Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of cancer (1,2), with >780,000 new cases and ~745,000
deaths annually worldwide (3).
Unfortunately, there are only a few treatment options, with
unsatisfactory efficacy. Tumor stage is a decisive factor in the
selection of treatment strategy. Liver transplantation and
hepatectomy are the best potentially curative treatment regimens
for patients with HCC. However, a number of patients with
advanced-stage HCC are not eligible for these treatments. The
5-year survival rate for advanced HCC currently remains poor.
Despite a number of studies attempting to elucidate the mechanism
underlying tumorigenesis, no method has been found to be suitable
for the entire patient population due to the lack of specificity
and sensitivity. Therefore, it is urgent to identify sensitive and
specific biomarkers for HCC progression and to elucidate the
molecular mechanisms involved in HCC progression to predict
prognosis and to develop a novel targeted therapeutic strategy.
Genome-wide expression profiles have recently been
used to identify differentially expressed genes (DEGs) during
disease progression (4,5), which enables the identification of
candidate biomarkers for the diagnosis, therapy and prognosis of
tumors. The high-throughput platforms of gene expression are the
base of genome-wide regulatory and interaction networks (5,6).
Recently, next-generation sequencing accompanied by higher
throughput developed rapidly, allowing more accurate and
comprehensive examination of global gene expression profiles
(7). However, to date, there have
only been a few studies with a small sample size that used RNA
sequencing data in the transcriptomic landscape of HCC (8,9), and it
is imperative to further elucidate the prominent role of
whole-transcriptome sequencing in HCC.
The Cancer Genome Atlas (TCGA) is a project that was
initiated in 2005 by the National Cancer Institute to identify
genetic mutations implicated in cancer using genome sequencing and
bioinformatics. A large number of tissue samples are stored in the
TCGA database and examined from multiple aspects, including genomic
expression. In addition, the data are freely available to all
researchers for their individual studies (7). Therefore, the large TCGA RNA sequencing
(RNAseq) dataset was used in the present study.
All RNAseq data and clinical data were extracted
from the TCGA database of HCC. Global gene expression changes were
compared between tumor tissues (T) and adjacent non-tumor tissues
(NT) and numerous DEGs were identified. Then, DEGs were subjected
to a gene enrichment analysis with an online functional annotation
tool to identify gene sets and signaling pathways that were
significantly enriched with DEGs, and to construct a
protein-protein interaction (PPI) network and obtain hub genes for
survival analysis. Then, the top hub gene, cyclin-dependent kinase
(CDK)1, which is crucial for the mitotic process, was further
analyzed and validated with Gene Expression Omnibus (GEO) data to
conduct a survival analysis, in order to determine whether CDK1
expression is directly associated with survival time and prognosis
in HCC.
Materials and methods
TCGA data of HCC
All available TCGA data on HCC were obtained from
the TCGA data portal (TCGA group). In September 2016, there were
RNAseq data on 424 HCC samples, including 324 single tumor samples,
50 pairs of HCC and adjacent non-tumor liver tissues, and clinical
data including survival time and survival status records of 370
patients (excluding 1 case without survival time and 3 cases of
recurrence). The data, which had been generated using the Illumina
HiSeq 2000 platform, were annotated to a reference transcript set
of Human GRCh38/hg38 gene standard track.
GEO data of HCC
We extracted a microarray expression profile (GEO
group) from the GEO database. All 247 patients with HCC included in
the GSE14520 profile were identified; 26 patients were excluded
from this study, including 22 patients on GPL571 and 5 without
outcome data (1 case was on GPL571 and had no outcome data).
Finally, 221 patients carried out on GPL3921 were included in the
present analysis. All liver tissues were obtained from patients who
underwent radical resection between 2002 and 2003 at the Liver
Cancer Institute and Zhongshan Hospital Affiliated to Fudan
University. Tumor sample processing and microarray analysis were
performed as reported by Roessler et al (10,11).
