Introduction
In our previous pre-clinical study, it was
identified that colorectal cancer patients, who have a high
expression level of growth hormone receptor (GHR), are
radioresistant (1), indicating
that GH/GHR signaling may be associated with cellular stress
responses to radiotherapy. Using Chinese hamster ovary cells
(CHO-4) in vitro, Madrid et al (2) found that growth hormone (GH) was able
to reduce the cell damage induced by radiotherapy through its
association with GHR on the cell surface. Isla et al
(3,4) also identified that GH is involved in
protection against the noxious effects of radiotherapy in the
spinal cords of rats and in cell cultures of the central nervous
systems. A recent study demonstrated that autocrine human growth
hormone is able to protect mammary carcinoma cells from the
induction of DNA double-strand breaks following chemotherapy
(5).
Based on these preliminary data, the current study
aimed to investigate whether recombinant human growth hormone
(rhGH) also has a role in the protection of colorectal cancer cells
post-radiotherapy, as it does in the protection of normal healthy
cells.
Materials and methods
Detection of GHR expression on the
colorectal cancer cell surface by flow cytometry
Nine colorectal cancer cell lines were provided by
Shanghai Institute of Biochemistry and Cell Biology (Shanghai,
China), including LOVO, HCT-8, SW480, Ls-174-T, HT-29,
CL187/CCL187, COLO320/DM, COLO205 and HCE8693. A mouse monoclonal
antibody against GHR was provided by Sigma-Aldrich (St. Louis, MO,
USA). phycoerythrin (PE)-labeled goat anti-mouse IgG1 secondary
antibody was provided by Caltag Laboratories (Carlsbad, CA, USA).
PE-labeled mouse IgG1 was provided by R&D Systems (Emeryville,
CA, USA) as the control. All of the cell lines were cultured in
RPMI-1640 medium at 37°C in 5% CO2. Flow cytometry was
conducted according to standard procedures.
Experimental grouping
GHR positive HCT-8 cells and GHR negative LOVO cells
were selected from nine colorectal cancer cell lines. Recombinant
human growth hormone (rhGH, Saizen) was provided by Serono (no.
1260411D04). The GHR antagonist (GHRA) is the goat anti-human GHR
antibody, which actively neutralizes human GH and was provided by
R&D Systems. The median neutralization concentration (ND50) of
GHRA was 0.125–0.5 μg/ml.
Serum starvation was conducted for the HCT-8 cells
(16 h) and LOVO cells (8 h) prior to the experiments. The cells
were radiated following 6 h treatment of rhGH and GHRA pretreatment
was conducted 1 h prior to rhGH treatment. Radiation was performed
by an Elekta1070 linear accelerator (Varian Medical Systems, Palo
Alto, CA, USA) under room temperature with a dose series of 2, 4
and 8 Gy, respectively and a dose rate of 100 cGy/min. Following
radiation, the cells were collected at various time points
according to different samples and each group had five replicates.
The experimental grouping is summarized in Table I.
 | Table IExperimental grouping for examining
the effects of rhGH on human colorectal cancer cell
radiosensitivity. |
Table I
Experimental grouping for examining
the effects of rhGH on human colorectal cancer cell
radiosensitivity.
| Group | HCT-8 (GHR+) | LOVO (GHR−) |
|---|
| Radiation | 1 | A |
| rhGHa + radiation | 2 | B |
| GHRAb + rhGH + radiation | 3 | C |
Colony forming assay
A colony forming assay is the gold standard for
determining cell proliferation (6). Following treatment and radiation, the
cells from the culture flask were digested into a single cell
suspension. The cells of the suspension were inoculated in a petri
dish with 10 ml pre-heated medium at a density of 500 cells/dish.
