Introduction
Prolonged exposure to high concentrations of oxygen
(>90% oxygen) can lead to either acute or chronic lung injury in
former premature infants. It leads to the production of free oxygen
radicals that exceed the cell defense capacity, leading to
inflammation, cell damage and gene overexpression that may result
in necrosis and apoptosis (1).
The alveolar epithelium is the major target of
oxidant injury (2) and repair of
the injury depends on the ability of its stem cells, the alveolar
epithelial type II cells, which are able to spread and proliferate.
Alveolar epithelial type II cells (AEC IIs) can spread, proliferate
and are important in the repair process of the alveolar epithelium.
(3).
Substance P (SP) is widely distributed in the airway
endothelial cell layer. It was revealed that SP is able to trigger
an exuberant neuroinflammatory response and regulate proliferation,
migration and differentiation of the impaired cells (4,5).
However, the molecular mechanisms that regulate the activities of
AEC IIs and attenuate oxidative stress injury are poorly
understood. It has been reported that the Sonic hedgehog (SHH)
pathway is critical in lung morphogenesis and organogenesis
(6,7). SHH signaling proteins, including SHH,
Patched, Smoothened (SMO) and Gli are important in a variety of
processes, including embryogenesis, tissue repair and wound healing
(8–10).
The aim of the present study was to investigate the
effects of SP on primary AEC IIs of premature rats exposed to
hyperoxia and its regulation of the SHH signaling pathway. In view
of this, the present study aimed to elucidate the associated
regulatory mechanism of SP in the damage to AEC IIs exposed to
hyperoxia.
Materials and methods
Experimental animals
The 180–200 g specific pathogen-free Sprague-Dawley
(SD) rats were obtained for the present study from the Experimental
Animal Center of Chongqing Medical University (Chongqing, China).
All the experiments were conducted in accordance with the National
Guidelines for the Care and Use of Laboratory Animals. The
experiment was approved by the Ethics Committee of Chongqing
Medical University (Chongqing, China).
Materials
SP was provided by Abcam (Cambridge, MA, USA) and
L703.606, a selective NK1R antagonist, was provided by Sigma (St.
Louis, MO, USA). The Annexin V fluorescein isothiocyanate
(FITC)-labeled apoptosis kit was provided by Kaiji Biotechnology
Co., Ltd. (Nanjing, China). SMO polyclonal antibody was purchased
from Abbiotec (San Diego, CA, USA) and the reactive oxygen species
kit was purchased from Beyotime Biotechnology Co., Ltd. (Shanghai,
China).
Culture and isolation of premature
rats
Healthy adult SD rats were raised in a cage with a
gender ratio of 1:1. Pregnancy was confirmed if a vaginal plug was
observed on the second morning following the mixed raising.
Uterine-incision delivery was employed to remove the premature rats
from the pregnant rats on day 19 (22 days for full term) and fetal
lungs were isolated. AEC IIs were isolated through digestion with
0.25% trypsin and 0.04 mg/ml DNAse for 20 min at 37°C, 0.1%
collagenase for 15 min at 37°C and centrifugation at 200 × g for 5
min. Subsequently, the isolated cells were stained with modified
Papanicolaou stains as previously described (11) and observed under a transmission
electron microscope (Hitachi, Ltd., Tokyo, Japan).
Establishment of the cellular oxidative
model
Following culturing for 24 h, the isolated and
purified cells were treated with air (21% oxygen), hyperoxia (95%
oxygen), SP + hyperoxia or SP + L703.606 + hyperoxia. The cells
were exposed to air and hyperoxia for 24 h in a closed oxygen
chamber; 1×10−8 mol/l SP and 1×10−7 mol/l
L703.606 were added in advance. Then, the cells were exposed to air
and hyperoxia for 24 h. The cells were collected 24 h after
exposure.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium brom ide (MTT)
cellular proliferation assay
The proliferation of treated AEC IIs was detected
using an MTT detection kit (Amresco LLC, Solon, OH, USA) according
to the manufacturer’s instructions.
