Introduction
Malignant peripheral nerve sheath tumors (MPNSTs)
are rare neoplasms, accounting for only 5% of all malignant
soft-tissue sarcomas (1,2). The most common sites of occurrence
are the torso, extremities and head and neck, whereas MPNSTs are
extremely rarely located in the mediastinum (3). This is the case report of a patient
with an MPNST in the anterior mediastinum, originating from the
right phrenic nerve.
Case report
A 28-year-old man underwent screening chest
radiography and was found to have an anterior mediastinal mass.
There was no abnormal shadow on a chest radiograph obtained the
previous year. The patient was asymptomatic, had no family history
of neurofibromatosis type 1 (NF1) and his medical history included
a right pneumothorax that was treated surgically, with pleurodesis,
performed 3 years prior. The results of blood testing were
unremarkable. A chest radiograph revealed a well-defined tumor, 10
cm in size, in the right anterior mediastinum (Fig. 1A). Contrast-enhanced computed
tomography of the chest revealed a heterogeneous mass with rim
enhancement; the mass compressed the diaphragm and the right atrium
(Fig. 1B). Magnetic resonance
imaging revealed an anterior mediastinal mass with heterogeneous
high intensity on T1 and T2-weighted images. The area of high
intensity persisted on fat saturation imaging and was therefore
suspected to represent hemorrhage.
Considering the rapid growth of the lesion, its
location and the possibility of malignancy, surgical resection of
the tumor was recommended. Surgical access was obtained via a right
posterolateral thoracotomy over the sixth rib. The tumor was
located in the interlobular space, appearing to originate from the
right phrenic nerve and was densely adherent to the middle and
lower lobes of the right lung, the pericardium and the diaphragm.
The tumor was completely resected and partial resection of the
pericardium, right phrenic nerve, diaphragm and the middle and
lower lobes of the right lung was also performed.
Macroscopically, the tumor was an encapsulated solid
mass, sized 10×10×8 cm, with central hemorrhage and necrosis.
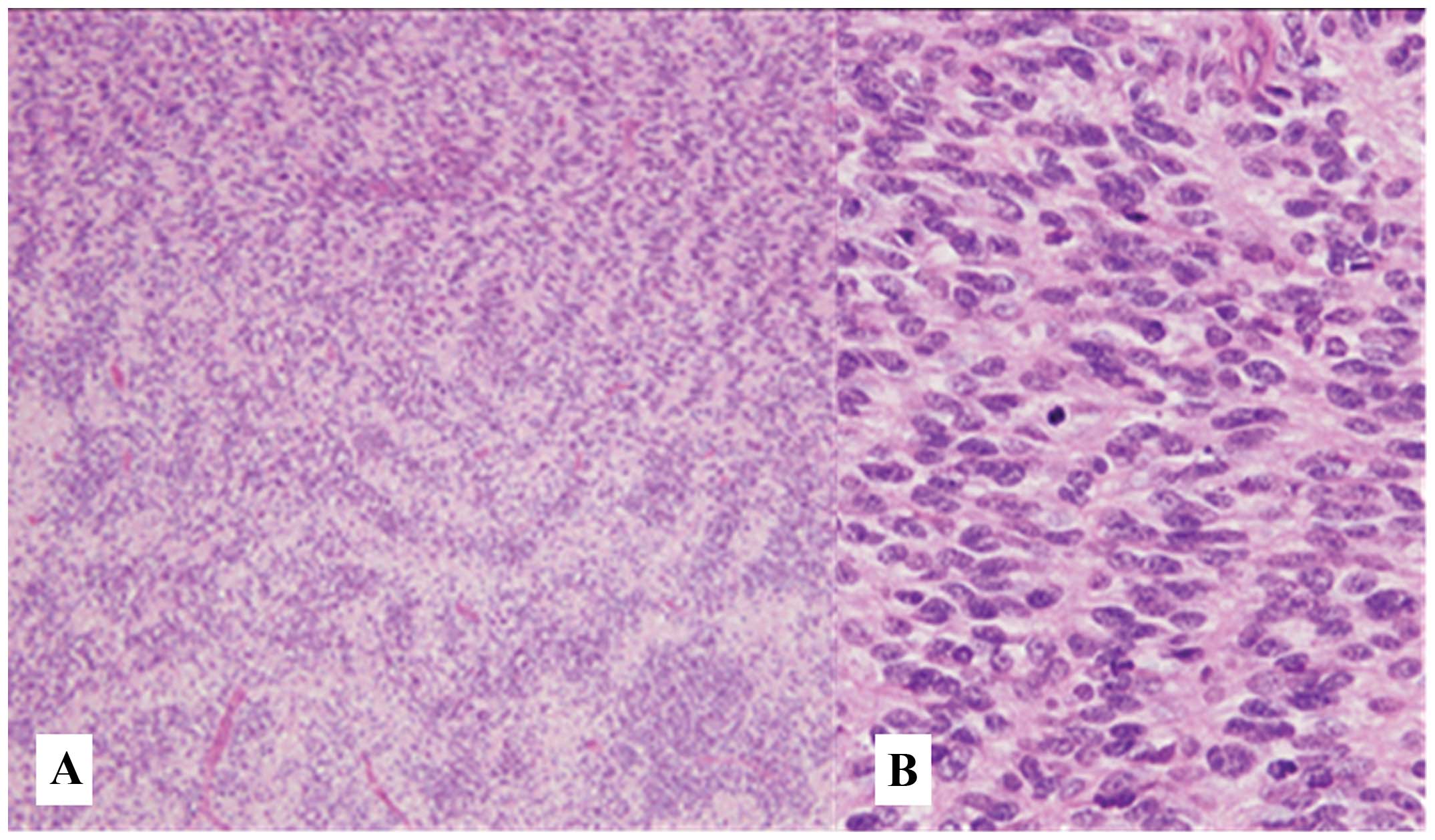
Microscopically, the tumor exhibited highly cellular as well as
loose regions, containing spindle-shaped tumor cells (Fig. 2A). Focal nuclear palisading was
present (Fig. 2B), with high
mitotic activity (30 mitotic figures per 10 high-power fields) and
foci of necrosis. There was no tumor cell invasion of the
neighboring organs.
