Introduction
Breast cancer bone metastasis causes serious
complications, including chronic pain and pathologic fractures,
which severely reduce quality of life (1). Bone metastasis secondary to breast
cancer is associated with a poor prognosis (2) and current therapies for the
management of metastasis and osteolysis are far from satisfactory.
Hence, it is necessary to develop novel alternative therapies with
improved efficacy and fewer side-effects.
Metastasis is a complicated process, which proceeds
through a sequence of cancer cell proliferation, adhesion, invasion
and migration (3,4). Matrix metalloproteinases (MMPs) are
thought to be critical to this process (5–7) and
MMP-9 is considered to be the most relevant for tumor invasion
(8). MMP regulation occurs at
multiple levels and a number of stimuli activate MMP-9, including
growth factors, cytokines and phorbol 12-myristate 13-acetate (PMA)
(9–11). The MMP-9 promoter contains multiple
DNA binding sites for transcription factors, including nuclear
factor κB (NF-κB) (12).
Therefore, the downregulation of MMP-9 expression may be a useful
strategy for tumor metastasis intervention. Plant-derived compounds
with a chemopreventive potential have been shown to inhibit the
invasiveness of several types of cancer by modifying MMP-9
expression (13,14).
Andrographolide (AP) is a diterpenoid lactone
isolated from the traditional Chinese and Indian medicinal plant
Andrographis paniculata and it is widely used for its
efficacy and favorable safety profile in a number of diseases
(15,16). AP has gained attention for its
anticancer (17,18), anti-inflammation (19,20),
hepatoprotection (21,22) and anti-infection (16) activities. Previous studies have
demonstrated the anti-cancer effect of AP in the MCF-7 and TD-47
breast cancer cell lines (23–25);
however, the effect of AP on the more aggressive MDA-MB-231 cancer
cell line and on breast cancer bone metastasis in vivo has
not been reported.
The aim of the present study was to identify
supplementary therapeutic strategies for the treatment of breast
cancer metastasis and osteolysis through the investigation of the
in vitro action of AP on the invasion and migration of
MDA-MB-231 cells. In addition, the efficacy of AP in the prevention
of breast cancer bone metastasis and osteolysis were investigated
in an in vivo mouse xenograft model.
Materials and methods
Media and reagents
AP and PMA were purchased from Sigma-Aldrich (St.
Louis, MO, USA). Minimum Essential Medium-α (α-MEM), fetal bovine
serum (FBS) and penicillin were obtained from Gibco-BRL
(Gaithersburg, MD, USA). The Cell Counting kit (CCK)-8 assay was
purchased from Dojindo Molecular Technology (Tokyo, Japan). Primary
antibodies (monoclonal rabbit antibody; species reactivity, human)
for β-actin, phospho-IκBα, IκBα and MMP-9 were purchased from Cell
Signaling Technology, Inc. (Beverly, MA, USA). The Luciferase Assay
system was from Promega (Sydney, Australia). Tris, glycine, NaCl,
SDS, and other reagents were from Sigma-Aldrich. The
Vybrant® Apoptosis Assay kit #2 was from Invitrogen
(Carlsbad, CA, USA).
Cell viability assay
MDA-MB-231 cells were cultured in L-15 Medium (Gibco
Life Technologies, Beijing, China) with 10% FBS and maintained in a
humidified atmosphere of 5% CO2 at 37°C. The complete
medium was changed every other day. The cells were treated with
increasing concentrations of AP (0, 7.5, 15, 30, 60 or 120 μM) for
two days prior to the cell viability assays. The anti-proliferative
effect of AP on MDA-MB-231 cells was assessed using CCK-8. Briefly,
following treatment, 10 μl CCK-8 solution was added to each well
and incubated for 4 h. The absorbance was measured at a wavelength
of 450 nm using a ELX800 absorbance microplate reader (BioTek
Instruments, Inc., Winooski, VT, USA) at a wavelength of 450 nm
(reference, 650 nm). The effect of AP on cell viability was
expressed as a percentage of cell viability, with the
vehicle-treated control cells set as 100%.
Apoptosis assay
AP induction of apoptosis in MBA-MD-231 cells was
determined with the Vybrant® Apoptosis Assay kit #2.
Following treatment, cells were washed twice with cold
phosphate-buffered saline (PBS) and resuspended in 1X
Annexin-binding buffer. Early apoptosis was detected via staining
with Alexa Fluor® 488 Annexin V and propidium iodide.
Fluorescence-activated cell sorting was performed using a FACScan™
flow cytometer and data were acquired using CellQuest software,
version 3.0 (BD Biosciences, Sunnyvale, CA, USA).
Migration assay
Transwell® Permeable Supports (Corning
Inc., Acton, MA, USA), 24-well chambers with 8-μm pore
polycarbonate filters, were used as described by the manufacturer.
MDA-MB-231 cells (5×104) were placed in 100 μl
serum-free medium in the presence or absence of AP and 600 μl
complete medium with 80 nM PMA was placed into the lower wells.
Following treatment, cells were fixed with 100% methanol for 20 min
and stained with Trypan blue for 30 min. Non-migrating cells on the
upper side of the filter were removed with cotton swabs. Migration
was quantified by counting the number of cells on the lower surface
of the filter.
Invasion assay
BioCoat™ Matrigel™ Invasion Chamber (BD
Biosciences), 24-well chambers with 8-μm pore polycarbonate
filters, were used according to the manufacturer’s instructions.
MDA-MB-231 cells (5×104) were placed in 100 μl
serum-free medium in the presence or absence of AP, and 600 μl
complete medium with 80 nM PMA was placed in the lower wells.