The cells were cultured for 2–3 weeks under the conditions of 5%
CO2, 37°C and saturated humidity. Following detection of
the colonies by the naked eye, the cells were stained and fixed by
Giemsa stain and dried in air for 10–30 min. The number of colonies
containing >50 cells were counted. The survival fraction (SF)
was calculated using the following formula: SF = number of
colonies/(number of inoculated colonies × PE/100), where PE is the
colony formation rate under certain conditions. The control group
without treatment was used as a standard.
Detection of DNA damage by comet
assay
A comet assay is also known as Single Cell Gel
Electrophoresis (SCGE) and is considered the standard method for
determining DNA damage (7). A
number of the steps of SCGE were modified according to our
laboratory conditions (Nanjing University of Traditional Chinese
Medicine, Nanjing, Jiangsu, China) (8), including cell separation and
treatment, constructing slides, cell lysis, DNA denaturation,
signal cell electrophoresis, neutralization, staining. The images
were analyzed by Comet Assay Software Project (CASP) Perceptive
Instruments (London, UK). A total of 50 cells were selected
randomly and detected at olive tail moment (OTM) from each
experimental group. OTM is a common index to determine the DNA
damage level in a comet assay (9)
and is defined as the product of the tail length and the fraction
of total DNA in the tail. The DNA repair process most commonly
requires 4–6 h following radiation and therefore the DNA damage was
detected at different time points of 0, 15, 30, 60, 60, 120 and 240
min following radiation. The concentration of rhGH and GHRA were
100 ng/ml and 0.2 μg/ml, respectively and the radiation dose was 8
Gy.
Detection of the gene expression level of
growth arrest and DNA damage 45 (GADD45) and apurinic/apyrimidinic
endonuclease (APEN) following rhGH treatment by western blot
analysis
GADD45 and APEN protein levels were detected in
GHR(+) HCT-8 cells 1, 3 and 6 h following rhGH treatment (100
ng/ml). Each experiment was performed as three replicates. GADD45
and APEN polyclonal antibodies were purchased from Santa Cruz
Biotechnology Inc. (Santa Cruz, CA, USA). Western blot analysis was
performed according to standard procedures.
Statistical analysis
The results are presented as the mean ± standard
deviation. The differences between the groups were analyzed by
single element variance analysis and P<0.05 was considered to
indicate a statistically significant difference. Data were analyzed
by statistics software SPSS 11.0 (SPSS, Inc., Chicago, IL,
USA).
Results
Expression level of GHR on the surface of
colorectal cancer cells
The expression level of GHR in human colorectal
cancer cells was investigated using flow cytometry. It was
identified that the cell lines, including LOVO, SW480, Ls-174-T,
HT-29, COLO320/DM, COLO205 and HCE8693 did not express or expressed
very low level of GHR. CL187/CCL187 expressed a certain level of
GHR (19.99% of cells expressed GHR) and HCT-8 expressed a high
level of GHR (58.23% of cells expressed GHR; Fig. 1). For the further experiments,
HCT-8 cells were selected as the GHR(+) cells and LOVO as the
GHR(−) cells, which acted as a control.