Global gene expression analysis
Differential gene expression analysis with RNAseq
data was performed using R package edgeR (7,12). As
suggested by edgeR, genes of low read counts are usually not of
interest in DGE analysis. Therefore, an average raw read count for
each gene >1 was applied to determine candidate genes that were
reasonably expressed. The T/NT expression fold change (FC) denotes
upregulation or downregulation according to the FC value.
Subsequently, logFC, logCPM, P-value and the corresponding false
discovery rate (FDR) were all reported by the R package. FDR
<0.05, logCPM >1 and |logFC| >2 were set as inclusion
criteria for DEG selection. The gene expression level based on
microarray data was calculated using R package limma with RMA
correction.
Gene ontology (GO) and pathway
enrichment analysis
GO analysis is a useful method for annotating genes
and gene sets with biological characteristics for high-throughput
genome or transcriptome data (13).
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway is a
knowledge base for systematic analysis of gene functions (14). The Database for Annotation,
Visualization and Integrated Discovery (DAVID) provides a
comprehensive set of efficient and concise annotation tools for
researchers to understand the biological meaning behind numerous
genes (15). GO and KEGG pathway
enrichment analysis were used for DEGs using the DAVID database.
FDR<0.05 was set as the cut-off criterion for the two
analyses.
PPI network
PPI networks can provide information on the
molecular mechanism underlying cellular activity. In the present
study, a PPI network of DEGs was constructed using an online
database, the Search Tool for the Retrieval of Interacting Genes
(STRING), which is designed for evaluating PPI information. STRING
(version 9.0) covers 1,133 organisms, including 5,214,234 proteins
(16). We mapped DEGs to STRING to
identify the interactive relationships among DEGs. A confidence
score of 0.4 was set as the cut-off criterion, and the top 10 DEGs
of node degrees were selected as hub genes.
Statistical analysis
All data analyses were performed in the R
programming environment (version 3.2.5) and Bioconductor (17). Statistical analyses were carried out
with GraphPad Prism 5.0 software (GraphPad Software, Inc., La
Jolla, CA, USA). Single comparisons between two groups were
performed with the Student's t-test. Survival analysis was
performed according to the Kaplan-Meier analysis and log-rank test.
Overall survival (OS) was defined as the time between the date of
surgery and date of death or the date of the last follow-up.
P-values <0.05 were considered to indicate statistically
significant differences.
Results
Identification of DEGs
In the present study, gene expression profiles from
TCGA were utilized to compare gene expression between T and NT. By
comparing the RNAseq read counts of the various genes and
subsequently applying the cut-off criteria, 610 genes were
identified as DEGs, including 444 upregulated and 166 downregulated
genes. High expression genes (logCPM>1) were included in the
volcano plot, with low expression genes excluded (Fig. 1). Subsequently, a heatmap of DEGs was
created; the mRNA expression profiles of T and NT resulted in
obviously separate clusters (Fig.
2).
Gene set enrichment analysis
To gain further insight into the function of
identified DEGs for HCC, gene enrichment analysis was performed
using DAVID, including GO and KEGG pathway enrichment analyses.