Following treatment, cells on the upper side of the filters were
removed. Invading cells on the underside of the filter were fixed
with 100% methanol for 2 min and stained with Liu’s stain for 2
min. Invasion was quantified by counting the number of cells on the
lower surface of the filter.
Intratibial xenograft model of breast
cancer bone metastasis
BALB/c nu/nu mice (Harlan, Indianapolis, IN, USA)
were housed in individual cages, maintained in an animal facility
under controlled temperature (22–24°C) and humidity (50–60%)
conditions and a 12 h light/dark cycle with free access to food and
water. Cultured MDA-MB-231 cells were resuspended in PBS at a
density of 5×106 cells/ml (26,27).
An aliquot (10 μl) of the cell suspension was slowly injected
through the anterior tuberosity of the proximal tibia in the right
limbs of 5- to 6-week-old female BALB/c nu/nu mice (Harlan,
Indianapolis, IN, USA). The mice were randomly assigned to vehicle
(0.9% NaCl, n=8) or AP (50 mg/kg body weight vehicle, n=8) groups
and treated via an intraperitoneal injection every other day. After
28 days, a bioluminescence assay was performed and fluorescence
intensity was quantified (Living Image v3.2, Caliper; Caliper Life
Sciences, Hopkinton, MA, USA). Radiographs using the Directview
Vita CR system. (Carestream Kodak, Rochester, NY, USA) of the
tibiae were obtained prior to euthanasia with ketamine,
administered by intraperitoneal injection (0.8 ml/100 g body
weight). The product from Carestream Kodak was. Tissues were
removed and fixed in 4% paraformaldehyde for 1 day at 4°C followed
by decalcification in 12% EDTA. Decalcified bones were
paraffin-embedded and sectioned. Samples were subjected to
tartrate-resistant acid phosphatase (TRAP) staining to identify
osteoclasts on the bone surface. Immunostaining for Ki67 (Dako,
Carpinteria, CA, USA) and terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling (TUNEL) were performed
as previously described (28,29).
Ki67- and TUNEL-positive tumor cells were counted and the
percentages of positive cells were calculated. This study was
approved by the ethics committee of Shanghai Ninth People’s
Hospital Affiliated to Shanghai Jiao Tong University School of
Medicine (Shanghai, China).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA isolation was performed as previously described
(30). Total RNA was extracted
using the Qiagen RNeasy Mini kit (Qiagen, Valencia, CA, USA)
following the manufacturer’s instructions. cDNA was synthesized
from 1 mg of total RNA using reverse transcriptase (TaKaRa
Biotechnology, Otsu, Japan). MMP-9 transcript expression levels
were determined using the MiniOpticon Real-Time PCR system (Bio-Rad
Laboratories, Hercules, CA, USA). qPCR was performed in a
thermocycler (Biometra, T-Gradient Thermoblock, Germany) with a
reaction volume of 10 μl containing 0.03 μg complementary DNA
product, 2 μM forward and reverse primers and the KAPA™
SYBR® FAST qPCR reagent (Kapa Biosystems, Wilmington,
MA, USA). The primers used were as follows: Forward,
5′-GAACCAATCTCACCGACAGG-3′, and reverse, 5′-GCCACCCGAGTGTAACCATA-3′
for MMP-9; and forward, 5′-TCTGCTGGAAGGTGGACAGT-3′, and reverse,
5′-CCTCTATGCCAACACAGTGC-3′ for β-actin. Cycling conditions were as
follows: 40 cycles of 95°C for 5 sec and 60°C for 34 sec. β-actin
was included as a reference control. The comparative
2−ΔΔCt method was used to calculate the relative
expression of each gene (30).
NF-κB-dependent luciferase reporter
assay
The effect of AP on PMA-induced NF-κB activation was
measured in MDA-MB-231 cells stably transfected with an NF-κB
luciferase reporter construct (13). MDA-MB-231 cells were maintained in
serum-free medium for 12 h, pretreated with AP for 1 h, followed by
stimulation with PMA for 20 h. Subsequently, the cell lysis was
incubated with substrate (Promega, Madison, WI, USA) at room
temperature for about 2min, luciferase activity was measured using
the Promega Luciferase Assay System (Promega, Madison, WI, USA).
Luciferase activity was measured and normalized to the internal
control. Results were obtained from three independent
experiments.
Western blotting
Western blotting was performed as previously
described (30). The vehicle- or
AP-treated cells were pretreated with PMA, washed twice in PBS and
lysed in ice-cold lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1%
Nonidet P-40, 0.1% SDS, 1% sodium deoxycholate) supplemented with
phenylmethanesulfonyl fluoride (Shen Neng Bo Cai Corp., Shanghai,
China). Lysates were maintained on ice for 30 min followed by
centrifugation at 12,000 × g for 10 min. Protein concentrations
were determined using a bicinchoninic acid (BCA) assay (Thermo
Scientific, Rockford, IL, USA). Equal amounts of protein were
separated by 10% SDS-PAGE and electroblotted onto polyvinylidene
fluoride membranes (Roche, Mannheim, Germany). The membranes were
blocked with 5% (w/v) skim milk solution for 1 h and probed with
primary antibodies (β-actin, 1:1,000; phospho-IκBa, 1:1,000; IκBa,
1:1,000; and MMP-9, 1:1,000) at room temperature for 4 h, followed
by incubation with horseradish peroxidase-conjugated secondary
antibodies (anti-human; Cell Signaling Technology, Inc.; 1:5,000)
for 1 h. Antibody reactivity was visualized using an
Odyssey® Infrared Imaging system (Li-Cor, Lincoln, NE,
USA).
