was used to analyze group differences, followed by
the Tukey's post-hoc test. All repeated experiments were carried
out at least in triplicate. *P<0.05 was considered to
indicate a statistically significant difference.
Results
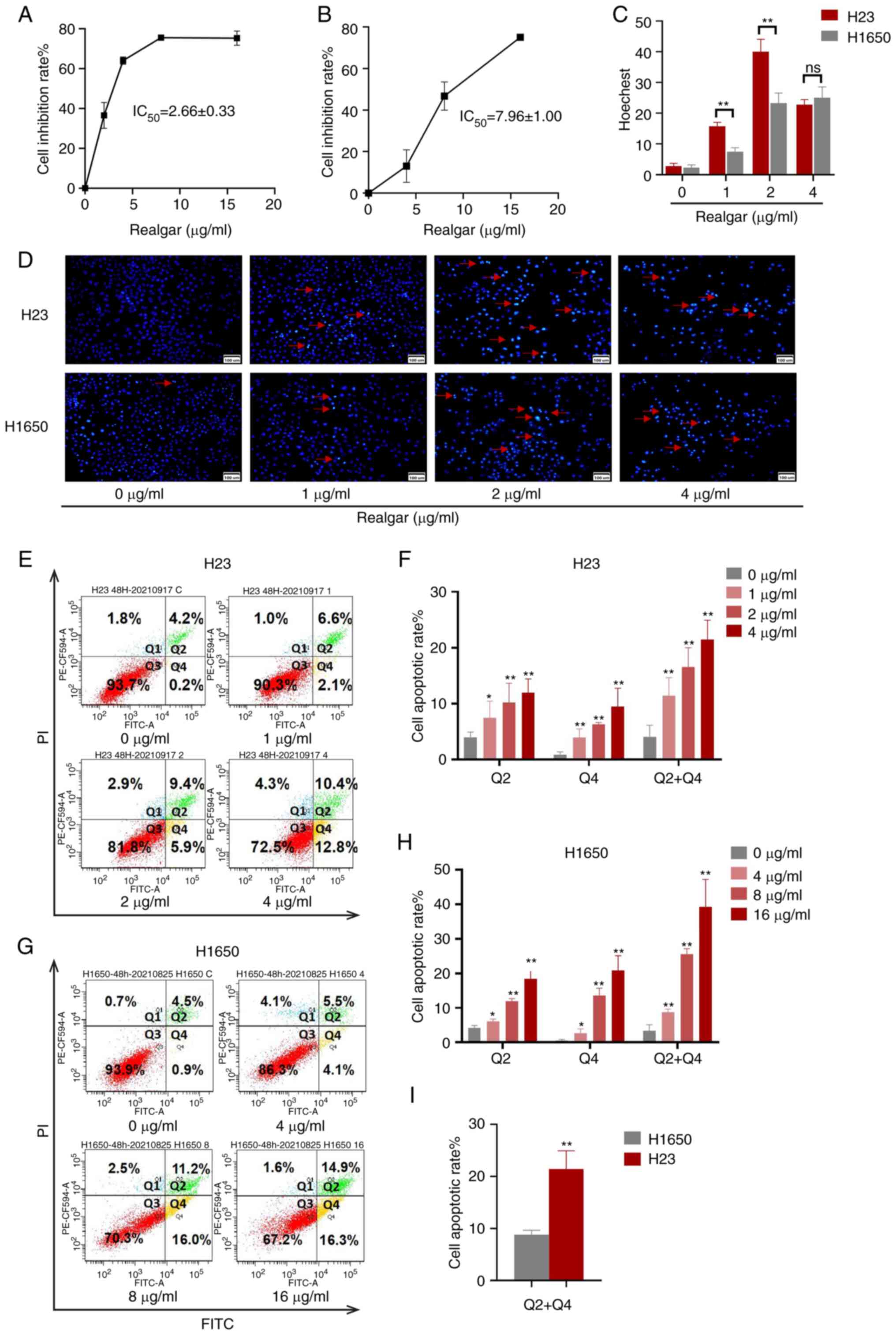
Realgar induces H23 cell apoptosis and
inhibition of H23 cell proliferation
A total of 4 NSCLC cell lines (H23, A549, H460 and
H1650) were applied to assess the cytotoxicity of realgar. Compared
with 24, 48 h of cell treatment resulted in a larger inhibition
rate which led to the selection of the 48 h time point for
subsequent experiments. Following 48 h of treatment, the half
inhibitory concentration (IC50) values for H23, A549,
and H460 were 2.66±0.33, 4.66±0.76, and 7.02±0.36 µg/ml,
respectively (Table SI). This
result indicated that H23 cells exhibited the higher sensitivity to
realgar among the three KRAS mutant cell lines.
H1650, a non-KRAS mutant cell line, was selected as
a control for the KRAS mutant cell line H23 in order to evaluate
the cytotoxicity of realgar following inhibition of KRAS. When
realgar was applied to both H23 and H1650 cells for 48 h, the
corresponding IC50 values were 2.66±0.33 and 7.96±1.00
µg/ml, respectively (P<0.01). Following 48 h treatment of
the cells with realgar, the cell morphology was assessed. It was
noted that the proliferation of H23 cells was strongly inhibited by
realgar, whereas the response of H1650 cells to realgar at the same
concentration and time period was not as profound as that of H23
(Fig. S1). The findings revealed
that realgar was highly effective in inhibiting the proliferation
on KRAS mutant cells in a concentration-dependent manner following
48 h of treatment (Fig. 1A and B).
Realgar was suggested to be a promising anticancer compound that
targets KRAS. Subsequently, the investigation of the induction of
apoptosis following treatment of the aforementioned cell lines with
realgar was performed.
When the cells undergo apoptosis, the apoptotic cell
nuclei can be densely stained from Hoechst 33258. In the present
study, the number of dense and sparkle-stained dots (red arrows)
was increased in realgar treatment compared with that of the
control group. Concomitantly, the numbers of H23 and H1650 cells
were reduced and the cells were irregularly arranged following
treatment with realgar for 48 h (Fig.
1C and D). Annexin V and PI staining was also utilized to
identify the number of apoptotic cells. Following 48 h of treatment
with realgar, the apoptotic rates of H1650 cells and H23 cells were
increased in a concentration-dependent manner (Fig. 1E-H). Furthermore, the apoptotic
rate of H1650 cells treated with 4 µg/ml realgar was 8.2%,
while that of H23 cells was 21.46% (P<0.01; Fig. 1I). The number of H1650 apoptotic
cells following treatment with 8 µg/ml realgar was
equivalent to that of H23 apoptotic cells following treatment with
4 µg/ml realgar. According to the results, realgar was more
potent in KRAS mutant cells (H23) than in non-KRAS mutant cells
(H1650). Therefore, KRAS mutant cells H23 were selected for further
analysis. It was evident that realgar not only inhibited the
proliferation of KRAS mutant cells, but also induced their
apoptosis, thereby exerting a considerable anticancer effect.