Apoptosis assay
The cellular apoptosis and necrosis were detected
using an Annexin V-FITC apoptosis detection kit on the basis of
Annexin V and propidium iodide staining. Apoptosis was assayed by
flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
Oxidation assay
To evaluate the damage caused by oxygen, reactive
oxygen species (ROS) were measured using a flow cytometer (BD
Biosciences, Franklin Lakes, NJ, USA) according to the
manufacturer’s instructions.
Quantitative polymerase chain reaction
(qPCR)
qPCR was performed using the SYBR-Green real-time
PCR method. Total RNA was extracted from AEC IIs. qPCR was
performed using an FTC2000 PCR instrument (Funglyn Biotech Inc.,
Toronto, Ontario, Canada) using two-stage program parameters
provided by the manufacturer, as follows: 4 min at 94°C and then 35
cycles of 20 sec at 94°C, 30 sec at 60°C and 30 sec at 70°C. The
specificity of the produced amplification product was confirmed by
the examination of dissociation reaction plots. A distinct single
peak indicated that a single DNA sequence was amplified during PCR.
PCR products were run on 2% agarose gels to confirm that the
correct molecular sizes were presented. Each sample was assessed in
triplicate and the samples obtained from three independent
experiments were used for the analysis of relative gene expression
using the 2−ΔΔCt method. The following primers were used
for qPCR: Forward: 5′-CCC ATC TAT GAG GGT TAC GC-3′ and reverse:
5′-TTT AAT GTC ACG CAC GAT TTC-3′ for glyceraldehyde-3-phosphate
dehydrogenase; and forward: 5′-ACC TCC AGC GAG ACC CTA TC-3′ and
reverse: 5′-TGA GGA CGA AGG GGA GTG AC-3′ for SMO.
Western blot analysis
Total cellular proteins were calculated using the
bicinchoninic acid protein assay and proteins (50 μg) from each
sample were loaded onto a 10% SDS/polyacrylamide gel and
electrophoretically transferred onto polyvinylidene fluoride