Statistical analysis
Significant differences were determined with the
Student’s t-test using SPSS v13.0 software (SPSS Inc., Chicago, IL,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
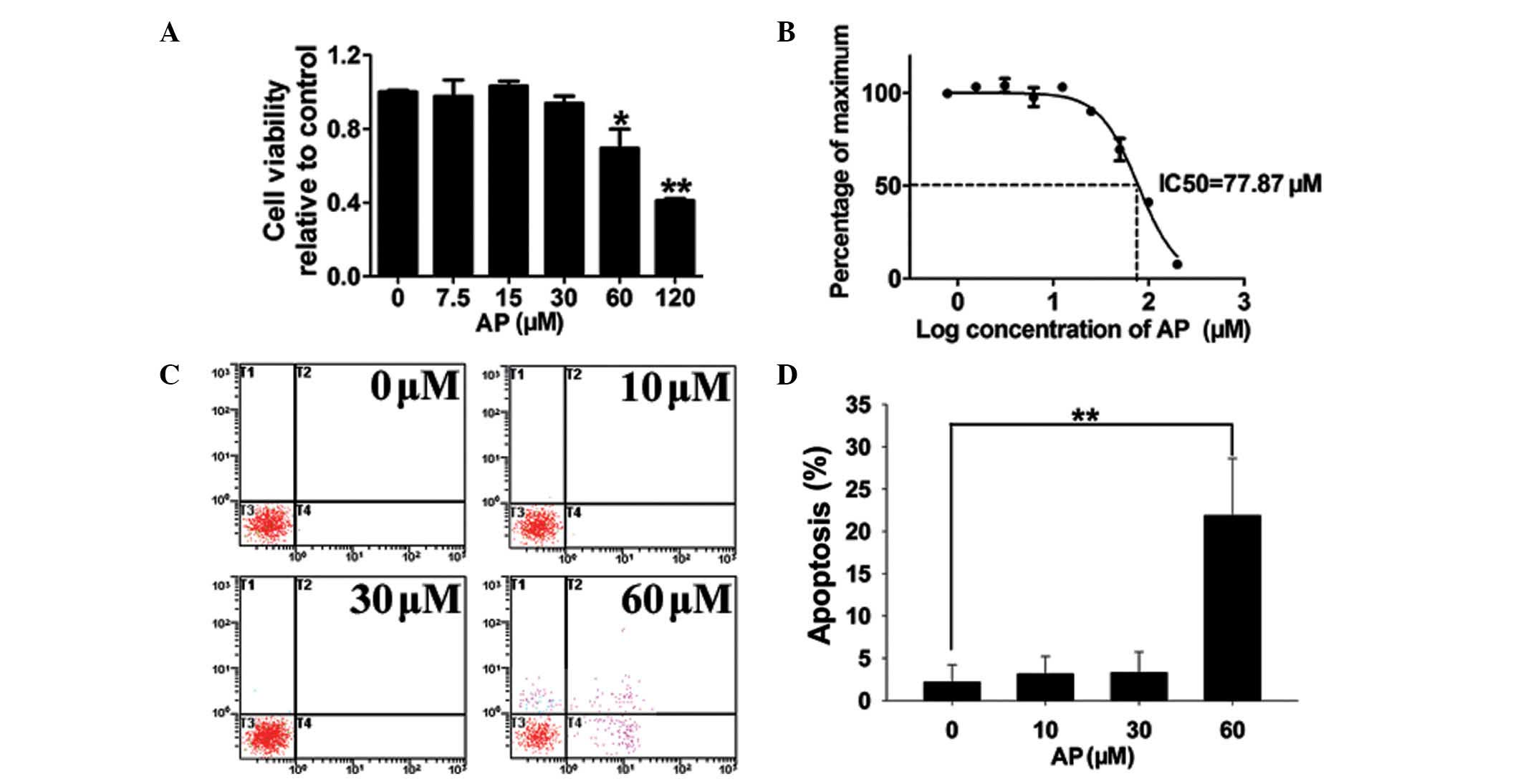
AP inhibits the proliferation of
MDA-MB-231 breast cancer cells and promotes apoptosis at high
concentrations
Following a 48-h culture, a CCK-8 proliferation
assay revealed that AP did not affect MDA-MB-231 cell proliferation
at concentrations ≤30 μM (Fig.
1A). AP significantly suppressed cell proliferation at
concentrations ≥60 μM. The calculated IC50 for AP is
77.87 μM (Fig. 1B). In cells
treated with 10 or 30 μM AP, the observed apoptotic effects were
similar to those of the vehicle control; however, the higher
concentration of 60 μM AP induced apoptosis in 22% of cells
(Fig. 1C and D). In order to
exclude AP-mediated apoptosis, non-lethal concentrations (≤30 μM)
were used in subsequent experiments.