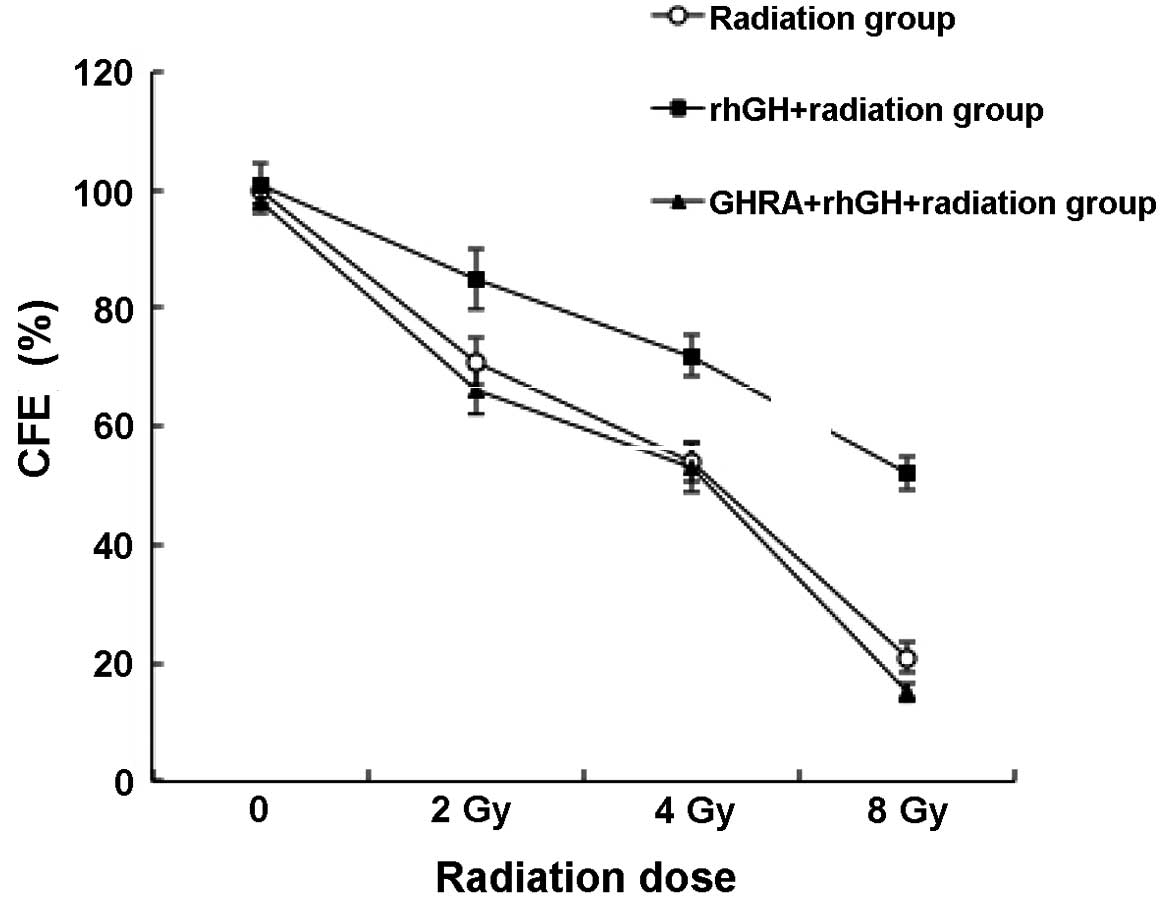
Effects of rhGH treatment on the colony
forming efficiency (CFE) of colorectal cancer cells following
radiation
As demonstrated in Fig.
2, when treated with 100 ng/ml rhGH, post-radiotherapy, HCT-8
cells had a significantly higher CFE than the control group,
particularly under a radiation dose of 8 Gy (52.1±2.9 vs. 21.0±2.7;
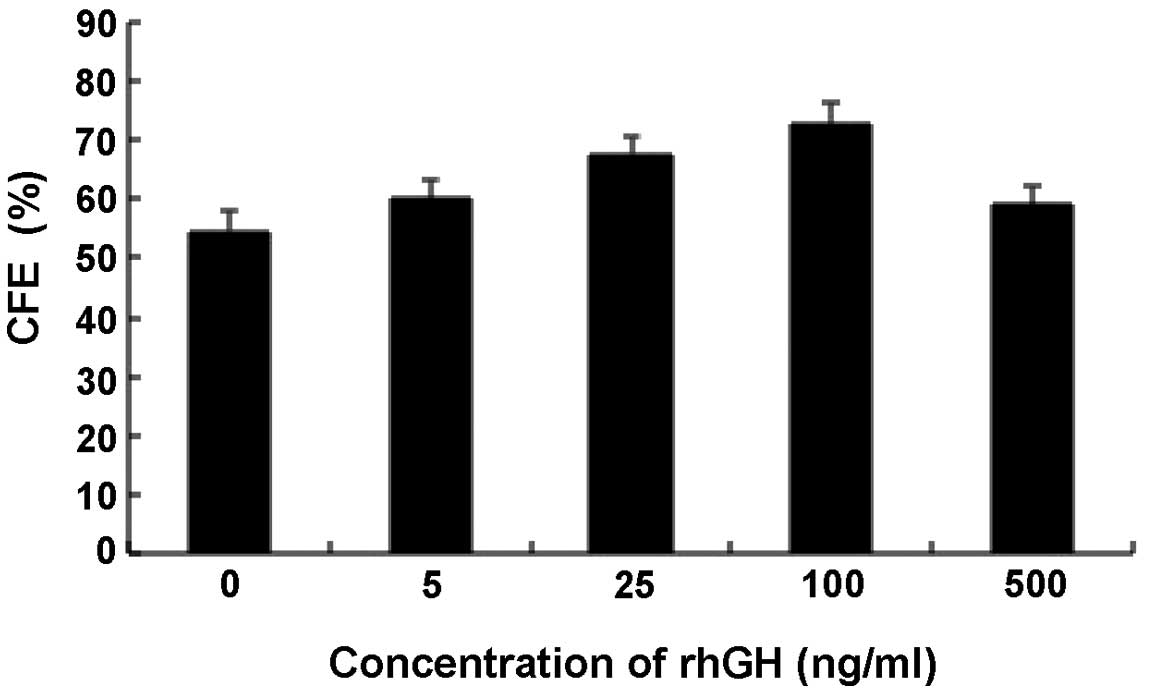
P<0.001). To investigate whether this effect was dependent on
the concentration of rhGH, the CFE of cells treated with various
concentrations of rhGH was examined. Notably, it was identified
that the CFE was only dependent on the concentration of rhGH within
a certain range (0–100 ng/ml; Fig.
3). Above a concentration of 100 ng/ml, rhGH began to reduce
the CFE of the cells.
To rule out the possibility that rhGH alone, rather
than the association of rhGH with GHR, increased the CFE, an
antagonist was utilized to prevent GHR from binding to rhGH
(Fig. 2). With the treatment of
GHRA, it was identified that the CFE decreased to a similar level
of the control even in the presence of rhGH, suggesting that the
protective function of rhGH was eliminated by pretreating GHR with
GHRA.
To further confirm that rhGH functions through GHR,
the CFE of GHR(−) LOVO cells post-radiotherapy was detected.
Consistent with the previous results, rhGH treatment did not
significantly increase the CFE of GHR(−) LOVO cells following
radiation (P>0.05; Fig. 4).
Following the GHRA treatment, no significant difference was found
between the GHRA and GHR treatment, and the control group
(P>0.05). Similarly, changing the concentration of rhGH did not
affect the level of CFE (data not presented), indicating rhGH only
functions in the presence of GHR.
Effects of rhGH treatment on DNA repair
in post-radiotherapy colorectal cancer cells
To further investigate the mechanism of the
protective function of rhGH, a comet assay was used to determine
the extent of DNA damage in the cells. As demonstrated in the
radiation alone group in Table II
and Fig. 5, the DNA damage level
of HCT-8 cells was highest immediately after radiation and
gradually reached the plateau phase within 120 min through
continuous DNA repair. Treatment of rhGH resulted in a
significantly lower level of DNA damage in the HCT-8 cells
(21.53±2.88 vs. 36.56±3.93 in the control group; P=0.003) and the
level in plateau phase was also significantly lower than those
cells without rhGH treatment (5.5±0.42 vs. 9.07±0.84 in the control
group, P=0.012). Following GHRA pretreatment, the DNA damage level
returned to a similar level compared with the control group.