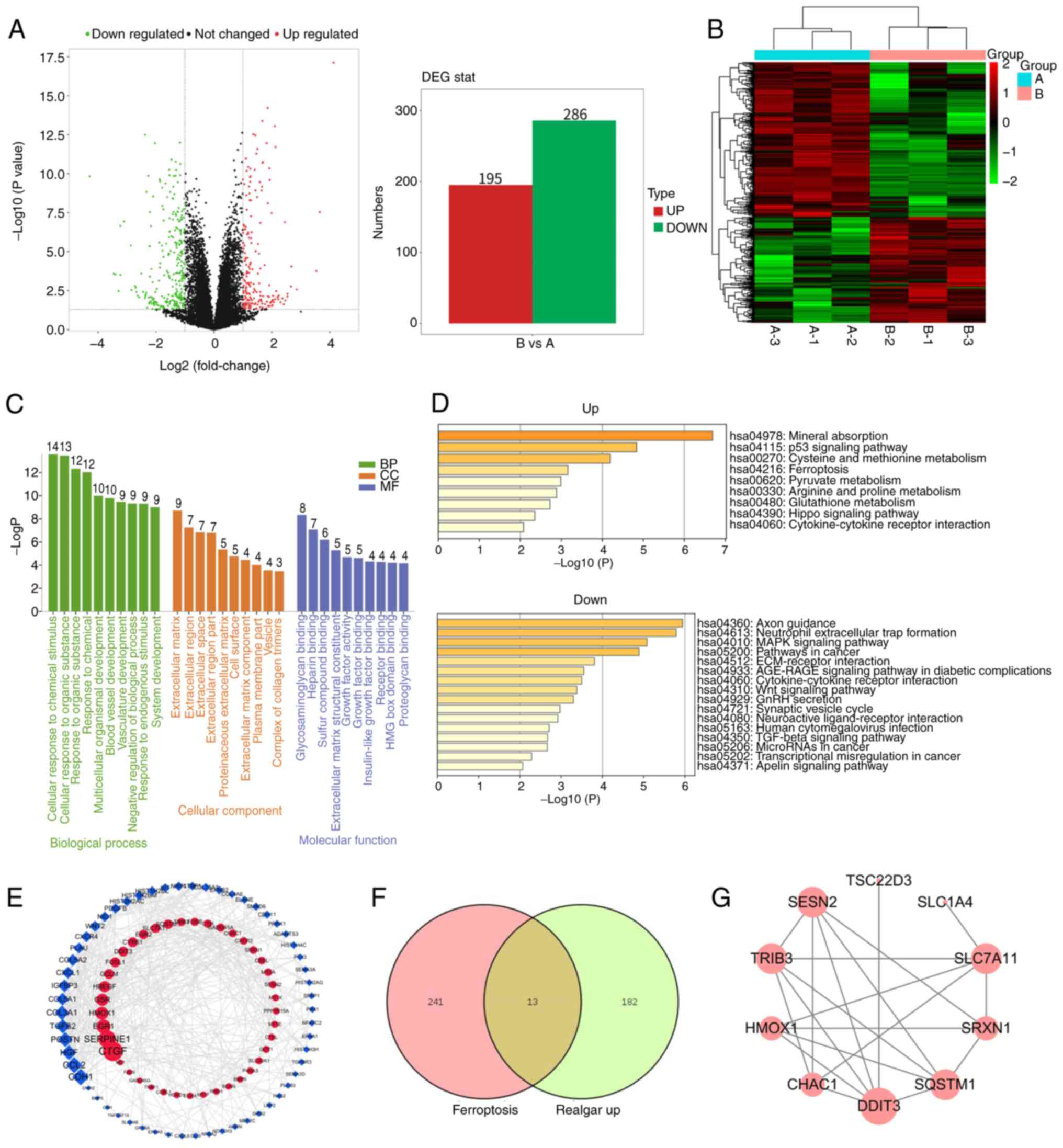
Realgar-mediated anticancer effect
involves induction of ferroptosis
Based on the aforementioned experiments, the data
indicated that realgar exhibited an anticancer effect on KRAS
mutant cells. To investigate further this effect, transcriptome
analysis was performed on realgar-treated (2 µg/ml) H23
cells. The analysis aimed to identify potential genes regulated by
realgar in order to gain insight into the molecular mechanism by
which realgar inhibits lung cancer progression.
In H23 cells, realgar resulted in a >2-fold
upregulation of 195 genes and >2-fold downregulation of 286
genes (Fig. 2A). Among them, the
top-ranked genes HMOX1, SESN2, CYR61, and TRIB3, the remaining
upregulated differentially expressed genes (DEGs), TNFRSF19,
SPOCK2, CTDSP1, TRIB3, as well as other downregulated DEGs were
involved in a variety of biological pathways important for cancer
development. To further clarify the DEGs, a hierarchical cluster
analysis was performed (Fig. 2B).
The results revealed a clear color contrast between the groups
examined; the color of the same cluster within a group was similar.
As a result, the results of the hierarchical cluster analysis were
trustworthy. Furthermore, the DAVID online platform was used to
identify gene ontology enrichments, such as those for biological
process, cellular component, and molecular function components. The
column diagram (Fig. 2C) depicts
all of their top 10 pathways (count ≥3) that are primarily related
to cellular response to chemical stimulus, extracellular matrix
regulation, and sulfur compound binding. The functional analysis
for genes in the key module was performed using Metascape and
revealed four pathways closely related to ferroptosis among the
upregulated pathways, namely the p53 signaling pathway, cysteine
and methionine metabolism (36),
glutathione metabolism and ferroptosis. GPX4 has been shown to be a
central regulator of ferroptosis and a key factor in the regulation
of glutathione metabolic pathways. Among the downregulated
pathways, four have been identified that are closely related to
cancer as follows: The MAPK signaling pathway, the Wnt signaling
pathway, the TGF-β signaling pathway, and cancer transcriptional
misregulation (Fig. 2D). Using
Cytoscape (degree >3), a protein-protein interaction (PPI)
network with 99 nodes and 366 edges was generated, with upregulated
and downregulated genes labeled in blue and red, respectively
(Fig. 2E). These results revealed
that the regulation of cell proliferation by realgar was closely
related with the ferroptosis pathway and the MAPK signaling pathway
in KRAS mutant cells.
FerrDb (http://www.zhounan.org/ferrdb/current/) is the first
worldwide database that contains information on ferroptosis
regulators and markers with 254 ferroptosis-related targets and
ferroptosis-related illness connections. In the present study, it
was found that 14 co-differentially expressed genes were identified
following treatment of realgar to H23 cells and induction of
ferroptosis (Fig. 2F), including
HMOX1, SLC7A11, SESN2, TRIB3, OLFM4, GOT1, and SLC1A4 (Table I). A PPI network was constructed
using STRING database (37)
(Fig. 2G). Based on the
aforementioned results, it was hypothesized that ferroptosis was
the main mechanism of anticancer action of realgar. However, this
hypothesis requires further validation.
 | Table IThe intersection genes identified
following comparison of the DEGs of realgar-associated treatment
and those of considered as ferroptosis targets. |
Table I
The intersection genes identified
following comparison of the DEGs of realgar-associated treatment
and those of considered as ferroptosis targets.
| Symbol | Features in
ferroptosis |
|---|
| SLC7A11 | Suppressor,
Marker |
| HMOX1 | Driver, Suppressor,
Marker |
| SESN2 | Suppressor,
Marker |
| TRIB3 | Marker |
| GOT1 | Driver |
| SLC1A4 | Marker |
| SQSTM1 | Suppressor |
| PCK2 | Marker |
| CHAC1 | Driver, Marker |
| TSC22D3 | Marker |
| DDIT3 | Marker |
| SLC2A8 | Marker |
| SRXN1 | Marker |
| AIFM2 | Suppressor |
Ferroptosis mediates realgar-induced H23
cell death
Transcriptome analysis indicated that realgar
exerted a significant effect on KRAS mutant lung cancer cells via
the induction of ferroptosis. Therefore, subsequent analysis was
performed to assess whether ferroptosis was actually involved in
the mechanism of action of realgar.
In order to examine the function of ferroptosis in
the inhibition of H23 cell proliferation caused by realgar, several
studies were previously carried out. The intracellular iron ions
are accumulated during ferroptosis (38). Excessive free iron boosts the
Fenton reaction, which produces ROS when the iron balance is
disrupted. This will in turn facilitate lipid peroxidation and
cause cell death (39). MDA is a
marker of lipid peroxidation and GSH is an intracellular
antioxidant; both play significant roles in the induction of cell
ferroptosis (40). The current
results indicated that Fe2+, GSH, MDA and ROS levels
were altered in a dose-dependent manner following treatment with
realgar. Treatment of the cells with realgar caused a 1.86-fold
increase in the intracellular Fe2+ (Fig. 3A) than the corresponding levels
noted in the control cells (P<0.01). ROS levels were increased
0.71-fold compared with those noted in the control cells (Fig. 3B). GSH levels were decreased
following realgar treatment (Fig.
3C), while the MDA levels (Fig.
3D) were elevated. Therefore, realgar could indeed cause
induction of ferroptosis of KRAS mutant cells.