Enrichment analyses of the upregulated and downregulated genes were
performed separately, as previously recommended (18). By subjecting the upregulated genes to
enrichment analysis, we observed numerous enriched gene sets. For
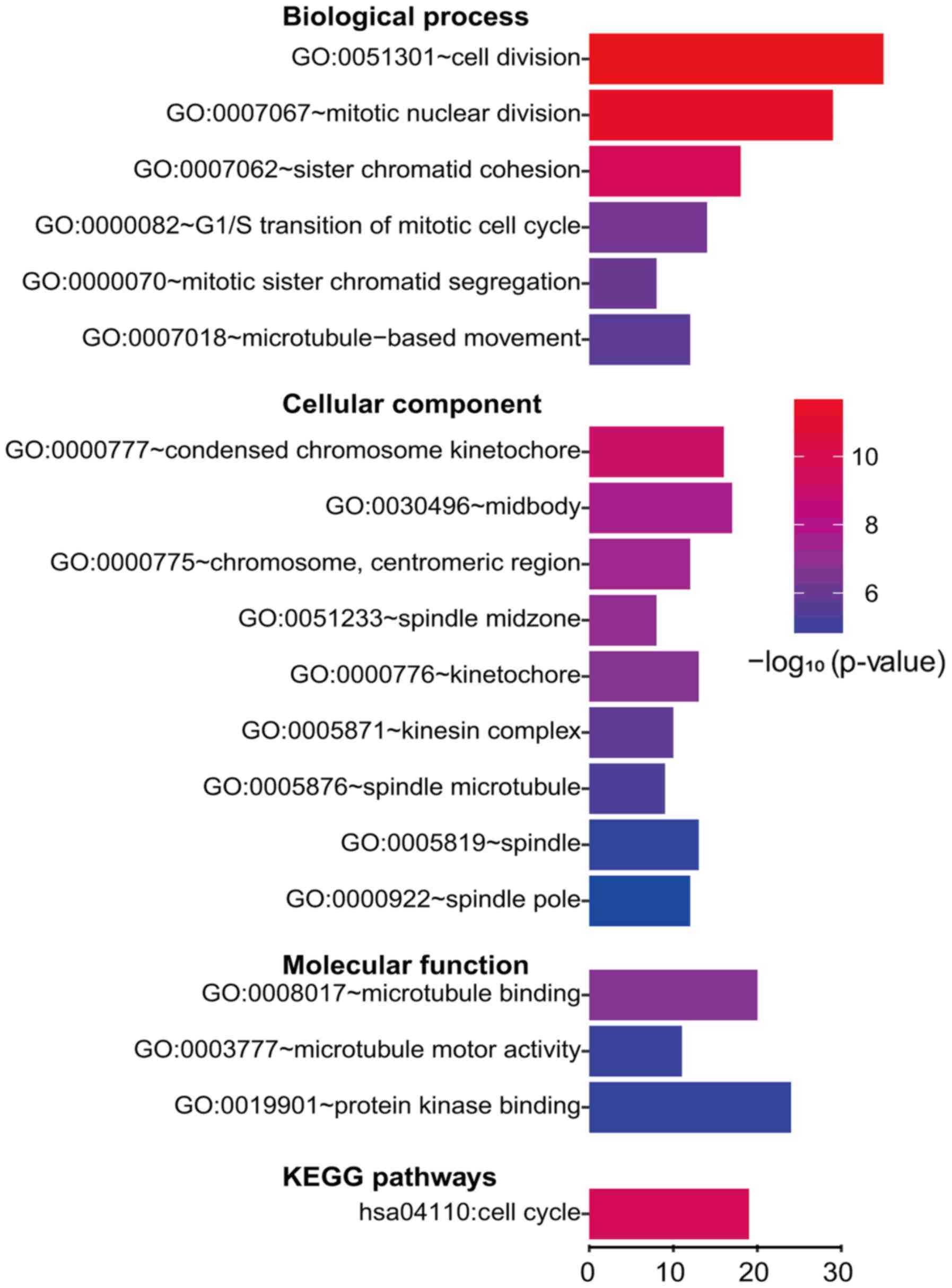
GO biological process, the genes were mainly enriched in cell
division. For GO cellular component, the gene enrichment mainly
involved condensed chromosome and spindle. For GO molecular
function, microtubule binding and protein kinase were implicated
(Fig. 3). A number of downregulated
genes were significantly enriched in various GO domains (Fig. 4). The biological process was
associated with negative regulation of growth and mineral ion
response, cellular component involved extracellular region and
organelle membrane, and molecular function was related to heme
binding and enzyme activity.
We further investigated the functional implications
of these DEGs in the development of HCC by KEGG pathway analysis. A
number of DEGs were enriched in four KEGG pathways, including Cell
cycle in upregulated DEGs and Retinol metabolism, Mineral
absorption and Chemical carcinogenesis in downregulated genes
(Figs. 3 and 4).