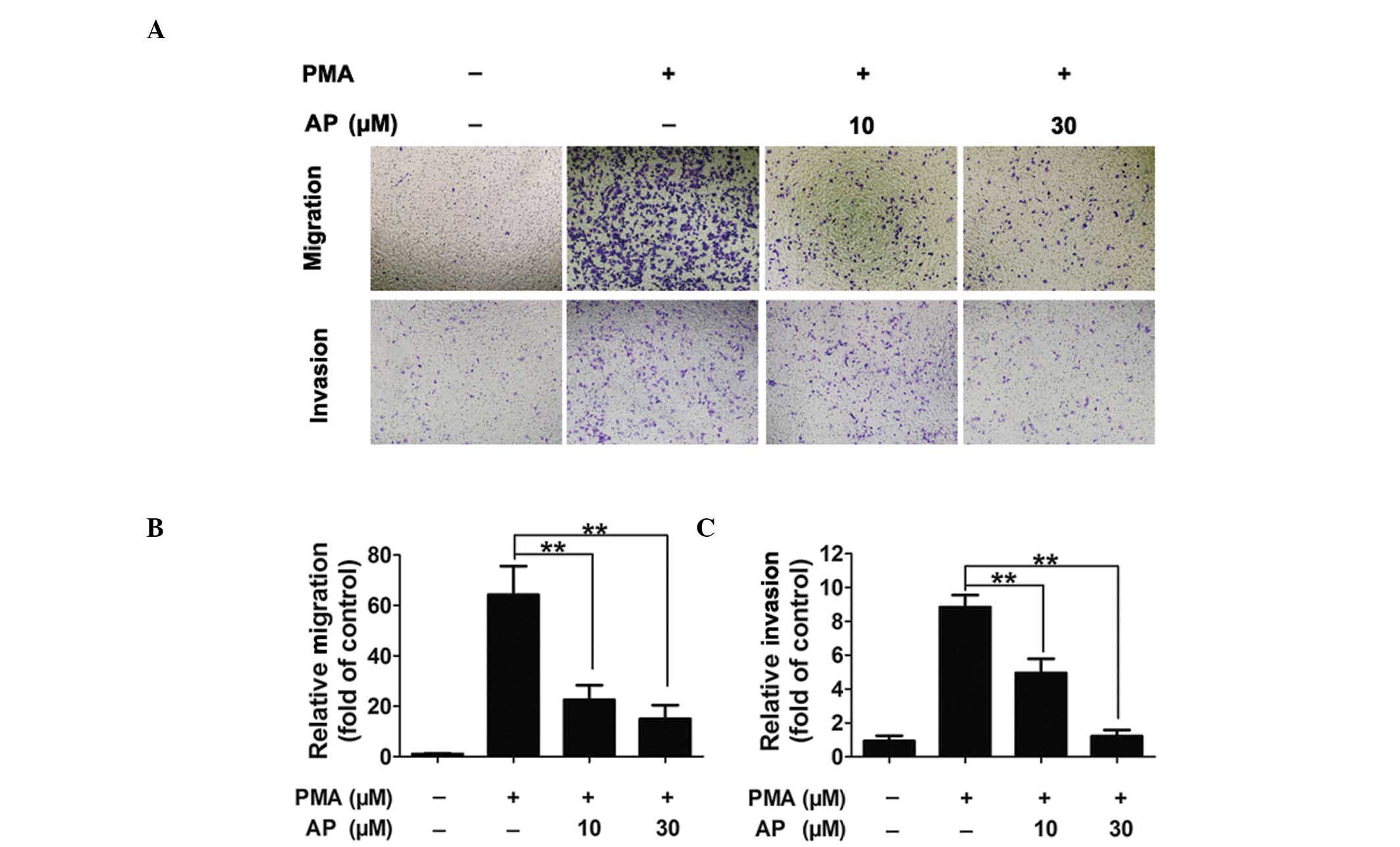
AP inhibits PMA-induced MDA-MB-231 cell
migration and invasion in a concentration-dependent manner
PMA (80 nM) induced increased levels of MDA-MB-231
cell migration and invasion compared with those observed in the
untreated cells; however, pretreatment with AP inhibited the
PMA-induced migration and invasion in a concentration-dependent
manner (Fig. 2A). Quantitative
analysis confirmed AP inhibition of cell migration and invasion at
concentrations as low as 10 μM (Fig.
2B and C).
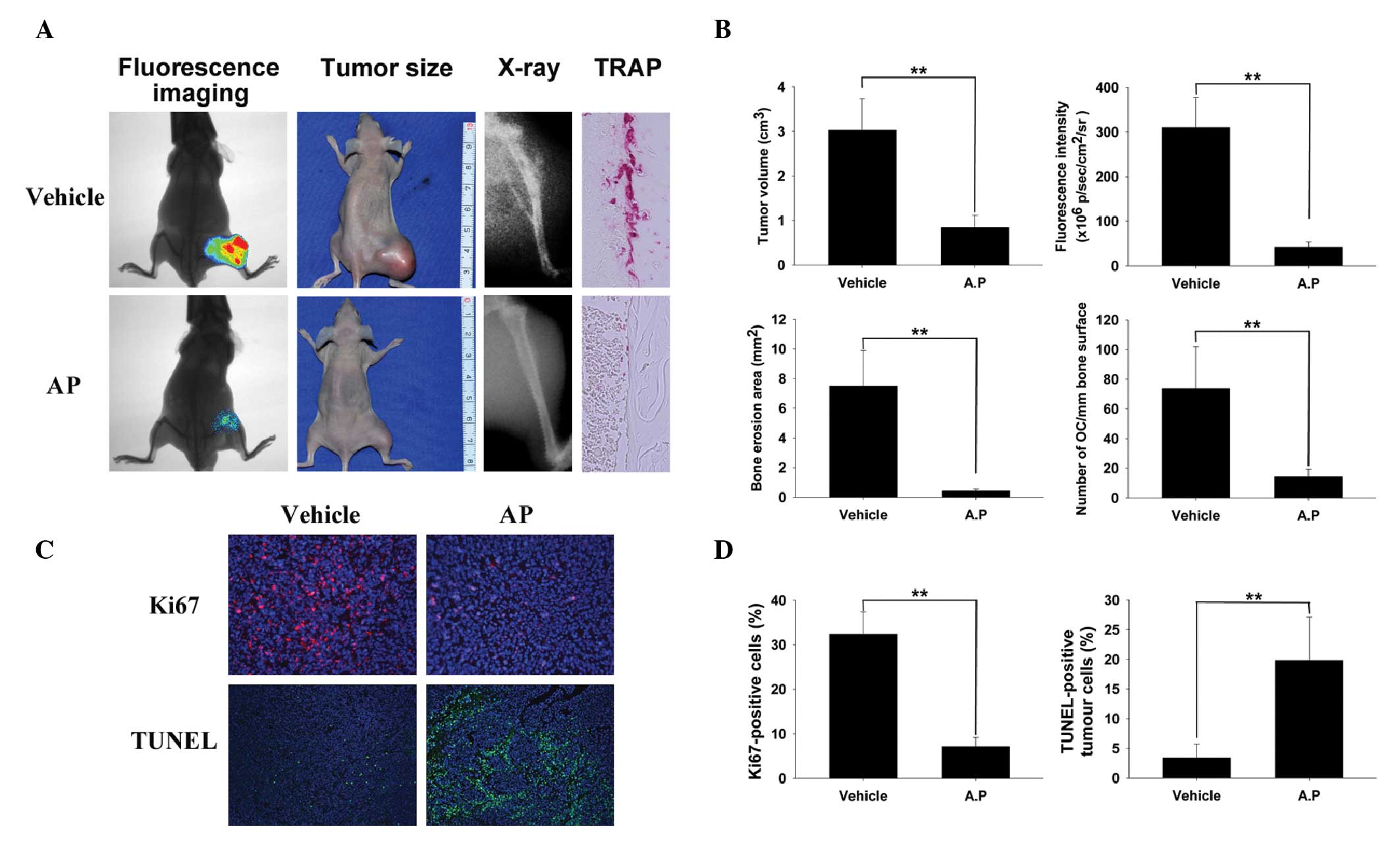
AP inhibits breast cancer bone metastasis
and osteolysis in vivo
To determine the effects of AP on breast cancer bone
metastasis and cancer cell-induced osteolysis in vivo, a
mouse xenotransplant model was used with human breast cancer cells
(luciferase-labeled MDA-MB-231) (26,31).
MDA-MB-231 cells were injected directly into the tibiae plateau via
a percutaneous approach. After 28 days, bioluminescence was
detected in the limbs of the control mice; however, the area and
density of bioluminescence were reduced in the AP group compared
with those in the control group (Fig.
3A), indicating that AP effectively suppressed breast cancer
bone metastasis and growth in vivo. These observations were
consistent with the results of the tumor volume assay (Fig. 3A). To confirm that osteolytic bone