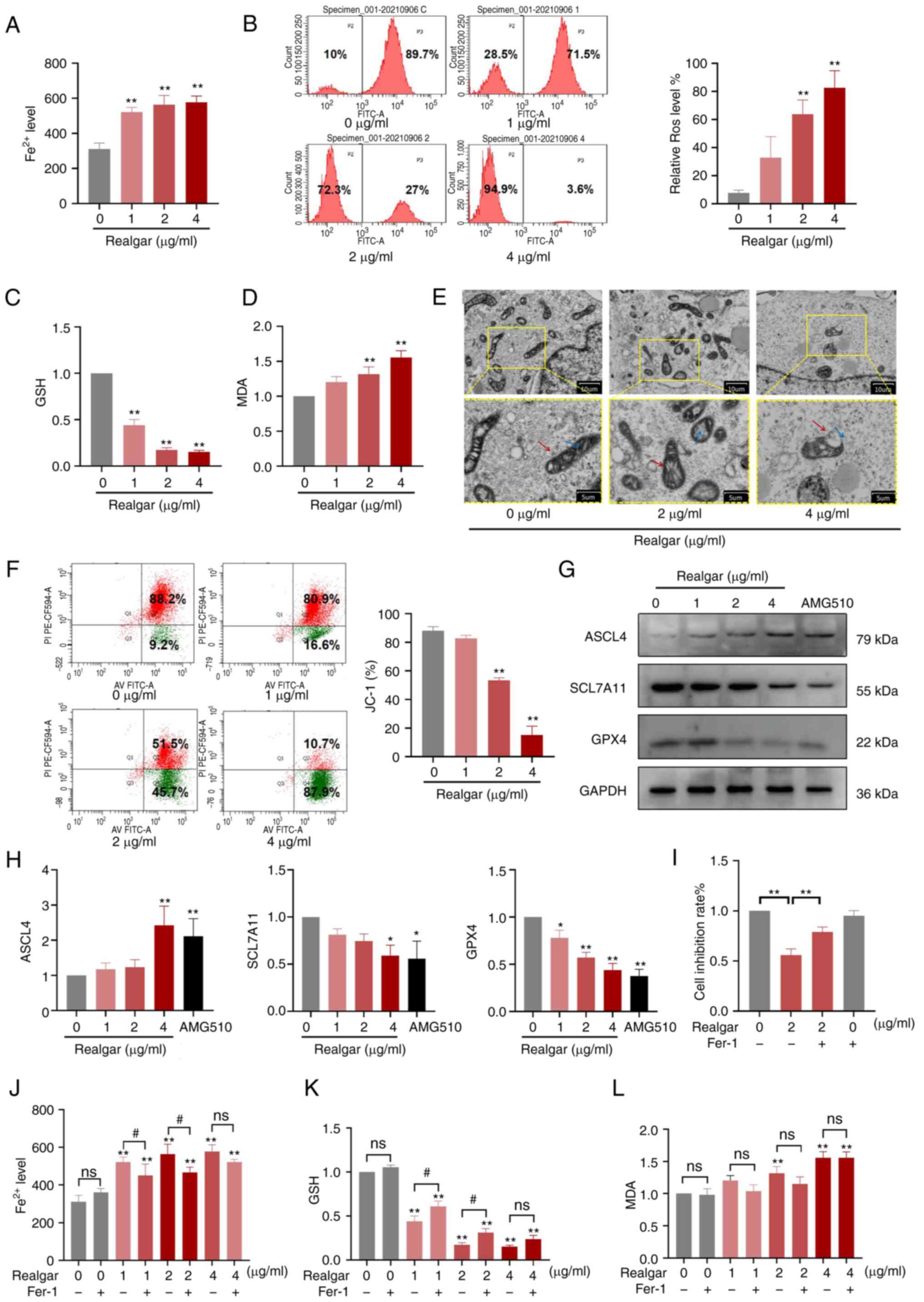 | Figure 3Realgar induces ferroptosis, which is
linked to cell death. (A) The intracellular Fe2+ levels in H23
cells were measured. (B) ROS levels in H23 cells following
treatment with different concentrations of realgar. (C) GSH
reduction in H23 cells treated with realgar. (D) Increase in MDA
levels of H23 cells treated with realgar. (E) Morphological changes
in mitochondria isolated from H23 cells following 48 h of realgar
treatment at 2 µg/ml or 4 µg/ml. Two images are
presented for each group and the scale is marked on the bottom
right of each image. The diagram is an expanded version of the
yellow box area. The red arrow represents the mitochondrial
membrane, while the blue arrow represents the cristae. (F) Flow
cytometry was used to measure the intracellular JC-1 levels. (G)
Western blot analysis of ferroptosis markers. (H) Indicating a
decrease in the expression levels of GPX4 and SCL7A11 and an
increase in the expression levels of ASCL4 in H23 cells following
realgar treatment. (I) H23 cells were incubated with 2 µg/ml
realgar for 48 h and subsequently pre-treated with Fer-1 for 4 h.
(J) The intracellular iron levels were determined in H23 cells by
using a Fe2+ iron probe known as Ferro Orange. Realgar
increased the concentration levels of Fe2+; this effect
was reversed by the ferroptosis rescue agent Fer-1 (1 µM).
(K) The reduction of GSH in H23 cells treated with realgar was
reversed by the ferroptosis rescue agent Fer-1 (1 µM). (L)
The increase in the concentration levels of MDA was detected in H23
cells following treatment with realgar. All data are presented as
the mean ± SD of three independent experiments.
*P<0.05 and **P<0.01 compared with the
control group. #P<0.05 compared with realgar
treatment at different concentrations. AMG-510=10 µg/ml.
GPX4, glutathione peroxidase 4; SCL7A11, recombinant solute carrier
family 7, member 11; ASCL4, acyl-CoA synthetase long-chain family
member 4; ROS, reactive oxygen species; GSH, glutathione; MDA,
malondialdehyde; Fer-1, ferrostatin-1; ns, not significant. |
Iron is metabolized primarily by the mitochondria
through the iron catabolic, anabolic, and utilization pathways, all
of which are connected to the ferroptosis process (41). The mitochondrial apoptosis process
depends on the mitochondrial membrane potential (MMP). MMP may be
downregulated concurrently with ferroptosis causing an alteration
in the mitochondrial membrane structure (42). Therefore, the mitochondrial
morphology and MMP serve as indicators of ferroptosis. TEM is the
gold standard for detecting mitochondrial abnormalities; therefore,
JC-1 labeling was employed to assess mitochondrial damage in H23
cells. The ultramorphological features showed that the cell
membrane was broken and blistered, the mitochondria became smaller,
the membrane density increased, the mitochondrial ridge decreased
or disappeared, the outer membrane of mitochondria was broken, the
size of nucleus was normal, but lack of chromatin condensation.
Specifically, mitochondria appeared smaller and the inner membrane
folding was disturbed (Fig. 3E).
However, as the concentration increases the cell membrane ruptures,
thus it was hypothesized that ferroptosis occurred under the action
of 2 µg/ml of realgar. Following treatment of H23 cells with
realgar, the aggregated JC-1 levels in the mitochondria were
decreased, whereas the monomeric JC-1 levels were increased
(Fig. 3F). The data implied that
realgar had the potential to alter the mitochondrial energy
metabolism, which was essential for the induction of
ferroptosis.
Western blotting (Fig.
3G) was used to compare the expression levels of specific
proteins in the control and realgar groups. The ferroptosis
negative regulatory proteins that have been identified to date are
GPX4 and cysteine/glutamate transporter (SLC7A11/xCT). GPX4
eliminates lipid peroxides by employing GSH as a reducing agent and
lipid ROS as the main substrate (43). SLC7A11/xCT facilitates the transfer
of cystine, a GSH precursor, and is a key negative regulatory
protein of ferroptosis (44).
ACSL4 is a crucial ferroptosis positive regulatory protein that can
facilitate the increase in lipid ROS levels and induce ferroptosis
(45). The results of the present
study indicated that GPX4 (0.65-fold, P<0.01) and SCL7A11
(0.75-fold, P<0.05) levels were significantly lower in cells
treated with 4 µg/ml realgar than those noted in the control
group, while ASCL4 levels were increased dose-dependently
(2.70-fold, P<0.01; Fig. 3H)
which were consistent with the RNA sequencing results.
These findings demonstrated that realgar increased
the key monitor of ferroptosis in KRAS mutant H23 cell lines. To
verify this hypothesis, Fer-1 was added, a ferroptosis inhibitor,
and results showed that Fer-1 abolished the increase in cell death
caused by realgar (Fig. 3I). The
levels of Fe2+, GSH and MDA were also reversed by Fer-1
by 29.17, 13.82, 16.62%, respectively in 2 µg/ml realgar
(Fig. 3J-L).