| Table IIEffects of rhGH on HCT-8 DNA repair
following radiation. |
Table II
Effects of rhGH on HCT-8 DNA repair
following radiation.
| Time after radiation
(min) |
|---|
|
|
|---|
| Group | 0 | 15 | 30 | 60 | 120 | 240 |
|---|
| Control | 0.78±0.08 | 1.47±0.15 | 0.95±0.10 | 1.24±0.07 | 1.09±0.09 | 0.75±0.10 |
| Radiationc | 36.56±3.93 | 22.4±1.91 | 14.68±1.30 | 10.69±0.99 | 9.07±0.84 | 9.49±0.94 |
| rhGHa + radiation | 21.53±2.88** | 13.78±1.25* | 9.35±1.05* | 6.05±0.79* | 5.5±0.42* | 5.65±0.62* |
| GHRAb + rhGH + radiation | 39.45±3.59 | 19.5±2.21 | 15.9±1.76 | 9.97±1.25 | 8.61±0.64 | 8.85±0.91 |
rhGH upregulates the protein expression
level of GADD45 and APEN in colorectal cancer cells
Since the mechanisms underlying the protective
effects of rhGH remain obscure, the correlation between rhGH and
two proteins that are associated with DNA repair was examined. As
demonstrated in Figs. 6 and
7, the protein expression levels
of GADD45 and APEN were significantly upregulated following 1 h of
treatment with rhGH (P<0.001) and the high expression level was
maintained for as long as 6 h (P<0.001).
Discussion
In the present study, it was demonstrated that
exogenous growth hormone significantly increased the CFE of HCT-8
following radiation and the level of CFE was dependent on the
concentration of rhGH within a specific concentration range (0–100
ng/ml). However, the same results were not observed in the GHR(−)
LOVO cells. Furthermore, when GHR was blocked by pretreatment of
GHRA, rhGH failed to increase the CFE level of HCT-8 cells. These
results suggest that GH has an important role in protecting GHR(+)
colorectal cancer cells from radiation and that this protective
mechanism is mediated through its interaction with GHR on the cell
surface. Of note, when applying rhGH at a markedly high
concentration, the protective effect was no longer evident, which
may be attributed to the inhibition of interaction between GH and
GHR (10). Under normal
conditions, one molecule of rhGH requires two GHRs in order to form
a dimer and transduce a signal into cells. When the concentration
of rhGH is particularly high, one rhGH molecule will only bind to
one GHR and therefore does not form a functional dimer and thus
fails to transduce a biological signal into cells.
DNA is the major target of the damaging effects of
radiation. The DNA damage caused by radiation and the ability of
DNA to repair in cells affects the cellular activity and overall
viability. Following exposure to radiation, cellular DNA repair
occurs within 4–6 h, during which the cells with mild DNA damage
enter mitosis while cells with severe damage are induced to enter
apoptosis (11). In the present
study, a comet assay was used to detect the DNA damage of GHR(+)
HCT-8 cells at various time points within 4 h following radiation.
It was identified that post-radiotherapy cells treated with rhGH
had lower levels of DNA damage compared with the control group and
the DNA damage level of rhGH-treated cells was also low at plateau
phase. However, the protective effect was inhibited by blocking the
cells with neutralizing antibody GHRA. This suggests that the
association of GH and GHR improves the DNA repair ability in HCT-8
colorectal cancer cells, which may increase the CFE following
radiation.
Previously, a study of the rat liver demonstrated
that GH induces the expression of two genes implicated in the
control of DNA damage and in cellular stress responses (12), namely, GADD45 and APEN. GADD45 is a
nucleoprotein closely correlated with cellular DNA repair
mechanisms. When DNA is damaged, GADD45 expression is increased and
then prevents the cell from entering the S phase for DNA
replication (13), while
simultaneously activating nucleotide excision repair (NER). In
addition, GADD45 is able to induce cell cycle arrest, predominantly
via activating the G2 checkpoint (14). APEN is a member of a family of DNA
repair enzymes and is ubiquitously expressed in various tissues.