metastasis was blocked by AP, the osteolysis in the long bones of
the hind legs was examined using radiography. AP significantly
inhibited cancer cell-induced osteolysis (represented by
radiolucency; Fig. 3A). TRAP
staining (red) revealed numerous osteoclasts with intense activity
in the vehicle-treated controls, however, in contrast, the number
of osteoclasts was markedly reduced at the boundary in the treated
mice (Fig. 3A), indicating that AP
suppressed tumor-related osteolysis by inhibiting osteoclasts in
vivo. All the results were confirmed using quantitative
analysis (Fig. 3B). The
proliferation-indicator Ki67 assay and the apoptosis-indicator
TUNEL assay were also performed. Treatment of MDA-MB-231 tumor
cells with AP (50 mg/kg) suppressed cellular proliferation compared
with that in the control cells (Fig.
3C). The percentage of Ki67-positive cell nuclei was 7.1% in
the AP-treated group and 32.4% in the vehicle-treated group
(Fig. 3D). The levels of apoptosis
were significantly increased in the AP-treated group of MDA-MB-231
cell-associated breast tumors compared with those of the
vehicle-treated group in the TUNEL assay (Fig. 3C and D). All in vivo data
were consistent with the in vitro data, demonstrating that
AP inhibits MDA-MB-231 cancer cell invasion and migration and
suppresses tumor-induced osteolysis, possibly via inhibited
osteoclast activity.