membranes. The membranes were inhibited in Tris-buffered saline
with Tween-20 (TBST) containing 0.05% Tween-20/5% non-fat dried
milk for 1 h, rinsed, and incubated with the appropriate smo
polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA) overnight at 4°C. Following washing in TBST, the membranes
were incubated with horseradish peroxidase secondary antibody
(GenScript Ltd., Piscataway, NJ, USA) for 1.5 h at room
temperature, followed by three washes in TBS. The immunoreactive
proteins were visualized with peroxidase and an enhanced
chemiluminescence system (ECL kit; Pierce Biotechnology, Inc.,
Rockford, IL, USA).
Statistical analysis
All the data are expressed as the mean ± standard
deviation. The statistical significance of the differences between
the means of the groups was determined by one-way analysis of
variance or two-tailed Student’s t-tests. P<0.05 was considered
to indicate a statistically significant difference.
Results
Effects of SP on cell growth and
apoptosis of AEC IIs following exposure to hyperoxia
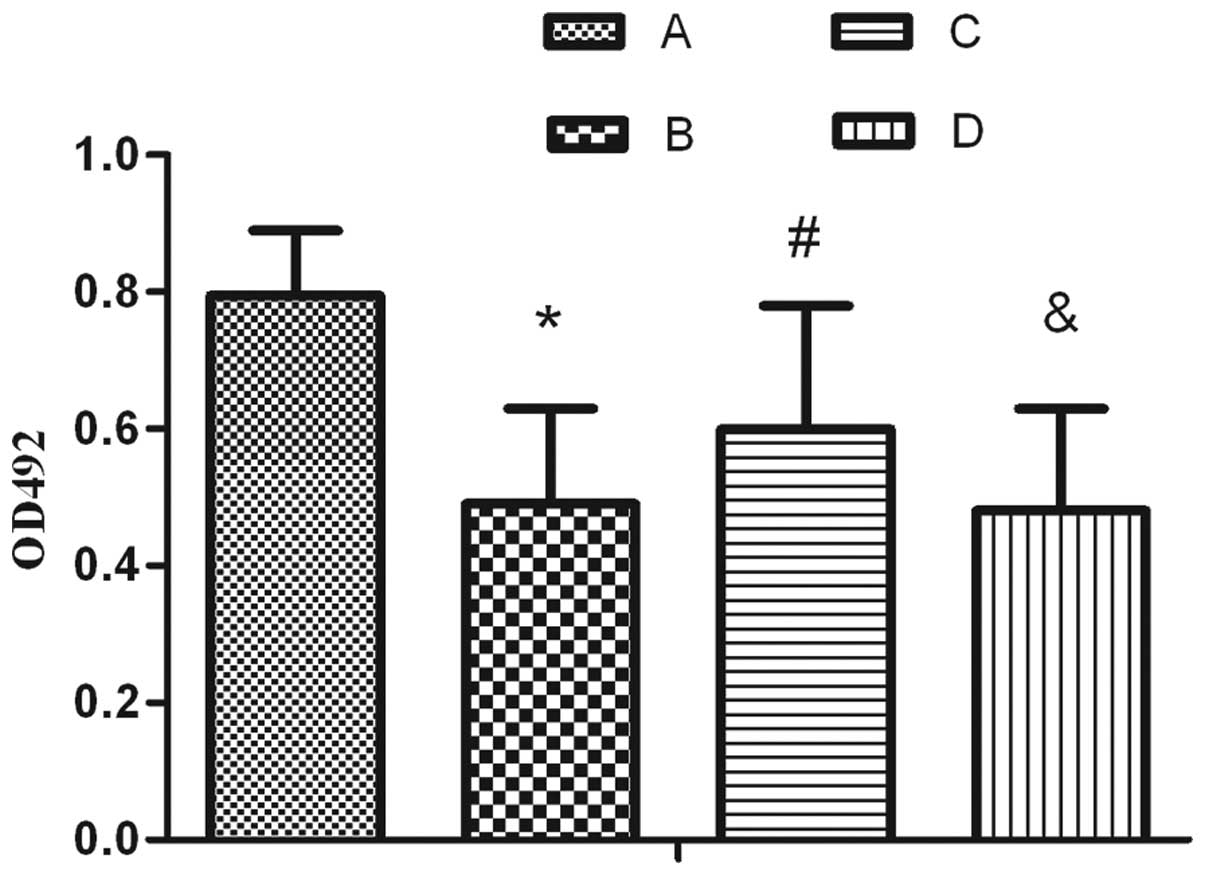
Following exposure to hyperoxia for 24 h and
compared with the air treatment, AEC II proliferation markedly
decreased and the percentage of apoptotic cells markedly increased
(Figs. 1–2). Furthermore, it was revealed that
following SP treatment, the proliferation activity increased
compared with simple exposure to hyperoxia, which was able to be
reversed by L703.606 (Fig. 1).
Accordingly, the number of apoptotic cells decreased markedly
following treatment with SP when compared with that in the
hyperoxia group. This decrease was inhibited by L703.606 (Fig. 2).
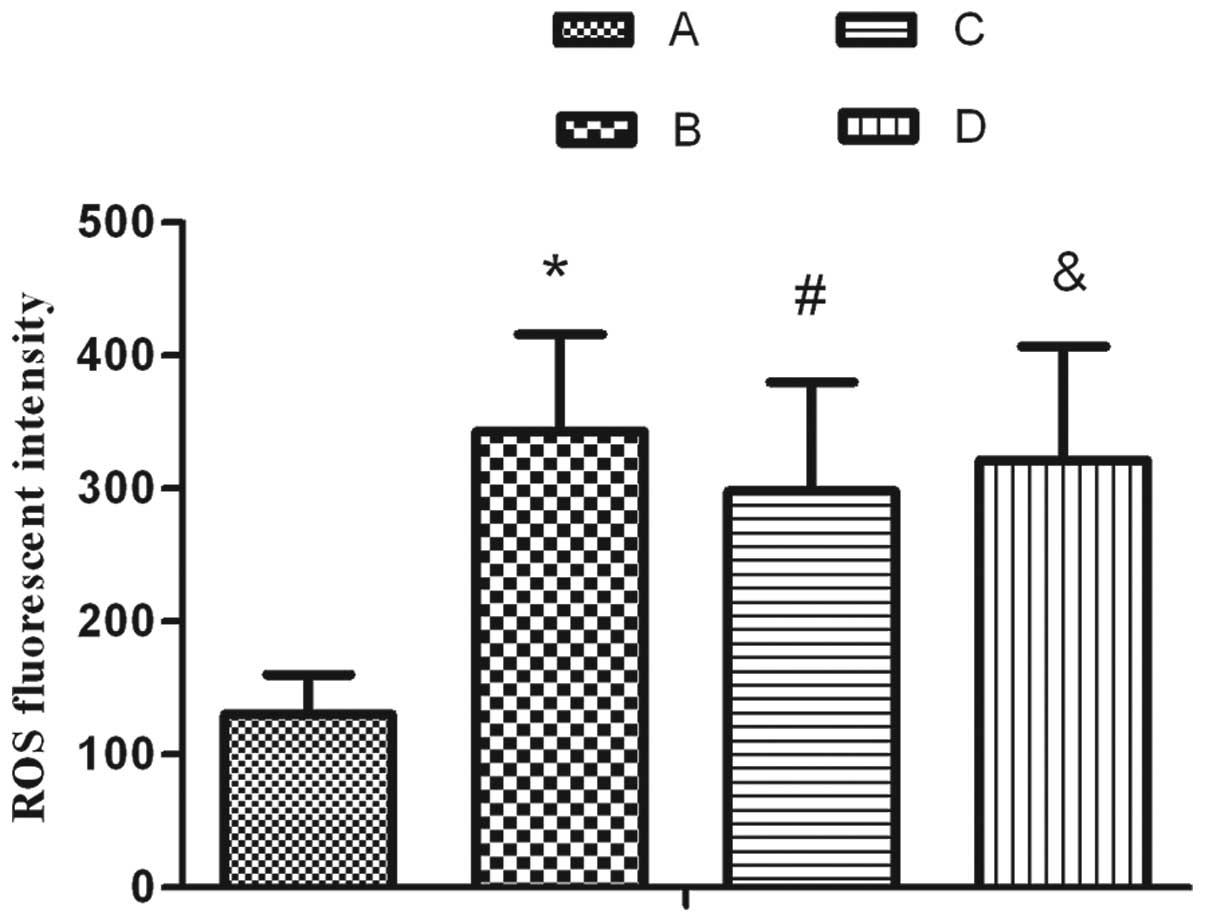
Effect of SP on the oxidative damage of
AEC IIs following exposure to hyperoxia
The results demonstrated that the content of ROS
significantly increased upon treatment with 95% oxygen for 24 h. SP
markedly inhibited the increase in ROS; however, L703.606 reversed