PPI network
The PPI network of DEGs was constructed, which
consisted of 568 nodes and 1,952 edges, with a mean node degree of
6.87. The top 10 genes were selected as hub genes by degree, such
as CDK1, TOP2A, CCNB1, CDC20, PLK1, BIRC5, CCNB2, FOS, AURKA and
AURKB (Fig. 5). Subsequently, the
hub genes were again submitted to STRING to verify the interaction
among them. The PPI network consisted of 10 nodes and 40 edges,
with a mean node degree of 8, and showed a closer protein
interaction among the hub genes (Fig.
6). Then, the top hub gene, CDK1, was selected as a candidate
gene for further analysis.
CDK1 overexpression and correlation
with hub genes
Next, to validate dysregulated expression of CDK1,
we analyzed its expression data separately with 50 pairs of T and
NT, and all samples in the TCGA group. These data confirmed that
CDK1 was significantly overexpressed in HCC compared with adjacent
NT tissues (Fig. 7A and B), with
>2-fold increase in CDK1 expression in 98% (49/50) of the tumors
(Fig. 8). A similar result was found
in the GEO group (Fig. 7C). To
further investigate the link between CDK1 and the other hub genes,
Pearson's correlation was used and revealed a statistically
significant correlation between CDK1 and TOP2A (R=0.96, P<0.01),
CCNB1 (R=0.91, P<0.01), CDC20 (R=0.90, P<0.01), PLK1 (R=0.91,
P<0.01), BIRC5 (R=0.93, P<0.01), CCNB2 (R=0.94, P<0.01),
FOS (R=−0.63, P<0.01), AURKA (R=0.81, P<0.01) and AURKB
(R=0.90, P<0.01) (Fig. 9).
 | Figure 9.Pearson's correlation analysis
confirmed that there was a statistically significant correlation
between CDK1 and the other hub genes: TOP2A (R=0.96, P<0.01),
CCNB1 (R=0.91, P<0.01), CDC20 (R=0.90, P<0.01), PLK1 (R=0.91,
P<0.01), BIRC5 (R=0.93, P<0.01), CCNB2 (R=0.94, P<0.01),
FOS (R=−0.63, P<0.01), AURKA (R=0.81, P<0.01) and AURKB
(R=0.90, P<0.01). CDK, cyclin-dependent kinase. |
Survival analysis
In the TCGA group, a total of 370 cases of HCC
patients were enrolled in the study. The patients were divided into
two groups according to gene expression, and expression levels
higher than the median were classified into the high expression
group; otherwise, they were classified into the low expression
group. OS was calculated based on gene expression. Subsequently,
survival analysis was performed to determine the association
between the gene expression level and patient OS. We found that the
expression level of the hub genes, except FOS, was negatively
correlated with OS, with a statistically significant difference
(P<0.01; Fig. 10A). We further
analyzed the data to validate our findings in the GEO group.
Similarly, Kaplan-Meier and log-rank test analysis revealed that
the gene expression level was negatively correlated with OS
(Fig. 10B). To summarize, high
expression of CDK1 was shown to predict a worse prognosis in
patients with HCC.
Discussion
Despite the surgical and medical advances in the
treatment of HCC patients, the overall mortality has remained
unchanged over the past decades (2)
and the molecular mechanism underlying the development of this
cancer has not been fully elucidated. HCC remains one of the most
common causes of cancer-related morbidity and mortality. HCC is
very difficult to detect at an early stage, and there are currently
no effective treatments for patients with advanced-stage disease.
Therefore, it is crucial to improve survival rate and prognosis
through understanding the etiological factors and molecular
mechanisms involved in HCC. Recently, microarray technology has
rapidly developed and has been widely applied to identify general
genetic alterations in malignant diseases, such as HCC (19,20).
With the recent technological advances, next-generation sequencing
enables a more comprehensive and accurate examination of global
gene expression profiles. High-throughput analyses are used to
identify gene expression signatures to improve the accuracy of
prognosis (21).