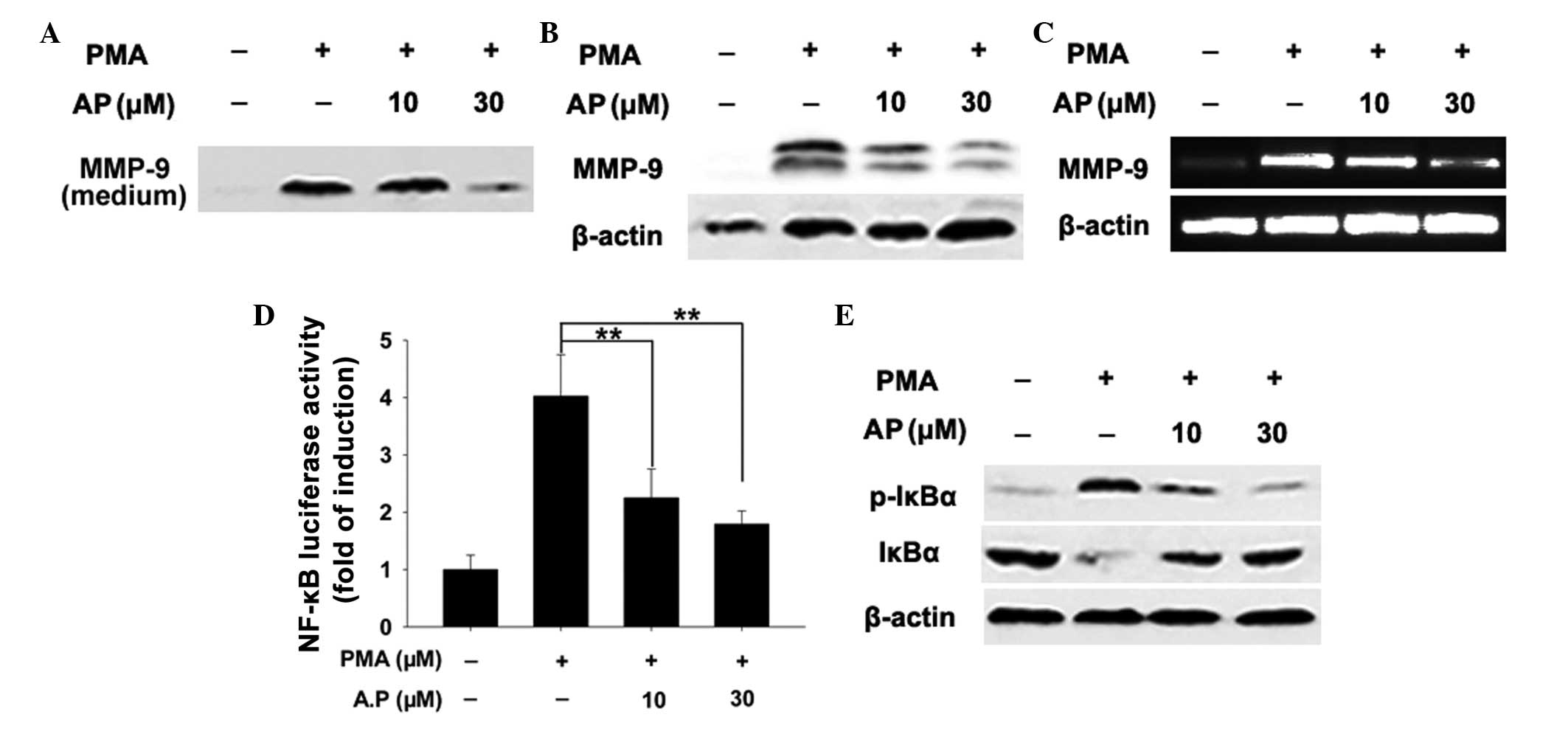
AP reduces PMA-stimulated MMP-9 secretion
and expression
MMP-9 mediates tumor invasion and migration. In the
current study, AP reduced the levels of MMP-9 secretion into the
medium compared with those observed in the control cells (Fig. 4A). Consistent with the
aforementioned findings, treatment of MDA-MB-231 cells with AP
reduced PMA-stimulated MMP-9 protein expression in a
concentration-dependent manner (Fig.
4B). qPCR revealed that PMA-induced MMP-9 mRNA expression
decreased with AP treatment, indicating that AP-mediated inhibition
of MMP-9 occurs at the transcriptional level (Fig. 4C).
AP suppresses NF-κB signaling
MMP-9 is highly inducible in response to various
stimuli and the MMP-9 promoter contains a binding site for
transcription factor NF-κB (12),
hence it can be used to detect NF-κB signaling. Measurement of
NF-κB-dependent luciferase activity in MDA-MB-231 cells revealed
that PMA-induced NF-κB transcriptional activity was suppressed by
AP (Fig. 4D). NF-κB is normally
sequestered in the cytoplasm in an inactive form associated with
NF-κ-B inhibitor α (IκBα). Upon stimulation, the NF-κB subunit is
released via the phosphorylation and proteasomal degradation of
IκBa and translocated to the nucleus to initiate target gene
transcription (32,33). AP was observed to prevent the
PMA-induced degradation of IκBa (Fig.
4E). As degradation of IκBα is primarily the result of IκBα
phosphorylation (33), it was
hypothesized that this effect may be due to the AP-induced
inhibition of IκBα phosphorylation. In the present study, AP caused
a concentration-dependent reduction in PMA-induced IκBα
phosphorylation (Fig. 4E). These
results indicate that the inhibitory effect of AP on NF-κB
signaling occurs via the inhibition of IκBa phosphorylation, which
in turn suppresses transcriptional activity. NF-κB signaling
activates MMP-9 transcription; thus, these results indicate that AP
attenuates MMP-9 expression by inhibiting NF-κB signaling.
Discussion
Previous studies have revealed the anti-cancer
activity of AP (17,18). The current study investigated the
utility of AP in fighting aggressive MDA-MB-231 breast cancer cell
invasion and bone metastasis. It was revealed that AP effectively
inhibits breast cancer cell migration and invasion in vitro.
In vivo, AP inhibits breast cancer bone metastasis,
suppresses tumor growth and induces tumor apoptosis in bone. This
inhibition was associated with the downregulation of MMP-9
expression levels.
MMP-9 expression levels are highly correlated with
breast cancer cell invasion (34)
and agents that downregulate MMP-9 have been observed to inhibit
tumor invasion (9,35). MMP-9 is inducible by a number of
stimuli; the MMP-9 promoter contains DNA-binding sites for NF-κB,
which regulates MMP-9 expression and secretion (36,37).
The transcription factor NF-κB regulates the transcription of genes
associated with cancer development, tumor invasion and
inflammation. It is a target for numerous biologically active
phytochemicals, including curcumin, resveratrol and
epigallocatechin gallate. Exposure of cells to stimuli such as PMA
leads to IκBa phosphorylation and degradation, allowing NF-κB to
translocate to the nucleus where it binds to the MMP-9 promotor and
activates transcription (9).