On immunohistochemical analysis, the tumor cells
were positive for vimentin, B-cell lymphoma 2 protein and cluster
of differentiation (CD) 56, weakly positive for CD99 and negative
for S-100 protein, synaptophysin, chromogranin A, h-caldesmon,
α-smooth muscle actin, desmin, calretinin, Wilms tumor protein 1
and CD34. Based on the these markers, the intraoperative findings
and the characteristic microscopic appearance indicating the
nervous origin of the tumor (highly cellular and loose regions
containing spindle-shaped tumor cells), the diagnosis of MPNST was
established.
The postoperative course of the patient was
uneventful and there was no need for adjuvant therapy. At 1 year
after the surgery, the patient remains free of recurrence.
Discussion
The majority of mediastinal neurogenic tumors arise
in the posterior mediastinum, with only 3% found in the anterior
mediastinum (4). To the best of
our knowledge, only 5 patients with MPNST of the anterior
mediastinum have been reported to date, including our patient
(Table I) (5–8). Of
all the reported cases, our patient is the only one to have a tumor
derived from the phrenic nerve.
 | Table ICase reports of MPNST in the anterior
mediastinum. |
Table I
Case reports of MPNST in the anterior
mediastinum.
| Author | Age/gender | Size, cm | NF1 | Invasiona | Resection | Recurrence | Metastasis | Outcome | (Refs.) |
|---|
| Ducatman and
Scheithauer | 31/F | Unknown | (+) | Unknown | Incomplete | Unknown | Unknown | DOD at 3 months | (5) |
| Otani et
al | 17/F | Unknown | (+) | (+) | Incomplete | Unknown | (+) | DOD at 7 months | (6) |
| Zisis et
al | 30/M | 6.5 | (−) | (−) | Complete | (+) | (−) | AWD at 1 year | (7) |
| Shimoyama et
al | 75/M | 17 | (−) | (−) | Complete | (−) | (+) | DOD at 7 years | (8) |
| Present study | 28/M | 10 | (−) | (−) | Complete | (−) | (−) | NED at 1 year | |
The World Health Organization (WHO) classification
system defines MPNST as originating from either a peripheral nerve
or from a pre-existing benign nerve sheath tumor (usually a
neurofibroma), or as occurring in a patient with NF1. The diagnosis
is based on a particular constellation of histological and
immunohistochemical findings; if these are absent, ultrastructural
characteristics suggesting Schwann-cell differentiation are used
for diagnosis. According to WHO, MPNST is positive for S-100 in
<50% of the cases (1). Our
patient was negative for S-100; however, we diagnosed the tumor as
MPNST since we were able to verify its origin from the right
phrenic nerve and since the tumor exhibited the characteristic
microscopic features that confirmed its nervous origin.
MPNST arising in the thoracic cavity is difficult to
diagnose in its early stages, as the patients are usually
asymptomatic. Therefore, tumors of large size, with invasion of the
surrounding organs, are occasionally discovered. As MPNST has a
very poor prognosis, complete surgical resection is the mainstay of
treatment. It is difficult, however, to achieve complete resection
with large tumors, as their removal sometimes requires resection of
adjacent organs. Of the reported patients with MPNST arising in the
anterior mediastinum, complete resection was not achieved in 2 and
these patients eventually succumbed to the disease (5,6).