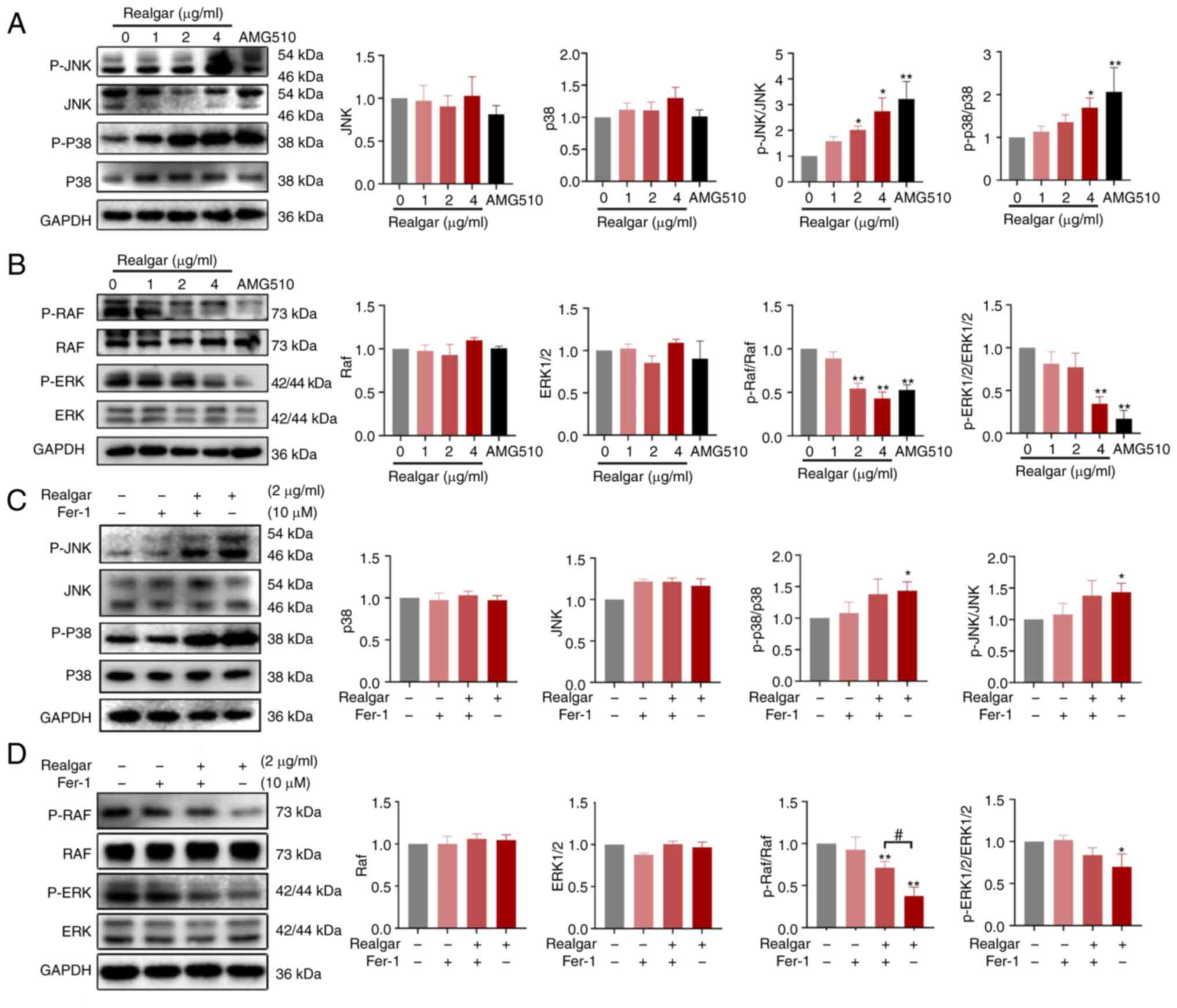
Ras/MAPK pathway plays a critical role in
realgar's anti-cancer activity of KRAS mutant cells
The initiation and development of ferroptosis is
influenced by a variety of signaling pathways and variables,
although it is predominantly caused by the metabolism of amino
acids and iron, the lipid peroxidation, and the levels of lipid ROS
(46). The ferroptosis pathway is
activated by redundant glutamate, which also phosphorylates certain
MAPKs, such as ERK, JNK, and p38 (47). Based on a previous study, it was
discovered that realgar could downregulate Ras expression via the
Ras/MAPK signaling pathway in a C. elegans model (15). To further investigate the mechanism
of realgar in H23 cells, western blotting was used to examine the
expression levels of the related signaling proteins following 48 h
of cell exposure to realgar. JNK and p38 levels were not
significantly altered, whereas p-JNK and p-p38 protein levels were
significantly increased (2.74- and 1.71-fold, respectively)
following treatment of H23 cells with realgar for 48 h (Fig. 4A). Realgar inhibited p-ERK1/2 and
p-Raf protein levels in H23 cells following 48 h of treatment
(Fig. 4B), whereas the levels of
ERK1/2 and Raf were not affected. These findings suggested that the
anticancer activity of realgar may be associated with the Ras/MAPK
pathway.
Realgar induces ferroptosis in
KRAS-mutated H23 cells by targeting Raf
The Ras/Raf/MEK/ERK pathway is crucial in
oncogenesis and cancer progression (18). Because all Raf family members act
directly downstream of Ras, they are also important factors in
oncogenesis, mediating the effects of mutated Ras (48). To further study the functional
involvement of ferroptosis in the realgar-activated Ras/Raf/JNK/ERK
pathway in KRAS-mutant NSCLC, H23 cells were exposed to Fer-1 for 4
h. The result showed that Fer-1 could abolish realgar-induced cell
death (Fig. 3I) and restore p-Raf
protein levels in realgar-treated H23 cells (Fig. 4D), while realgar enhanced p-p38 and
p-JNK protein levels but had no effect on Fer-1 (Fig. 4C and D). It was hypothesized that
realgar-induced ferroptosis was primarily related to Raf and not
JNK/ERK.
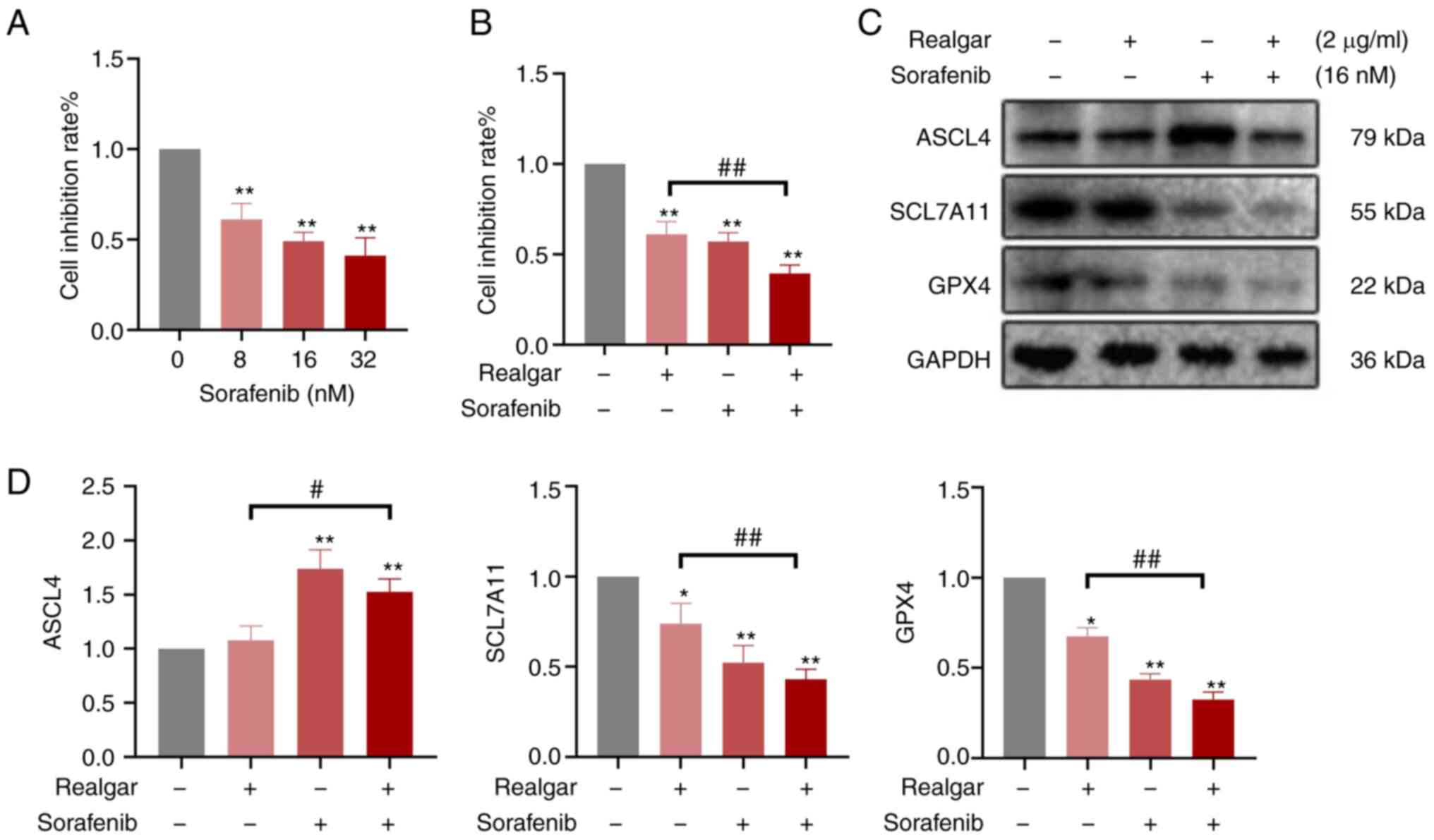
Then, Sorafenib was added, a Raf inhibitor, to
examine the role of Raf in realgar-induced ferroptosis. Using the
MTT assay, the effects of Sorafenib on the growth of H23 cells were
analyzed (Fig. 5A). H23 cells were
treated either with 16 nM sorafenib or 2 µg/ml realgar alone
or in combination for 48 h. Cell viability was decreased by an
average of 61.09±0.07 and 57.10±0.05% in Sorafenib and realgar,
respectively. However, after the addition of Sorafenib, realgar
reduced cell viability more effectively to 39.40±0.05% (Fig. 5B). Simultaneously, Sorafenib also
altered the expression of ferroptosis-related proteins SCL7A11,
GPX4 and ASCL4. These results (Fig. 5C
and D) showed that after treatment with Sorafenib, realgar
significantly decreased the expression of SCL7A11 and GPX4 proteins
while increasing the expression of ASCL4 in H23 cells compared with
cells treated with realgar alone.