the ROS level to a similar level as that in the hyperoxia treatment
group, as shown in Fig. 3.
mRNA levels of SMO in AEC II cells are
activated by hyperoxia and enhanced by SP treatment
qPCR revealed that SMO mRNA was expressed in normal
AEC IIs. The mRNA expression of SMO in the hyperoxia-exposed group
significantly increased compared with the air-exposed group. By
contrast, the expression of SMO in the hyperoxia-exposed group was
elevated by stimulation with SP. SP was able to significantly
increase the expression of SMO. The aforementioned upregulation
effect was attenuated following treatment with L703.606 (Fig. 4).
Protein levels of SMO in AEC II cells are
activated by hyperoxia and enhanced by SP treatment
Western blot analysis revealed that SMO proteins
were expressed in normal AEC IIs. The protein expression of SMO in
the hyperoxia-exposed group was significantly increased compared
with that in the air-exposed group. In addition, the expression of
SMO proteins in the hyperoxia-exposed group was elevated by
stimulation with SP. SP was able to significantly increase the
expression of SMO. The upregulation effect aforementioned was
attenuated following treatment with L703.606 (Fig. 5).
Discussion
Hyperoxia-induced lung injury is considered to be a
bimodal process resulting from direct oxygen toxicity and the
accumulation of inflammatory mediators within the lungs (12). Either reducing lung oxidative
stress injury or improving the survival of alveolar epithelial
cells may be a way to ameliorate or prevent hyperoxia-induced lung
injury. To date, however, no therapy has been identified to
potently or consistently prevent or reverse hyperoxia-induced lung
injury. AEC IIs may serve as ‘alveolar stem cells’ (13) and, consequently, the protection of
type II epithelial cells during oxygen therapy may be the key to
preventing hyperoxia-induced lung injury.