The standard surgical approach for anterior
mediastinal tumors is a median sternotomy. However, we performed a
posterolateral thoracotomy in our patient in order to gain access
to the inferior pulmonary vein. Intraoperatively, the tumor was
seen to invade the phrenic nerve and was densely adherent to the
middle and lower lobes of the right lung, the pericardium and the
diaphragm. We considered it likely that the tumor invaded these
surrounding organs and we therefore performed partial resection of
these structures. Fortunately, there was no evidence of invasion.
Of the 4 priorly reported patients with MPNST arising in the
anterior mediastinum, 1 patient exhibited tumor invasion of the
surrounding organs, which is associated with a poor prognosis
(6).
In a case series reported by Ducatman et al
(9), 52% of the patients with
MPSNT had a diagnosis of NF1 and the majority of MPNSTs arose
either from neurofibromas or de novo, from normal peripheral
nerves. No prior reports were able to identify the nerve origin of
the MPNST; however, we were able to confirm that our patient’s
tumor originated from the right phrenic nerve.
MPNST has a poor prognosis. A case series by Wong
et al (10) reported a
5-year survival rate of 52%, with a 49% risk of local recurrence
and a 49% risk of distant metastasis. In their retrospective
analysis, tumor size, location, patient history of NF1, tumor grade
and the integrity of the surgical margins were found to be
prognostic factors. Chemotherapy and radiotherapy are often
ineffective for patients with MPNST; therefore, the mainstay of
treatment is complete surgical resection. However, it was recently
demonstrated that adjuvant irradiation is associated with improved
local disease control (10). In a
series by Carli et al (11), the overall response rate to primary
chemotherapy in group 3 patients (using the Intergroup
Rhabdomyosarcoma Study grouping system) was 45% and it was
demonstrated that chemotherapy may be effective for patients with
unresectable tumors. However, the optimal treatment for MPNST has
not been clearly determined and further studies are required to
improve the prognosis of patients with MPNST.
In conclusion, our patient presented with an MPNST
in the anterior mediastinum originating from the phrenic nerve.
Although were able to achieve complete resection, careful follow-up
is required for our patient due to the poor prognosis associated
with MPNST.
Acknowledgements
We would like to thank Dr Aki Mitsuda, Department of
Surgical Pathology, Toho University, School of Medicine, for her
assistance with pathological diagnosis.
References
|
1
|
Nielsen GP, Antonescu CR and Lothe RA:
Malignant peripheral nerve sheath tumour. WHO Classification of
Tumours of Soft Tissue and Bone. Fletcher CDM, Bridge JA,
Hogendoorn PCD and Mertens F: 5. 4th edition. IARC Press; Lyon: pp.
187–189. 2013
|
|
2
|
Italiano A, Delva F, Mathoulin-Pelissier
S, et al: Effect of adjuvant chemotherapy on survival in FNCLCC
grade 3 soft tissue sarcomas: a multivariate analysis of the French
Sarcoma Group Database. Ann Oncol. 21:2436–2441. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sugio K, Inoue T, Inoue K, et al:
Neurogenic tumors of the mediastinum originated from the vagus
nerve. Eur J Surg Oncol. 21:214–216. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ingels GW, Campbell DC Jr, Giampetro AM,
Kozub RE and Bentlage CH: Malignant schwannomas of the mediastinum.
Report of two cases and review of the literature. Cancer.
27:1190–1201. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ducatman BS and Scheithauer BW: Malignant
peripheral nerve sheath tumors with divergent differentiation.
Cancer. 54:1049–1057. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Otani Y, Morishita Y, Yoshida I, et al: A
malignant Triton tumor in the anterior mediastinum requiring
emergency surgery: report of a case. Surg Today. 26:834–836. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zisis C, Fragoulis S, Rontogianni D,
Stratakos G and Bellenis I: Malignant triton tumour of the anterior
mediastinum as incidental finding. Monaldi Arch Chest Dis.
65:222–224. 2006.PubMed/NCBI
|
|
8
|
Shimoyama T, Yoshida K, Yamoto Y, Koike T
and Honma K: Long-term survival after removal of a malignant
peripheral nerve sheath tumor originating in the anterior
mediastinum. Gen Thorac Cardiovasc Surg. 57:310–314. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ducatman BS, Scheithauer BW, Piepgras DG,
Reiman HM and Ilstrup DM: Malignant peripheral nerve sheath tumors.
A clinicopathologic study of 120 cases. Cancer. 57:2006–2021. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wong WW, Hirose T, Scheithauer BW, Schild
SE and Gunderson LL: Malignant peripheral nerve sheath tumor:
analysis of treatment outcome. Int J Radiat Oncol Biol Phys.
42:351–360. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carli M, Ferrari A, Mattke A, et al:
Pediatric malignant peripheral nerve sheath tumor: the Italian and
German soft tissue sarcoma cooperative group. J Clin Oncol.
23:8422–8430. 2005. View Article : Google Scholar : PubMed/NCBI
|