In the current study, AP was revealed to inhibit
PMA-induced MMP-9 expression. The specific response of MMP-9
indicates that its downregulation by AP is mediated through an
upstream event. Concurrently, PMA was observed to increase the
levels of NF-κB transcriptional activity, whilst AP inhibited
PMA-induced NF-κB transcriptional activity. These results confirm
that NF-κB signaling is the molecular target for AP-induced
inhibition of MMP-9 expression. Furthermore, the AP-induced
reduction of PMA-stimulated NF-κB transcriptional activity was
identified to be due to the inhibition of IκBa phosphorylation and
IκBa proteasomal degradation. However, the mechanisms by which AP
inhibits the phosphorylation of IκBa remain unclear.
In conclusion, AP-induced inhibition of IκBa
phosphorylation was revealed to be the underlying mechanism of its
effect on PMA-stimulated MDA-MB-231 cancer cell invasion. At
sub-lethal concentrations, AP inhibits breast cancer cell migration
and invasion via the downregulation of MMP-9 expression levels. The
molecular mechanism by which AP inhibits MMP-9 expression involves
the suppression of NF-κB activation. Tumor metastasis is often
associated with poor prognosis and high mortality in breast cancer,
prompting the requirement for the discovery and development of
novel therapeutic strategies that target early tumor invasiveness
and/or metastasis. AP reduces the invasiveness of highly aggressive
MDA-MB-231 breast cancer cells in vitro, inhibits breast
cancer bone metastasis, tumor growth, and tumor-induced osteolysis,
and induces tumor apoptosis in vivo. It is thus a promising
candidate therapeutic agent against breast cancer invasion and
metastasis.
Acknowledgements
This study was supported by the Program for
Innovative Research Team of Shanghai Municipal Education Commission
(Phase I), a grant awarded for innovative research from Shanghai
Municipal Education Commission (grant no. 13YZ031), a grant for
scientific research from the National Natural Science Foundation
for the Youth of China (grant no. 81201364), and a grant awarded by
the Scientific Research Foundation for Returned Overseas Chinese
Scholars from the State Human Resource Ministry, as well as the Key
National Basic Research Program of China (grant no. 2012CB619101),
the Major Basic Research of Science and Technology Commission of
Shanghai Municipality (grant no. 11DJ1400303) and the Doctoral
Innovation Foundation from Shanghai Jiaotong University School of
Medicine (grant no. BXJ201330).
References
|
1
|
American Cancer Society. Breast cancer
facts & figures 2007–2008. American Cancer Society, Inc;
Atlanta, GA: pp. 1–13. 2007
|
|
2
|
Gonzalez-Angulo AM, Morales-Vasquez F and
Hortobagyi GN: Overview of resistance to systemic therapy in
patients with breast cancer. Breast Cancer Chemosensitivity. Yu D
and Hung MC: Springer; New York, NY: pp. 1–22. 2007
|
|
3
|
Lin CW, Shen SC, Hou WC, Yang LY and Chen
YC: Heme oxygenase-1 inhibits breast cancer invasion via
suppressing the expression of matrix metalloproteinase-9. Mol
Cancer Ther. 7:1195–1206. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gialeli C, Theocharis AD and Karamanos NK:
Roles of matrix metalloproteinases in cancer progression and their
pharmacological targeting. FEBS J. 278:16–27. 2011. View Article : Google Scholar
|
|
5
|
Kessenbrock K, Plaks V and Werb Z: Matrix
metalloproteinases: regulators of the tumor microenvironment. Cell.
141:52–67. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Deryugina EI and Quigley JP: Pleiotropic
roles of matrix metalloproteinases in tumor angiogenesis:
contrasting, overlapping and compensatory functions. Biochim
Biophys Acta. 1803:103–120. 2010. View Article : Google Scholar :
|
|
7
|
Shuman Moss LA, Jensen-Taubman S and
Stetler-Stevenson WG: Matrix metalloproteinases: changing roles in
tumor progression and metastasis. Am J Pathol. 181:1895–1899. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brinckerhoff CE and Matrisian LM: Matrix
metalloproteinases: a tail of a frog that became a prince. Nat Rev
Mol Cell Biol. 3:207–214. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Overall CM and López-Otín C: Strategies
for MMP inhibition in cancer: innovations for the post-trial era.
Nat Rev Cancer. 2:657–672. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chakraborti S, Mandal M, Das S, Mandal A
and Chakraborti T: Regulation of matrix metalloproteinases: an
overview. Mol Cell Biochem. 253:269–285. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Egeblad M and Werb Z: New functions for
the matrix metalloproteinases in cancer progression. Nat Rev
Cancer. 2:161–174. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sato H and Seiki M: Regulatory mechanism
of 92 kDa type IV collagenase gene expression which is associated
with invasiveness of tumor cells. Oncogene. 8:395–405.
1993.PubMed/NCBI
|
|
13
|
Ling H, Zhang Y, Ng KY and Chew EH:
Pachymic acid impairs breast cancer cell invasion by suppressing
nuclear factor-κB-dependent matrix metalloproteinase-9 expression.
Breast Cancer Res Treat. 126:609–620. 2011. View Article : Google Scholar
|
|
14
|
Weng CJ, Chau CF, Hsieh YS, Yang SF and
Yen GC: Lucidenic acid inhibits PMA-induced invasion of human
hepatoma cells through inactivating MAPK/ERK signal transduction
pathway and reducing binding activities of NF-kappaB and AP-1.