Consequently, realgar-induced ferroptosis may be
targeted to regulate Raf kinase, thereby further regulating the
downstream JNK/ERK signaling cascade to suppress KRAS cells and
exert an anticancer activity.
Discussion
KRAS mutant lung cancer remains to find effective
therapeutic treatment. Realgar has demonstrated an optimal
anticancer effect (49,50). One of the main goals of the present
study was to assess the effect of this compound on lung cancer
cells with KRAS mutations. According to our findings presented,
realgar exhibited selectivity for the KRAS mutant H23 cells
compared with non-KRAS mutant H1650 cells. The inhibition ratio
followed a dose-dependent manner in both of these cell lines and
realgar induced apoptosis more efficiently in H23 cells. JNK and
p38 have been shown to regulate a number of transcription factors,
increasing the production of pro-apoptotic proteins while
decreasing the expression of anti-apoptotic proteins (8). Through both transcriptional and
post-translational mechanisms, ERK can be anti-apoptotic by
upregulating anti-apoptotic proteins and downregulating
pro-apoptotic proteins (51). On
controlling cell survival and apoptosis, they have been shown to
have opposite effects. According to Huang et al (52), arsenic trioxide (ATO)-induced p38
MAPK overexpression promotes apoptosis in K562 and MEG-01 cells.
According to Wu et al (53), activated p38 MAPK is a
pro-apoptotic signal for curcumin-induced mortality of
chemoresistant human lung cancer cells A549. Theabrownin, a
component of green tea, suppresses human NSCLC in a xenograft model
via activating the MAPK/JNK signaling pathway, according to Xiao
et al (54). Liu et
al (55) found that elevated
KLHL17 in NSCLC promotes tumor growth and migration via activating
the Ras/MAPK signaling pathway. The MEK inhibitor, on the other
hand, enhances growth inhibition and cell death in Calu-6 lung
cancer cells treated with ATO (56). JNK and p38 will be activated while
ERK is suppressed in order to cause apoptosis in cancer cells, as
evidenced by the effects of dominant-interfering or constitutively
active versions of particular JNK-p38 and ERK signaling pathway
components (57). Lee et al
(58) reported that
As4S4 could decrease the expression levels of
specific proteins, such as Bcl-2 and p-ERK in the KRAS mutant cell
lines A549 and H460 and exert an antiangiogenic effect, which led
to the inhibition of tumor cell growth, proliferation, invasion,
and metastasis. However, since tumor cells exhibit antiapoptotic
properties, novel molecular techniques were used in the current
study to assess their multiple mechanisms of action.
It is known that ferroptosis levels in tumor tissues
are typically higher than those noted in adjacent normal tissues.
Moreover, these levels are related to drug sensitivity, cancer
metastasis, clinical characteristics, and clinical outcomes
(59). Transcriptomic sequencing
analysis has been widely used to identify novel pathways so as to
improve the diagnosis and therapy of a number of disorders,
including cancer, immune system diseases, and infectious diseases
(60). Research revealed that
realgar may exert an anticancer effect by modulation of the p53
signaling pathway (61), cysteine,
methionine, and glutathione metabolic pathways. It is interesting
to note that all of these pathways are considered to be active
during ferroptosis. Our results also showed that ferroptosis
pathway was involved in the impact of realgar on KRAS mutant H23
cells. Therefore, it was proposed that ferroptosis plays an
important role in the effect of realgar on cancer cells.
Recent studies have revealed that ferroptosis is
essential for the development of malignancies, particularly lung
cancer (26), and is considered to
be a potential therapeutic target for Ras-mutant malignancies as
the Ras mutation has been shown to enhance the iron excess in cells
and make them more vulnerable to substances that promote
ferroptosis (32). The activation
of xCT transcription enables oncogene KRAS to shield cells from
oxidative stress by increasing intracellular GSH levels (62). This therapy-resistant tumor may
respond to xCT preserving redox balance and aiding oncogenic
KRAS-mediated transformation (63). As a crucial structural element of
the Xc cell system and a potential biological marker, SLC7A11 has
been to shown to be highly expressed in NSCLC (64). The inhibition of SLC7A11 mediates
KRAS mutant lung cancer cell death by decreasing lung cancer cell
growth and metastasis in vitro and in vivo (65). According to Zhang et al
(66), upregulation of SLC7A11
expression reduces ferroptosis in A549 and H1299 cells, promoting
the growth and spread of the tumors. Liu et al (67), found that capsaicin reduces the
growth of the KRAS mutant lung cancer cell lines A549 and NCI-H23
and induces ferroptosis by silencing SLC7A11/GPX4 signaling. Zhao
et al indicated that Fuzheng Kang'ai decoction induced
ferroptosis in NSCLC by inhibiting GPX4. Our data also showed the
expression levels of the ferroptosis-promoting signaling pathway
protein ACSL4 were substantially increased in the presence of 4
µg/ml realgar, whereas the expression levels of the
ferroptosis-inhibiting signaling pathway proteins GPX4 and xCT were
notably downregulated. Thus, regulating the SLC7A11/GPX4 pathway
and increasing the protein level of ACSL4 is one of the crucial
mechanisms for realgar-mediated ferroptosis to play an anticancer
role. In addition, realgar may result in a large decrease in
intracellular GSH and an increase in MDA content and intracellular
ROS levels as well as cell mitochondrial shrinkage. As revealed in
the present study, the number of mitochondrial ridges was
dramatically reduced compared with that of the control group. These
findings confirmed once again that realgar-induced ferroptosis was
the cell death-mediated mechanism of KRAS mutant H23 cells. The
improvement of the expression levels of the aforementioned
ferroptosis-related markers (Fe2+, GSH and MDA) by Fer-1
suggests that further confirming that ferroptosis occurs in KRAS
mutant H23 cells exposed to realgar.