In order to identify potential biomarkers for HCC
prognosis and therapy, we used data from TCGA to access valuable
information on liver cancer. A total of 610 DEGs were identified,
including 444 upregulated and 166 downregulated genes. To further
elucidate the underlying function of DEGs, functional enrichment
analysis based on GO and pathway enrichment analysis based on KEGG
were performed using DAVID. These upregulated genes were mainly
enriched in the GO and pathways related to proliferation, such as
cell division and cell cycle, while the downregulated genes were
mainly enriched in negative regulation of growth, immune response,
redox reactions and signal transmission. Numerous abnormally
modified GO and KEGG pathways were closely associated with cancer.
Redox reactions and biological metabolism are important for
maintaining normal life activities. Recent studies have
demonstrated that normal immune function is crucial for the
prevention and treatment of tumors (22,23).
Reprogramming energy metabolism and evading immune destruction are
considered to be two newly emerging hallmarks, which are as
important as uncontrolled proliferation and evasion of apoptosis in
tumorigenesis and tumor progression (24,25). Our
results demonstrated a liver cancer cell state of enhanced
proliferation and division, reduced negative growth regulation,
redox electron transport and immune function.
Among these DEGs, a closely interacting PPI network
was found, including 10 genes, namely CDK1, TOP2A, CCNB1, CDC20,
PLK1, BIRC5, CCNB2, FOS, AURKA and AURKB, which were again analyzed
by DAVID and were found to be associated with cell division and
proliferation. CDK1 had a highest degree in the PPI network and was
considered as the top hub gene. Pearson's correlation analysis
revealed that the expression of other hub genes exhibited a
significantly positive correlation with CDK1, except FOS, the
expression of which had a significantly negative correlation with
tumors.
The cell cycle is the series of events that occur
during cell division and DNA duplication, and it is an
evolutionarily conserved process necessary for mammalian cell
growth and development. For cells to accurately duplicate their
contents and divide, they must proceed through the steps of the
cell cycle in a specific order. Loss of normal cell cycle control
is a hallmark of human cancer (26).
Tumor cells accumulate genetic alterations that lead to unscheduled
cell proliferation and genomic instability. Chromosomal instability
is correlated with poor prognosis in multiple solid tumors,
indicating that increasing genetic diversity contributes to altered
tumor cell survival and chemoresistance (27). At present, several cell cycle-related
genes have been reported to be involved in HCC initiation and
progression (28,29). Our results demonstrated that a large
number of DEGs were significantly enriched in the molecular process
related to cell cycle, cell division and growth regulation,
demonstrating that cell cycle and cell regulation disorders are
crucial for tumorigenesis in HCC.
CDKs are master regulatory kinases and coordinate
all cell cycle events. During the entire cell cycle, cyclins, which
are expressed periodically, and CDKs, which are relatively stable,
combine to form a complex, which activates the kinase activity of
CDKs and can precisely regulate the cell cycle at different phases
through inducing or inhibiting the expression of several genes
necessary for cells to enter mitosis. CDK1 is a catalytic subunit
of M phase-promoting factor, which acts mainly towards the end of
the G2 phase and can direct cells into the mitotic M phase. Cyclin
B synthesis begins in the late G1 phase, increases in the S phase,
peaks in the late G2 and M phase, then enters the nucleus and binds
to CDK1. When the cells exit the M phase, cyclin B is degraded, the
kinase activity of CDK1 is inactivated, and the cells are guided
into the next cell cycle (30).
CCNB1 overexpression promotes cell proliferation and tumor growth
in human colorectal cancer (31),
and is a poor prognostic factor for breast cancer (32).
PLK1 plays an extremely important role in the
replication of hepatitis B virus, tumor metastasis and autophagy
(33–35).
TOP2A, as a common predictor of chemotherapy
efficacy, exhibits a significant correlation between its
amplification or deletion and the reactivity to anthracyclines
(36). Patients with high expression
of TOP2A were found to be more sensitive to anthracyclines, while
patients with low expression were resistant to these agents
(37).
FOS, an AP-1 transcription factor subunit, is
involved in mediating a number of biological processes, such as
cell proliferation, differentiation and death (38).