Carcinogenesis. 29:147–156. 2008. View Article : Google Scholar
|
|
15
|
Coon JT and Ernst E: Andrographis
paniculata in the treatment of upper respiratory tract infections:
a systematic review of safety and efficacy. Planta Med. 70:293–298.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang X, Yu P, Jiang J, et al: Synthesis
and evaluation of antibacterial activities of andrographolide
analogues. Eur J Med Chem. 44:2936–2943. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang LJ, Zhou X, Wang W, et al:
Andrographolide inhibits oral squamous cell carcinogenesis through
NF-κB inactivation. J Dental Res. 90:1246–1252. 2011. View Article : Google Scholar
|
|
18
|
Rajagopal S, Kumar RA, Deevi DS,
Satyanarayana C and Rajagopalan R: Andrographolide, a potential
cancer therapeutic agent isolated from Andrographis paniculata. J
Exp Ther Oncol. 3:147–158. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chiou WF, Lin JJ and Chen CF:
Andrographolide suppresses the expression of inducible nitric oxide
synthase in macrophage and restores the vasoconstriction in rat
aorta treated with lipopolysaccharide. Br J Pharmacol. 125:327–334.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shen YC, Chen CF and Chiou WF:
Andrographolide prevents oxygen radical production by human
neutrophils: possible mechanism(s) involved in its
anti-inflammatory effect. Br J Pharmacol. 135:399–406. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Trivedi NP, Rawal UM and Patel BP: Potency
of andrographolide as an antitumor compound in BHC-induced liver
damage. Integr Cancer Ther. 8:177–189. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Handa SS and Sharma A: Hepatoprotective
activity of andrographolide against galactosamine & paracetamol
intoxication in rats. Indian J Med Res. 92:284–292. 1990.PubMed/NCBI
|
|
23
|
Harjotaruno S, Widyawaruyanti A,
Sismindari and Zaini NC: Apoptosis inducing effect of
andrographolide on TF-47 human breast cancer cell line. Afr J
Tradit Complement Altern Med. 4:345–351. 2007.
|
|
24
|
Kumar S, Patil SH, Sharma P, et al:
Andrographolide inhibits osteopontin expression and breast tumor
growth through down regulation of PI3 kinase/Akt signaling pathway.
Curr Mol Med. 12:952–966. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chao CY, Lii CK, Hsu YT, et al: Induction
of heme oxygenase-1 and inhibition of TPA-induced matrix
metalloproteinase-9 expression by andrographolide in MCF-7 human
breast cancer cells. Carcinogenesis. 34:1843–1851. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ooi LL, Zhou H, Kalak R, et al: Vitamin D
deficiency promotes human breast cancer growth in a murine model of
bone metastasis. Cancer Res. 70:1835–1844. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zheng Y, Zhou H, Modzelewski JR, et al:
Accelerated bone resorption, due to dietary calcium deficiency,
promotes breast cancer tumor growth in bone. Cancer Res.
67:9542–9548. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Traxler P, Allegrini PR, Brandt R, et al:
AEE788: a dual family epidermal growth factor receptor/ErbB2 and
vascular endothelial growth factor receptor tyrosine kinase
inhibitor with antitumor and antiangiogenic activity. Cancer Res.
64:4931–4941. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu W, Onn A, Isobe T, et al: Targeted
therapy of orthotopic human lung cancer by combined vascular
endothelial growth factor and epidermal growth factor receptor
signaling blockade. Mol Cancer Ther. 6:471–483. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li H, Zhai Z, Liu G, et al: Sanguinarine
inhibits osteoclast formation and bone resorption via suppressing
RANKL-induced activation of NF-κB and ERK signaling pathways.
Biochem Biophys Res Commun. 430:951–956. 2013. View Article : Google Scholar
|
|
31
|
Ooi LL, Zheng Y, Zhou H, et al: Vitamin D
deficiency promotes growth of MCF-7 human breast cancer in a rodent
model of osteosclerotic bone metastasis. Bone. 47:795–803. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ghosh S and Hayden MS: New regulators of
NF-kappaB in inflammation. Nat Rev Immunol. 8:837–848. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ghosh S and Baltimore D: Activation in
vitro of NF-kappa B by phosphorylation of its inhibitor I kappa B.
Nature. 344:678–682. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Duffy MJ, Maguire TM, Hill A, McDermott E
and O’Higgins N: Metalloproteinases: role in breast carcinogenesis,
invasion and metastasis. Breast Cancer Res. 2:252–257. 2000.
View Article : Google Scholar
|
|
35
|
Park JM, Kim A, Oh JH and Chung AS:
Methylseleninic acid inhibits PMA-stimulated pro-MMP-2 activation
mediated by MT1-MMP expression and further tumor invasion through
suppression of NF-kappaB activation. Carcinogenesis. 28:837–847.
2007. View Article : Google Scholar
|
|
36
|
Takada Y, Ichikawa H, Badmaev V and
Aggarwal BB: Acetyl-11-keto-beta-boswellic acid potentiates
apoptosis, inhibits invasion, and abolishes osteoclastogenesis by
suppressing NF-kappa B and NF-kappa B-regulated gene expression. J
Immunol. 176:3127–3140. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang Q, Shen HM and Ong CN: Inhibitory
effect of emodin on tumor invasion through suppression of activator
protein-1 and nuclear factor-kappaB. Biochem Pharmacol. 68:361–371.
2004. View Article : Google Scholar : PubMed/NCBI
|