In a number of diseases, ferroptosis is intimately
correlated with MAPK signaling (68). Ferroptosis may be selectively
produced in cells overexpressing mutant Ras oncoproteins. The
recovery effect of p-Raf was the most obvious when the ferroptosis
inhibitor Fer-1 was used to interfere with the changes in the MAPK
signaling pathway of H23 cells caused by realgar. According to Ma
et al (69), lidocaine
prevented ferroptosis and barrier failure in
hypoxia-reoxygenation-induced A549 cells by inhibiting the p38 MAPK
signaling pathway. In addition, Yang et al (70) demonstrated that in KRAS mutant
colorectal cancer cells, cetuximab increased Ras-selective lethal
(RSL3)-induced ferroptosis via the activation of p38 MAPK. L-F001
can prevent RSL3-induced ferroptosis by preserving iron homeostasis
and suppressing JNK in HT22 cells (71). This demonstrated the powerful
MAPK-mediated crosstalk mechanism between apoptosis and ferroptosis
(72). Meanwhile, the
realgar-induced ferroptosis was also aggravated by adding to Raf
inhibitor Sorafenib. However, while Sorafenib has tumor-suppressing
efficacy as a single agent, its clinical application is limited by
numerous complex drug resistance mechanisms and side effects. Our
findings indicated that the concomitant administration of realgar
and Sorafenib enhanced ferroptosis in H23 cells, suggesting that
realgar may be used as a synergistic drug for Sorafenib.
The current study contains significant limitations.
Not only protein expression, but also protein knockdown or
knock-out model should be applied to show the unique role of
realgar. The present study examined only the regulation of
ferroptosis by realgar in KRAS mutant NSCLC cells, and our
experimental methods can only prove that Raf plays a partial, not a
full, role in realgar-induced ferroptosis. Thus, it is anticipated
that different cell death pathways may be implicated in the
mechanism of anticancer action of realgar. In future studies, the
causal connection between ferroptosis and apoptosis and their
potential to occur concurrently following treatment of lung cancer
cells with realgar will be investigated. In addition, in
vivo tests will also be performed in the future to support
these findings.
The findings of the present study included
confirmation of the role of realgar in inducing ferroptosis in KRAS
mutant NSCLC tumors and a preliminary investigation of the putative
mechanisms regulating ferroptosis via the Ras/MAPK pathway
(Fig. 6). In conclusion, the
present study demonstrated that realgar could prevent the
proliferation of lung cancer cells with a KRAS mutation and exert
an anticancer function by regulating ferroptosis via targeting
Raf-mediated Ras/MAPK pathway. These data provided a novel
medication for NSCLC, moreover, realgar could be a co-treatment
with Sorafenib for clinical therapeutic strategy.
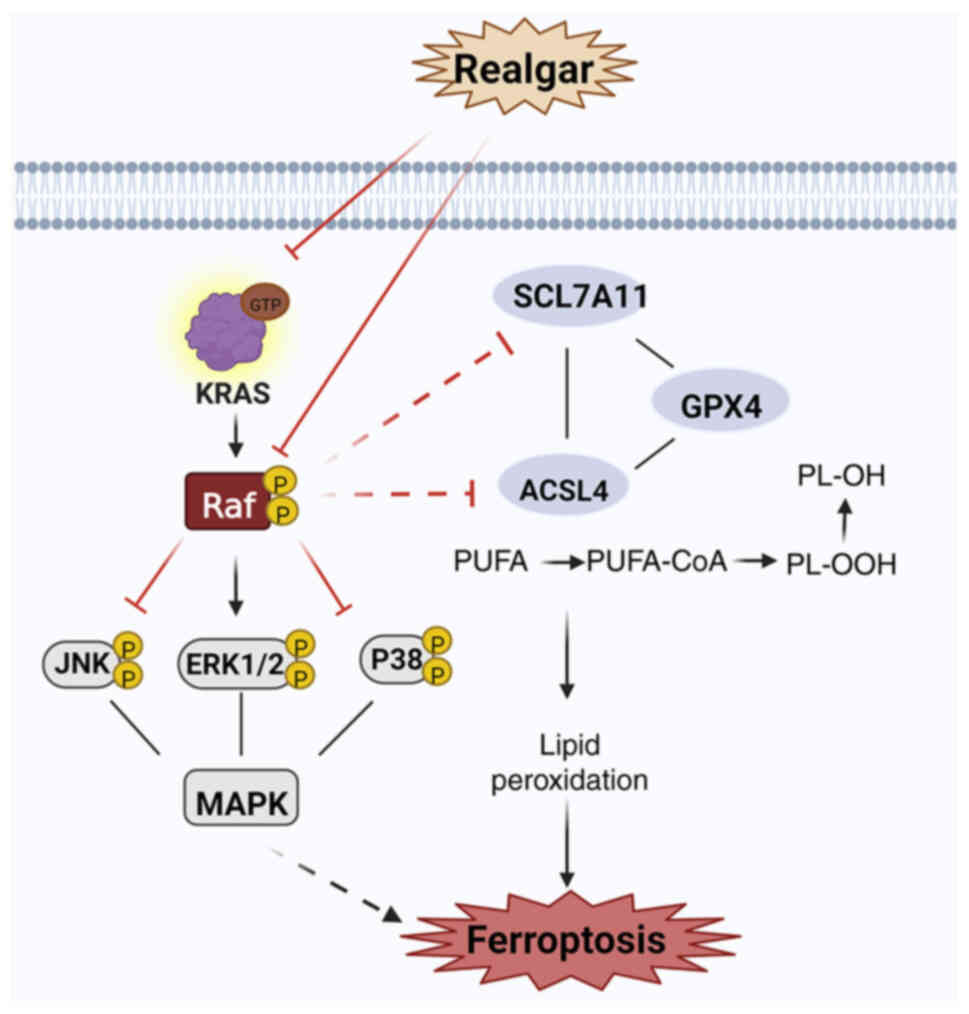 | Figure 6Realgar-induced ferroptosis may be
mediated via KRAS/Raf/MAPK. Realgar may be targeted to regulate Raf
kinase, thereby further regulating the downstream JNK/ERK signaling
cascade to suppress KRAS cells and exert an anticancer activity. At
the same time, realgar inactivating GPX4, the ability of realgar to
impair Xc- or suppress GSH synthesis can result in a buildup in the
lipid peroxide levels and in an increase in ferroptotic cell death
by targeting Raf-mediated Ras/MAPK pathway. Two enzymes, notably
ACSL4, which are required for lipid remodeling, can convert PUFAs
into PUFA-PEs. Created in BioRender.com. GPX4, glutathione peroxidase 4; ROS,
reactive oxygen species; GSH, glutathione; PUFA, polyunsaturated
fatty acid; PE, phosphatidylethanolamine. |
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study are
included in this published article and its Supporting Information
files. The transcriptomic sequencing datasets have been submitted
to a public database (NCBI). This is the associated accession
numbers and password: geoftp; rebUzyi1. (ftp-private.ncbi.nlm.nih.gov).
Authors' contributions
Among the authors listed, XL and YH did the majority
of the work in this study, writing the manuscript and
conceptualization. JD assisted with the western blot analysis and
methodology. LX revised the statistical findings and performed
visualization. WH and JS assisted with the cell experiments and
edited. WR concentrated primarily on the preliminary investigation
and cell screening. DL designed the study, oversaw the work, and
provided the study's facilities. XL and YH confirm the authenticity
of all the raw data. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 81760789 and 82074419) and Research
Center of Traditional Chinese Medicine, Gansu Province (grant no.
zyzx-2020-21).
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
SCL7A11
|
recombinant solute carrier family 7,
Member 11
|
|
GPX4
|
glutathione peroxidase 4
|
|
ACSL4
|
acyl-CoA synthetase long-chain family
member 4
|
|
IC50
|
The half inhibitory concentration
|
|
ROS
|
reactive oxygen species
|
|
MDA
|
malondialdehyde
|
|
GSH
|
glutathione
|
|
p-
|
phosphorylated
|
|
Tfr1
|
transferrin receptor 1
|
|
TEM
|
transmission electron microscopy
|
|
Fer-1
|
ferrostatin-1
|
|
xCT
|
anti-glutamate-cystine antiporter
|
References
|
1
|
Huang J, Deng Y, Tin MS, Lok V, Ngai CH,
Zhang L, Lucero-Prisno DE III, Xu W, Zheng ZJ, Elcarte E, et al:
Distribution, risk factors, and temporal trends for lung cancer
incidence and mortality: A global analysis. Chest. 161:1101–1111.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ryan MB and Corcoran RB: Therapeutic
strategies to target RAS-mutant cancers. Nat Rev Clin Oncol.