The aurora kinases (AURK) are an evolutionarily
conserved family of serine/threonine kinases related to mitosis and
meiosis, and most mitotic cells express two AURK isoforms (AURKA
and AURKB). These kinases, as molecular switches, regulate multiple
processes in cell division, including spindle organization,
chromosome alignment, the spindle assembly checkpoint and
cytokinesis, among others (39).
CDC20 is an important cofactor of the
anaphase-promoting complex or cyclosome (APC/C) E3 ubiquitin ligase
by regulating APC/C ubiquitin activity on specific substrates for
their subsequent degradation by the proteasome. It plays an
important role in chromosome segregation and mitotic exit as a
target of the spindle assembly checkpoint (40).
BIRC5, a member of the inhibitor of apoptosis
protein (IAP) family, plays an important role in apoptosis,
proliferation and angiogenesis, and is an important prognostic
marker and survival factor (41,42).
In the present study, the expression of the hub
genes, except FOS, was found to be significantly increased in HCC,
and the expression level was negatively correlated with OS, with a
statistically significant difference. These results were similar to
the expression in other solid tumors (4,43–45),
showing the characteristics of the hub genes as oncogenes and the
key role of the PPI network in tumorigenesis and tumor
progression.
Taken together, the results of the present study
demonstrated that several pathways are altered and numerous hub
genes, including CDK1, are overexpressed in HCC, with the
expression level being significantly associated with survival time;
they may be indicative of poor prognosis and may be valuable as
prognostic markers for HCC patients. It may be preferable to study
these genes as a whole in the context of a PPI network for further
analysis, as this will hopefully provide new insights into the
molecular mechanisms, prevention and treatment of HCC.
There were certain limitations to the present study,
as the results from the RNASeq and bioinformatics lack
corresponding experiments in vitro and in vivo.
Functional research is necessary to uncover the molecular
mechanisms interlinking DEGs in HCC and their role in prognosis and
therapy.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
This study was supported in part by the Wenzhou
Science and Technology Project (grant no. Y20170096), the Zhejiang
Provincial Natural Science Foundation of China (grant no.
LY18H160056) and the Science Technology Department of Zhejiang
Province Project (grant no. 2016C37127). Fund body only provided a
part of the financial support for our study, but did not
participate in or interfere with our research design, data
collection, data interpretation, or manuscript writing.
Authors' contributions
QiaZ wrote the manuscript and interpreted the data.
HQ designed the study as director. YS performed data mining. QinZ
helped correct the manuscript. QH performed data analysis and
statistical analysis. All the authors have read and approved the
final version of this manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Availability of data and materials
The datasets generated and analysed in the present
study are available from the corresponding author on reasonable
request.
Glossary
Abbreviations
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
DEGs
|
differentially expressed genes
|
|
TCGA
|
The Cancer Genome Atlas
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
GEO
|
Gene Expression Omnibus
|
|
FC
|
fold change
|
|
FDR
|
false discovery rate
|
|
PPI
|
protein-protein interaction
|
|
OS
|
overall survival
|
|
CDKs
|
cyclin-dependent kinases
|
References
|
1
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Villanueva A and Llovet JM: Liver cancer
in 2013: Mutational landscape of HCC-the end of the beginning. Nat
Rev Clin Oncol. 11:73–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN. Int J Cancer. 136:E359–E386. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shi YX, Zhu T, Zou T, Zhuo W, Chen YX,
Huang MS, Zheng W, Wang CJ, Li X, Mao XY, et al: Prognostic and
predictive values of CDK1 and MAD2L1 in lung adenocarcinoma.
Oncotarget. 7:85235–85243. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liang B, Li C and Zhao J: Identification
of key pathways and genes in colorectal cancer using bioinformatics
analysis. Med Oncol. 33:1112016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee JS and Thorgeirsson SS: Comparative
and integrative functional genomics of HCC. Oncogene. 25:3801–3809.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ho DW, Kai AK and Ng IO: TCGA
whole-transcriptome sequencing data reveals significantly
dysregulated genes and signaling pathways in hepatocellular
carcinoma. Front Med. 9:322–330. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang Q, Lin B, Liu H, Ma X, Mo F, Yu W,
Li L, Li H, Tian T, Wu D, et al: RNA-Seq analyses generate
comprehensive transcriptomic landscape and reveal complex
transcript patterns in hepatocellular carcinoma. PLoS One.