SP is an important sensory neuropeptide. Subsequent
to its release, SP binds primarily to NK-1 receptors (NK-1R)
regulating airway smooth muscle responses, airway inflammation, and
epithelial migration and proliferation (14,15).
Oslund et al (16) found
that SP is an important mediator in airway epithelial cell death
and subsequent proliferation following ozone exposure. Scott et
al (5) revealed that SP levels
were greater in hypertrophic scars than in uninjured skin, and may
result in an exuberant neuroinflammatory response, which is
associated with cell proliferation and regeneration. Yaraee et
al (17) found that SP was
able to directly modulate the release of TGF-β from the human
bronchial epithelial cell line and thereby is involved in various
lung functions or pathological conditions. Dib et al
(18) found that sensory
neurotransmitters were able to protect acute hyperoxic lung injury
from aggravation by activating NK1R-mediated functions.
The present study demonstrated that AEC IIs exposed
to 95% oxygen for 24 h may cause cellular injury, increase the
production of ROS, induce apoptosis and inhibit cellular
proliferation. However, SP was able to decrease the inhibition of
hyperoxia on cell proliferation, reduce AEC II apoptosis and
necrosis, and improve cell survival sequentially. In addition, the
aforementioned protective effect was attenuated following treatment
with L703.606. The data suggested that SP was able to decrease
oxidative damage in AEC IIs and that SP interference may be a
protective strategy for AEC IIs under oxidative stress. In our
previous study, SP protected against hyperoxia-induced lung damage
by upregulating the SHH signaling pathway (19).
Information has emerged regarding the mechanisms and
significance of the SHH-SMO-Gli pathway in lung morphogenesis.
Bellusci et al (20) found
that the SHH signaling pathway is important in lung morphogenesis
and SHH overexpression in transgenic mice results in increased
mesenchymal and epithelial cell proliferation. Litingtung et
al (21) reported that
SHH-deficient mice resulted in a phenotype with defects in
tracheoesophageal and lung morphogenesis, including hypoplastic
lung buds, loss of lung symmetry and a single tracheoesophageal
tube. Pepicelli et al (22)
also found that the ectopic expression of SHH increases
proliferation and in SHH null mutants, the trachea and esophagus do
not separate properly and the lungs form a rudimentary sac.
Overexpression of SHH in the lung results in increased levels of
Gli mRNA and GLI1 is the principal effector of SHH signaling
(23). It has been demonstrated
that initiation of lung morphogenesis is instructed by SHH
signaling (24).
In order to elucidate the effects of SP on the SHH
signaling pathway, the mRNA and protein expression levels of SMO
were determined. The results from the present study demonstrated
that exposure to hyperoxia significantly activated SMO transiently
and supplementary SP improved cell survival and decreased apoptosis
accompanied by enhancing the expression of SMO. To further confirm
that the activation of the SHH pathway was associated with the
protection cells against hyperoxia by SP, L703.606 was added. The
cells in the L703.606 group showed increased ROS activity, enhanced
apoptosis and decreased proliferation, accompanied by the
decreasing expression of SMO, compared with the SP group.
It is suggested that SP interference, a protective
regulatory factor, had a protective effect on AEC IIs exposed to
hyperoxia, which may be associated with the promotion of the
activation of SHH pathways. SP may therefore have a therapeutic
effect on patients with hyperoxia-induced lung injury.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 30973218).
References
|
1
|
Gordo-Vidal F, Calvo-Herranz E,
Abella-Alvarez A, et al: Hyperoxia-induced pulmonary toxicity. Med
Intensiva. 34:134–138. 2010.(In Spanish).
|
|
2
|
Buccellato LJ, Tso M, Akinci OI, et al:
Reactive oxygen species are required for hyperoxia-induced Bax
activation and cell death in alveolar epithelial cells. J Biol
Chem. 279:6753–6760. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miyake Y, Kaise H, Isono K, et al:
Protective role of macrophages in noninflammatory lung injury
caused by selective ablation of alveolar epithelial type II Cells.