15:709–720. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nagasaka M, Li Y, Sukari A, Ou SI,
Al-Hallak MN and Azmi AS: KRAS G12C game of thrones, which direct
KRAS inhibitor will claim the iron throne? Cancer Treat Rev.
84:1019742020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rodak O, Peris-Díaz MD, Olbromski M,
Podhorska-Okołów M and Dzięgiel P: Current landscape of non-small
cell lung cancer: Epidemiology, histological classification,
targeted therapies, and immunotherapy. Cancers (Basel).
13:47052021. View Article : Google Scholar
|
|
7
|
Skoulidis F, Li BT, Dy GK, Price TJ,
Falchook GS, Wolf J, Italiano A, Schuler M, Borghaei H, Barlesi F,
et al: Sotorasib for lung cancers with KRAS G12C-mutation. N Engl J
Med. 384:2371–2381. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ryan MB, Coker O, Sorokin A, Fella K,
Barnes H, Wong E, Kanikarla P, Gao F, Zhang Y, Zhou L, et al:
KRASG12C-independent feedback activation of wild-type
RAS constrains KRASG12C inhibitor efficacy. Cell Rep.
39:1109932022. View Article : Google Scholar
|
|
9
|
Xiaoxia X, Jing S, Dongbin X, Yonggang T,
Jingke Z, Yanying Z and Hulai W: Realgar nanoparticles inhibit
migration, invasion and metastasis in a mouse model of breast
cancer by suppressing matrix metalloproteinases and angiogenesis.
Curr Drug Deliv. 17:148–158. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang L, Zhou GB, Liu P, Song JH, Liang Y,
Yan XJ, Xu F, Wang BS, Mao JH, Shen ZX, et al: Dissection of
mechanisms of Chinese medicinal formula realgar-indigo naturalis as
an effective treatment for promyelocytic leukemia. Proc Natl Acad
Sci USA. 105:4826–4831. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu DP, Qiu JY, Jiang B, Wang Q, Liu KY,
Liu YR and Chen SS: Tetra-arsenic tetra-sulfide for the treatment
of acute promyelocytic leukemia: A pilot report. Blood.
99:3136–3143. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang SL, Guo AX, Xiang Y, Wand XB, Lin HX
and Fu L: Clinical study on the treatment of acute promyelocytic
leukemia with composite indigo naturalis tablets. Chin J Hematol.
16:26–28. 1995.
|
|
13
|
Shi G and Shan G: Effects of yellow loquat
on changes in immune function, hemorheology and coagulation
function in patients with lung cancer. China Tradit Chin Med Sci
Technol. 20:115–116. 2013.
|
|
14
|
Yang FR, Zhao YF, Hu XW, Liu ZK, Yu XD, Li
CY, Li XR and Li HJ: Nano-realgar suppresses lung cancer stem cell
growth by repressing metabolic reprogramming. Gene. 788:1456662021.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu D, Zhi D, Zhou T, Yu Q, Wan F, Bai Y
and Li H: Realgar bioleaching solution is a less toxic arsenic
agent in suppressing the Ras/MAPK pathway in Caenorhabditis
elegans. Environ Toxicol Pharmacol. 35:292–299. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Drosten M and Barbacid M: Targeting the
MAPK Pathway in KRAS-driven tumors. Cancer Cell. 37:543–550. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moore AR, Rosenberg SC, McCormick F and
Malek S: RAS-targeted therapies: Is the undruggable drugged? Nat
Rev Drug Discov. 19:533–552. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Karoulia Z, Gavathiotis E and Poulikakos
PI: New perspectives for targeting RAF kinase in human cancer. Nat
Rev Cancer. 17:676–691. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Karnoub AE and Weinberg RA: Ras oncogenes:
Split personalities. Nat Rev Mol Cell Biol. 9:517–531. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leevers SJ, Paterson HF and Marshall CJ:
Requirement for Ras in Raf activation is overcome by targeting Raf
to the plasma membrane. Nature. 369:411–414. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stokoe D, Macdonald SG, Cadwallader K,
Symons M and Hancock JF: Activation of Raf as a result of
recruitment to the plasma membrane. Science. 264:1463–1467. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peng SB, Henry JR, Kaufman MD, Lu WP,
Smith BD, Vogeti S, Rutkoski TJ, Wise S, Chun L, Zhang Y, et al:
Inhibition of RAF isoforms and active dimers by LY3009120 leads to
anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell.
28:384–398. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Awad MM, Liu S, Rybkin II, Arbour KC,
Dilly J, Zhu VW, Johnson ML, Heist RS, Patil T, Riely GJ, et al:
Acquired resistance to KRASG12C inhibition in cancer. N
Engl J Med. 384:2382–2393. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J,
Kiedrowski LA, Michel AG, Syed MU, Fella KA, Sakhi M, et al:
Clinical acquired resistance to KRASG12C inhibition
through a novel KRAS switch-II pocket mutation and polyclonal
alterations converging on RAS-MAPK reactivation. Cancer Discov.
11:1913–1922. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yagoda N, von Rechenberg M, Zaganjor E,
Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM,
Boniface JJ, et al: RAS-RAF-MEK-dependent oxidative cell death
involving voltage-dependent anion channels. Nature. 447:864–868.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen X, Kang R, Kroemer G and Tang D:
Broadening horizons: The role of ferroptosis in cancer. Nat Rev
Clin Oncol. 18:280–296. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen P, Li X, Zhang R, Liu S, Xiang Y,
Zhang M, Chen X, Pan T, Yan L, Feng J, et al: Combinative treatment
of β-elemene and cetuximab is sensitive to KRAS mutant colorectal
cancer cells by inducing ferroptosis and inhibiting
epithelial-mesenchymal transformation. Theranostics. 10:5107–5119.
2020. View Article : Google Scholar :
|
|
28
|
Balihodzic A, Prinz F, Dengler MA, Calin
GA, Jost PJ and Pichler M: Non-coding RNAs and ferroptosis:
Potential implications for cancer therapy. Cell Death Differ.
29:1094–1106. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alemán MR, Santolaria F, Batista N, de La
Vega M, González-Reimers E, Milena A, Llanos M and Gómez-Sirvent
JL: Leptin role in advanced lung cancer. A mediator of the acute
phase response or a marker of the status of nutrition? Cytokine.
19:21–26. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang C, Zhang X, Yang M and Dong X:
Recent progress in ferroptosis inducers for cancer therapy. Adv
Mater. 31:19041972019. View Article : Google Scholar
|
|
31
|
Gammella E, Buratti P, Cairo G and
Recalcati S: The transferrin receptor: The cellular iron gate.
Metallomics. 9:1367–1375. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang WS and Stockwell BR: Synthetic lethal
screening identifies compounds activating iron-dependent,
nonapoptotic cell death in oncogenic-RAS-harboring cancer cells.
Chem Biol. 15:234–245. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen X, Yu C, Kang R, Kroemer G and Tang
D: Cellular degradation systems in ferroptosis. Cell Death Differ.
28:1135–1148. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Deng S, Wu D, Li L, Liu T, Zhang T, Li J,
Yu Y, He M, Zhao YY, Han R and Xu Y: miR-324-3p reverses cisplatin
resistance by inducing GPX4-mediated ferroptosis in lung
adenocarcinoma cell line A549. Biochem Biophys Res Commun.
549:54–60. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kukulj S, Jaganjac M, Boranic M, Krizanac
S, Santic Z and Poljak-Blazi M: Altered iron metabolism,
inflammation, transferrin receptors, and ferritin expression in
non-small-cell lung cancer. Med Oncol. 27:268–277. 2010. View Article : Google Scholar
|
|
36
|
Jaune-Pons E and Vasseur S: Role of amino
acids in regulation of ROS balance in cancer. Arch Biochem Biophys.
689:1084382020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Szklarczyk D, Gable AL, Nastou KC, Lyon D,
Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al:
The STRING database in 2021: Customizable protein-protein networks,
and functional characterization of user-uploaded gene/measurement
sets. Nucleic Acids Res. 49(D1): D605–D612. 2021. View Article : Google Scholar
|
|
38
|
Nie Q, Hu Y, Yu X, Li X and Fang X:
Induction and application of ferroptosis in cancer therapy. Cancer
Cell Int. 22:122022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W and
Wang J: Molecular mechanisms of ferroptosis and its role in cancer
therapy. J Cell Mol Med. 23:4900–4912. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sun L, Dong H, Zhang W, Wang N, Ni N, Bai
X and Liu N: Lipid peroxidation, GSH depletion, and SLC7A11
inhibition are common causes of EMT and ferroptosis in A549 cells,
but different in specific mechanisms. DNA Cell Biol. 40:172–183.