6:e261682011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin KT, Shann YJ, Chau GY, Hsu CN and
Huang CY: Identification of latent biomarkers in hepatocellular
carcinoma by ultra-deep whole-transcriptome sequencing. Oncogene.
33:4786–4794. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roessler S, Jia HL, Budhu A, Forgues M, Ye
QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX and Wang XW: A
unique metastasis gene signature enables prediction of tumor
relapse in early-stage hepatocellular carcinoma patients. Cancer
Res. 70:10202–10212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roessler S, Long EL, Budhu A, Chen Y, Zhao
X, Ji J, Walker R, Jia HL, Ye QH, Qin LX, et al: Integrative
genomic identification of genes on 8p associated with
hepatocellular carcinoma progression and patient survival.
Gastroenterology. 142:957–966, e12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gene Ontology Consortium: The Gene
Ontology (GO) project in 2006. Nucleic Acids Res. 34(Database
Issue): D322–D326. 2006.PubMed/NCBI
|
|
14
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
da Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:(Database Issue). D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gentleman RC, Carey VJ, Bates DM, Bolstad
B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al:
Bioconductor: Open software development for computational biology
and bioinformatics. Genome Biol. 5:R802004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hong G, Zhang W, Li H, Shen X and Guo Z:
Separate enrichment analysis of pathways for up- and downregulated
genes. J R Soc Interface. 11:201309502013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen P, Zheng X, Zhou Y, Xu Y, Zhu L and
Qian Y: Talin-1 interaction network promotes hepatocellular
carcinoma progression. Oncotarget. 8:13003–13014. 2017.PubMed/NCBI
|
|
20
|
Sun H, Peng Z, Tang H, Xie D, Jia Z, Zhong
L, Zhao S, Ma Z, Gao Y, Zeng L, et al: Loss of KLF4 and
consequential downregulation of Smad7 exacerbate oncogenic TGF-β
signaling in and promote progression of hepatocellular carcinoma.
Oncogene. 36:2957–2968. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ko JH, Ko EA, Gu W, Lim I, Bang H and Zhou
T: Expression profiling of ion channel genes predicts clinical
outcome in breast cancer. Mol Cancer. 12:1062013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kulkarni A, Natarajan SK, Chandrasekar V,
Pandey PR and Sengupta S: Combining immune checkpoint inhibitors
and kinase-inhibiting supramolecular therapeutics for enhanced
anticancer efficacy. Acs Nano. 2016. View Article : Google Scholar
|
|
23
|
Hughes PE, Caenepeel S and Wu LC: Targeted
therapy and checkpoint immunotherapy combinations for the treatment
of cancer. Trends Immunol. 37:462–476. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dominguez-Brauer C, Thu KL, Mason JM,
Blaser H, Bray MR and Mak TW: Targeting mitosis in cancer: Emerging
strategies. Mol Cell. 60:524–536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Visconti R, Della Monica R and Grieco D:
Cell cycle checkpoint in cancer: A therapeutically targetable
double-edged sword. J Exp Clin Cancer Res. 35:1532016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cai H, Li H, Li J, Li X, Li Y, Shi Y and
Wang D: Sonic hedgehog signaling pathway mediates development of
hepatocellular carcinoma. Tumour Biol. 2016. View Article : Google Scholar
|
|
29
|
Jin B, Wang W, Du G, Huang GZ, Han LT,
Tang ZY, Fan DG, Li J and Zhang SZ: Identifying hub genes and
dysregulated pathways in hepatocellular carcinoma. Eur Rev Med
Pharmacol Sci. 19:592–601. 2015.PubMed/NCBI
|
|
30
|
Bertoli C, Skotheim JM and de Bruin RA:
Control of cell cycle transcription during G1 and S phases. Nat Rev
Mol Cell Biol. 14:518–528. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fang Y, Yu H, Liang X, Xu J and Cai X:
Chk1-induced CCNB1 overexpression promotes cell proliferation and
tumor growth in human colorectal cancer. Cancer Biol Ther.