The main function of APEN is to excise apurinic/apyrimidinic (AP)
bases on DNA via base excision repair (BER), thus maintaining
genetic stability (15).
The interaction of GH with its receptor induces the
activation of signaling cascades, including the janus kinase 2
(JAK2) tyrosine kinase (16),
mitogen-activated protein kinase (MAPK) (17,18),
insulin receptor substrate (IRS)-1 and IRS-2(19,20),
phosphatidylinositol 3-kinase (21), Src homologous and collagen-like
protein, growth factor receptor-bound protein 2 (22), protein kinase C and phospholipase
A2 (23) and others. A number of
these pathways, particularly those driven by JAK2 and MAPK,
activate transcription mediated by signal transducer and activator
of transcription (STAT)3 and STAT5 as well as SRF and c-fos
(24–26), and possibly stimulate the
expression of genes, such as GADD45 and APEN, which are involved in
radiation-induced DNA repair. In the present study, it was also
demonstrated that the association of GH and GHR significantly
upregulated the expression level of GADD45 and APEN in HCT-8
colorectal cancer cells, which may enhance the ability of DNA to
repair. These data may partly explain the molecular mechanism
underlying the protective effects of GH.
Currently, whether rhGH may be used in patients with
colorectal cancer remains controversial. A number of studies have
combined GH and radiotherapy in order to improve the radiation
resistance of cancer patients and enhance the efficacy. However, as
indicated by the results of the present study, the protective
effects of GH on colorectal cancer cells may, conversely, prevent
the radiation-induced apoptosis of cancer cells. Therefore, the use
of rhGH as a therapeutic approach may be limited in colorectal
cancer. By contrast, the competition of GHRAs (such as Pegvisomant)
(27) with GHR, blocks GH/GHR
signaling and may increase the radiosensitivity of colorectal
cancer cells.
In conclusion, the present study demonstrates that
rhGH has an important role in the protection of colorectal cancer
cells from radiation, and this protection may be associated with
elevated DNA repair ability. Further studies are required to
determine how GH/GHR signal transduction regulates feedback to
radiotherapy-mediated cellular stress.
References
|
1
|
Wu X, Wan M, Li G, Xu Z, et al: Growth
hormone receptor overexpression predicts response of rectal cancers
to pre-operative radiotherapy. Eur J Cancer. 42:888–894. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Madrid O, Varea S, Sanchez-Perez I, et al:
Growth hormone protects against radiotherapy-induced cell death.
Eur J Endocrinol. 147:535–541. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Isla A, Budke M, García-Grande A, et al:
Protective effects of the growth hormone (GH) on the irradiated
spinal cord in rats. Neurocirugia (Astur). 18:89–94. 2007.(In
Spanish).
|
|
4
|
Isla A, Budke M, Cacicedo L, et al:
Protective effects of growth hormone in cell cultures of the
central nervous system. Rev Neurol. 34:208–211. 2002.(In
Spanish).
|
|
5
|
Bougen NM, Steiner M, Pertziger M, et al:
Autocrine human GH promotes radioresistance in mammary and
endometrial carcinoma cells. Endocr Relat Cancer. 19:625–644. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dunne AL, Price ME, Mothersill C, et al:
Relationship between clonogenic radiosensitivity, radiation-induced
apoptosis and DNA damage/repair in human colon cancer cells. Br J
Cancer. 89:2277–2283. 2003. View Article : Google Scholar
|
|
7
|
Rojas E, Lopez MC and Valverde M: Single
cell gel electrophoresis assay: methodology and applications. J
Chromatogr B Biomed Sci Appl. 722:225–254. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Collins AR: The comet assay for DNA damage
and repair: principles, applications, and limitations. Mol
Biotechnol. 26:249–261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee E, Oh E and Lee J, Sul D and Lee J:
Use of the tail moment of the lymphocytes to evaluate DNA damage in
human biomonitoring studies. Toxicol Sci. 81:121–132. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fuh G, Cunningham BC, Fukunaga R, et al:
Rational design of potent antagonists to the human growth hormone
receptor. Science. 256:1677–1680. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Willers H, Dahm-Daphi J and Powell SN:
Repair of radiation damage to DNA. Br J Cancer. 90:1297–1301. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thompson BJ, Shang CA and Waters MJ:
Identification of genes induced by growth hormone in rat liver
using cDNA arrays. Endocrinology. 141:4321–4324. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang XW, Zhan Q, Coursen JD, et al: GADD45
induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci USA.