2021. View Article : Google Scholar
|
|
41
|
Wang H, Liu C, Zhao Y and Gao G:
Mitochondria regulation in ferroptosis. Eur J Cell Biol.
99:1510582020. View Article : Google Scholar
|
|
42
|
Gao M, Yi J, Zhu J, Minikes AM, Monian P,
Thompson CB and Jiang X: Role of mitochondria in ferroptosis. Mol
Cell. 73:354–363.e3. 2019. View Article : Google Scholar :
|
|
43
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar :
|
|
46
|
Su LJ, Zhang JH, Gomez H, Murugan R, Hong
X, Xu D, Jiang F and Peng ZY: Reactive oxygen species-induced lipid
peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med
Cell Longev. 2019:50808432019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chang WT, Bow YD, Fu PJ, Li CY, Wu CY,
Chang YH, Teng YN, Li RN, Lu MC, Liu YC and Chiu CC: A marine
terpenoid, heteronemin, induces both the apoptosis and ferroptosis
of hepatocellular carcinoma cells and involves the ROS and MAPK
pathways. Oxid Med Cell Longev. 2021:76890452021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhao J and Luo Z: Discovery of Raf family
is a milestone in deciphering the Ras-mediated intracellular
signaling pathway. Int J Mol Sci. 23:51582022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu G, Song Y, Li C, Liu R, Chen Y, Yu L,
Huang Q, Zhu D, Lu C, Yu X, et al: Arsenic compounds: The wide
application and mechanisms applied in acute promyelocytic leukemia
and carcinogenic toxicology. Eur J Med Chem. 221:1135192021.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lin CC, Huang YK, Cho CF, Lin YS, Lo CC,
Kuo TT, Tseng GC, Cheng WC, Chang WC, Hsiao TH, et al: Targeting
positive feedback between BASP1 and EGFR as a therapeutic strategy
for lung cancer progression. Theranostics. 10:10925–10939. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cuenda A and Rousseau S: p38 MAP-kinases
pathway regulation, function and role in human diseases. Biochim
Biophys Acta. 1773:1358–1375. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Huang CH, Lee YC, Chiou JT, Shi YJ, Wang
LJ and Chang LS: Arsenic trioxide-induced p38 MAPK and Akt mediated
MCL1 downregulation causes apoptosis of BCR-ABL1-positive leukemia
cells. Toxicol Appl Pharmacol. 397:1150132020.Epub ahead of print.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wu MF, Huang YH, Chiu LY, Cherng SH, Sheu
GT and Yang TY: Curcumin induces apoptosis of chemoresistant lung
cancer cells via ROS-regulated p38 MAPK phosphorylation = Arsenic
trioxide-induced p38 MAPK and Akt mediated MCL1 downregulation
causes apoptosis of BCR-ABL1-positive leukemia cells. Int J Mol
Sci. 23:82482022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Xiao X, Guo L, Dai W, Yan B, Zhang J, Yuan
Q, Zhou L, Shan L and Efferth Y: Green tea-derived theabrownin
suppresses human non-small cell lung carcinoma in xenograft model
through activation of not only p53 signaling but also MAPK/JNK
signaling pathway. J Ethnopharmacol. 291:1151672022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu Z, Zhao M, Jiang X, Zhang Y, Zhang S,
Xu Y, Ren H, Su H, Wang H and Qiu X: Upregulation of KLHL17
promotes the proliferation and migration of non-small cell lung
cancer by activating the Ras/MAPK signaling pathway. Lab Invest.
Aug 17–2022.Epub ahead of print. View Article : Google Scholar
|
|
56
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: The effect of MAPK inhibitors on arsenic
trioxide-treated Calu-6 lung cells in relation to cell death, ROS
and GSH levels. Anticancer Res. 29:3837–3844. 2009.PubMed/NCBI
|
|
57
|
Yang X, Wu X, Wu X, Huang L, Song J, Yung
C, He Z and Li Y: The flavagline compound 1-(2-(dimethylamino)
acetyl)-rocaglaol induces apoptosis in K562 cells by regulating the
PI3K/Akt/mTOR, JAK2/STAT3, and MAPK pathways. Drug Des Devel Ther.
16:2545–2557. 2022. View Article : Google Scholar :
|
|
58
|
Lee H, Lee HJ, Bae IJ, Kim JJ and Kim SH:
Inhibition of STAT3/VEGF/CDK2 axis signaling is critically involved
in the antiangiogenic and apoptotic effects of arsenic herbal
mixture PROS in non-small lung cancer cells. Oncotarget.
8:101771–101783. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu Z, Zhao Q, Zuo ZX, Yuan SQ, Yu K,
Zhang Q, Zhang X, Sheng H, Ju HQ, Cheng H, et al: Systematic
analysis of the aberrances and functional implications of
ferroptosis in cancer. iScience. 23:1013022020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yong WP, Rha SY, Tan IB, Choo SP, Syn NL,
Koh V, Tan SH, Asuncion BR, Sundar R, So JB, et al: Real-time tumor
gene expression profiling to direct gastric cancer chemotherapy:
Proof-of-concept '3G' trial. Clin Cancer Res. 24:5272–5281. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lim JKM and Leprivier G: The impact of
oncogenic RAS on redox balance and implications for cancer
development. Cell Death Dis. 10:9552019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lim JKM, Delaidelli A, Minaker SW, Zhang
HF, Colovic M, Yang H, Negri GL, von Karstedt S, Lockwood WW,
Schaffer P, et al: Cystine/glutamate antiporter xCT (SLC7A11)
facilitates oncogenic RAS transformation by preserving
intracellular redox balance. Proc Natl Acad Sci USA. 116:9433–9442.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Baek S, Choi CM, Ahn SH, Lee JW, Gong G,
Ryu JS, Oh SJ, Bacher-Stier C, Fels L, Koglin N, et al: Exploratory
clinical trial of (4S)-4-(3-[18F]fluoropropyl)-L-glutamate for
imaging xC-transporter using positron emission tomography in
patients with non-small cell lung or breast cancer. Clin Cancer
Res. 18:5427–5437. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hu K, Li K, Lv J, Feng J, Chen J, Wu H,
Cheng F, Jiang W, Wang J, Pei H, et al: Suppression of the
SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant
lung adenocarcinoma. J Clin Invest. 130:1752–1766. 2020. View Article : Google Scholar :
|
|
66
|
Zhang N, Huang J, Xu M and Wang Y: LncRNA
T-UCR Uc.339/miR-339/SLC7A11 axis regulates the metastasis of
ferroptosis-induced lung adenocarcinoma. J Cancer. 13:1945–1957.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Liu XY, Wei DG and Li RS: Capsaicin
induces ferroptosis of NSCLC by regulating SLC7A11/GPX4 signaling
in vitro. Sci Rep. 12:119962022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang
D and Dai E: Ferroptosis is a lysosomal cell death process. Biochem
Biophys Res Commun. 503:1550–1556. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ma X, Yan W and He N: Lidocaine attenuates
hypoxia/reoxygenation-induced inflammation, apoptosis and
ferroptosis in lung epithelial cells by regulating the p38 MAPK
pathway. Mol Med Rep. 25:1502022. View Article : Google Scholar :
|
|
70
|
Yang J, Mo J, Dai J, Ye C, Cen W, Zheng X,
Jiang L and Ye L: Cetuximab promotes RSL3-induced ferroptosis by
suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant
colorectal cancer. Cell Death Dis. 12:10792021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhu K, Zhu X, Sun S, Yang W, Liu S, Tang
Z, Zhang R, Li J, Shen T and Hei M: Inhibition of TLR4 prevents
hippocampal hypoxic-ischemic injury by regulating ferroptosis in
neonatal rats. Exp Neurol. 345:1138282021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wang Y, Zhang L, Yao C, Ma Y and Liu Y:
Epithelial membrane protein 1 promotes sensitivity to RSL3-induced
ferroptosis and intensifies gefitinib resistance in head and neck
cancer. Oxid Med Cell Longev. 2022:47506712022.PubMed/NCBI
|