15:1268–1279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ding K, Li W, Zou Z, Zou X and Wang C:
CCNB1 is a prognostic biomarker for ER+ breast cancer. Med
Hypotheses. 83:359–364. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Diab A, Foca A, Fusil F, Lahlali T,
Jalaguier P, Amirache F, N'Guyen L, Isorce N, Cosset FL, Zoulim F,
et al: Polo-like-kinase 1 is a proviral host factor for hepatitis B
virus replication. Hepatology. 66:1750–1765. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Wang R, Gannon OJ, Rezey AC, Jiang
S, Gerlach BD, Liao G and Tang DD: Polo-like Kinase 1 regulates
vimentin phosphorylation at ser-56 and contraction in smooth
muscle. J Biol Chem. 291:23693–23703. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ruf S, Heberle AM, Langelaar-Makkinje M,
Gelino S, Wilkinson D, Gerbeth C, Schwarz JJ, Holzwarth B,
Warscheid B, Meisinger C, et al: PLK1 (polo like kinase 1) inhibits
MTOR complex 1 and promotes autophagy. Autophagy. 13:486–505. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Slamon DJ and Press MF: Alterations in the
TOP2A and HER2 genes: Association with adjuvant anthracycline
sensitivity in human breast cancers. J Natl Cancer Inst.
101:615–618. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brase JC, Schmidt M, Fischbach T, Sültmann
H, Bojar H, Koelbl H, Hellwig B, Rahnenführer J, Hengstler JG and
Gehrmann MC: ERBB2 and TOP2A in breast cancer: A comprehensive
analysis of gene amplification, RNA levels, and protein expression
and their influence on prognosis and prediction. Clin Cancer Res.
16:2391–2401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
de Los Fayos Alonso Garces I, Liang HC,
Turner SD, Lagger S, Merkel O and Kenner L: The role of activator
protein-1 (AP-1) family members in cd30-positive lymphomas. Cancers
(Basel). 10:pii: E932018. View Article : Google Scholar
|
|
39
|
Carmena M and Earnshaw WC: The cellular
geography of aurora kinases. Nat Rev Mol Cell Biol. 4:842–854.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kapanidou M, Curtis NL and Bolanos-Garcia
VM: Cdc20: At the crossroads between chromosome segregation and
mitotic exit. Trends Biochem Sci. 42:193–205. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hingorani P, Dickman P, Garcia-Filion P,
White-Collins A, Kolb EA and Azorsa DO: BIRC5 expression is a poor
prognostic marker in Ewing sarcoma. Pediatr Blood Cancer. 60:35–40.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Q, Shu R, He H, Wang L, Ma Y, Zhu H,
Wang Z, Wang S, Shen G and Lei P: Co-silencing of Birc5 (survivin)
and Hspa5 (Grp78) induces apoptosis in hepatoma cells more
efficiently than single gene interference. Int J Oncol. 41:652–660.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang W, Cho H, Shin HY, Chung JY, Kang ES,
Lee EJ and Kim JH: Accumulation of cytoplasmic Cdk1 is associated
with cancer growth and survival rate in epithelial ovarian cancer.
Oncotarget. 7:49481–49497. 2016.PubMed/NCBI
|
|
44
|
Willder JM, Heng SJ, McCall P, Adams CE,
Tannahill C, Fyffe G, Seywright M, Horgan PG, Leung HY, Underwood
MA and Edwards J: Androgen receptor phosphorylation at serine 515
by Cdk1 predicts biochemical relapse in prostate cancer patients.
Br J Cancer. 108:139–148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sung WW, Lin YM, Wu PR, Yen HH, Lai HW, Su
TC, Huang RH, Wen CK, Chen CY, Chen CJ and Yeh KT: High
nuclear/cytoplasmic ratio of Cdk1 expression predicts poor
prognosis in colorectal cancer patients. BMC Cancer. 14:9512014.
View Article : Google Scholar : PubMed/NCBI
|