96:3706–3711. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liebermann DA and Hoffman B: Gadd45 in
stress signaling. J Mol Signal. 3:152008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fishel ML and Kelley MR: The DNA base
excision repair protein Ape1/Ref-1 as a therapeutic and
chemopreventive target. Mol Aspects Med. 28:375–395. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Argetsinger LS and Carter-Su C: Growth
hormone signalling mechanisms: involvement of the tyrosine kinase
JAK2. Horm Res. 45(Suppl 1): 22–24. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Love DW, Whatmore AJ, Clayton PE and Silva
CM: Growth hormone stimulation of the mitogen-activated protein
kinase pathway is cell type specific. Endocrinology. 139:1965–1971.
1998.PubMed/NCBI
|
|
18
|
Möller C, Hansson A, Enberg B, et al:
Growth hormone (GH) induction of tyrosine phosphorylation and
activation of mitogen-activated protein kinases in cells
transfected with rat GH receptor cDNA. J Biol Chem.
267:23403–23408. 1992.PubMed/NCBI
|
|
19
|
Argetsinger LS, Hsu GW, Myers MG Jr, et
al: Growth hormone, interferon-gamma, and leukemia inhibitory
factor promoted tyrosyl phosphorylation of insulin receptor
substrate-1. J Biol Chem. 270:14685–14692. 1995. View Article : Google Scholar
|
|
20
|
Argetsinger LS, Norstedt G, Billestrup N,
et al: Growth hormone, interferon-gamma, and leukemia inhibitory
factor utilize insulin receptor substrate-2 in intracellular
signalling. J Biol Chem. 271:29415–29421. 1996. View Article : Google Scholar
|
|
21
|
Jeay S, Sonenshein GE, Kelly PA, et al:
Growth hormone exerts antiapoptotic and proliferative effects
through two different pathways involving nuclear factor-kappaB and
phosphatidylinositol 3-kinase. Endocrinology. 142:147–156.
2001.
|
|
22
|
VanderKuur J, Allevato G, Billestrup N, et
al: Growth hormone-promoted tyrosyl phosphorylation of SHC proteins
and SHC association with Grb2. J Biol Chem. 270:7587–7593. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Argetsinger LS and Carter-Su C: Mechanism
of signalling by growth hormone receptor. Physiol Rev.
76:1089–1107. 1996.PubMed/NCBI
|
|
24
|
Wang YD and Wood WI: Amino acids of the
human growth hormone receptor that are required for proliferation
and Jak-STAT signalling. Mol Endocrinol. 9:303–311. 1995.PubMed/NCBI
|
|
25
|
Pircher TJ, Flores-Morales A, Mui AL, et
al: Mitogen-activated protein kinase inhibition decreases growth
hormone stimulated transcription mediated by STAT5. Mol Cell
Endocrinol. 133:169–176. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ihle JN, Witthuhn BA, Quelle FW, et al:
Signalling through the hematopoietic cytokine receptors. Ann Rev
Immunol. 13:369–398. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Drake WM and Trainer PJ: Clinical use of
pegvisomant for the treatment of acromegaly. Treat Endocrinol.
2:369–374. 2003. View Article : Google Scholar : PubMed/NCBI
|