J Immunol. 178:5001–5009. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scott JR, Muangman P and Gibran NS: Making
sense of hypertrophic scar: a role for nerves. Wound Repair Regen.
15(Suppl 1): S27–S31. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scott JR, Muangman PR, Tamura RN, et al:
Substance P levels and neutral endopeptidase activity in acute burn
wounds and hypertrophic scar. Plast Reconstr Surg. 115:1095–1102.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang M, Wang H, Teng H, et al: Expression
of SHH signaling pathway components in the developing human lung.
Histochem Cell Biol. 134:327–335. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Y, Zhang H, Choi SC, et al: Sonic
hedgehog signaling regulates Gli3 processing, mesenchymal
proliferation, and differentiation during mouse lung organogenesis.
Dev Biol. 270:214–231. 2004. View Article : Google Scholar
|
|
8
|
Le H, Kleinerman R, Lerman OZ, et al:
Hedgehog signaling is essential for normal wound healing. Wound
Repair Regen. 16:768–773. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luo JD, Hu TP, Wang L, et al: Sonic
hedgehog improves delayed wound healing via enhancing cutaneous
nitric oxide function in diabetes. Am J Physiol Endocrinol Metab.
297:e525–e531. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kusano KF, Pola R, Murayama T, et al:
Sonic hedgehog myocardial gene therapy: tissue repair through
transient reconstitution of embryonic signaling. Nat Med.
11:1197–1204. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Corti M, Brody AR and Harrison JH:
Isolation and primary culture of murine alveolar type II cells. Am
J Respir Cell Mol Biol. 14:309–315. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pagano A and Barazzone-Argiroffo C:
Alveolar cell death in hyperoxia-induced lung injury. Ann N Y Acad
Sci. 1010:405–416. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bishop AE: Pulmonary epithelial stem
cells. Cell Prolif. 37:89–96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hökfelt T, Pernow B and Wahren J:
Substance P: a pioneer amongst neuropeptides. J Intern Med.
249:27–40. 2001.PubMed/NCBI
|
|
15
|
Hafstrom I, Ringertz B, Lundeberg T and
Palmblad J: The effect of endothelin, neuropeptide Y, calcitonin
gene-related peptide and substance P on neutrophil functions. Acta
Physiol Scand. 148:341–346. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oslund KL, Hyde DM, Putney LF, et al:
Activation of neurokinin-1 receptors during ozone inhalation
contributes to epithelial injury and repair. Am J Respir Cell Mol
Biol. 39:279–288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yaraee R and Ghazanfari T: Substance P
potentiates TGFβ-1 production in lung epithelial cell lines. Iran J
Allergy Asthma Immunol. 8:19–24. 2009.PubMed/NCBI
|
|
18
|
Dib M, Zsengeller Z, Mitsialis A, et al: A
paradoxical protective role for the proinflammatory peptide
substance P receptor (NK1R) in acute hyperoxic lung injury. Am J
Physiol Lung Cell Mol Physiol. 297:L687–L697. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang L, Liu C, Dang H, et al: Substance P
attenuates hyperoxia-induced lung injury in neonatal rats. Mol Med
Rep. 9:595–599. 2014.PubMed/NCBI
|
|
20
|
Bellusci S, Furuta Y, Rush MG, et al:
Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth
and morphogenesis. Development. 124:53–63. 1997.PubMed/NCBI
|
|
21
|
Litingtung Y, Lei L, Westphal H and Chiang
C: Sonic hedgehog is essential to foregut development. Nat Genet.
20:58–61. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pepicelli CV, Lewis PM and McMahon AP:
Sonic hedgehog regulates branching morphogenesis in the mammalian
lung. Curr Biol. 8:1083–1086. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grindley JC, Bellusci S, Perkins D and
Hogan BL: Evidence for the involvement of the Gli gene family in
embryonic mouse lung development. Dev Biol. 188:337–348. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Warburton D, Zhao J, Berberich MA, et al:
Molecular embryology of the lung: then, now, and in the future. Am
J Physiol. 276:L697–L704. 1999.PubMed/NCBI
|