Introduction
Parkinson’s disease (PD) is the second most common
progressive neurodegenerative disorder following Alzheimer’s
disease, caused by the relatively selective loss of dopaminergic
neurons in the substantia nigra. The pathogenic hallmark of PD is
the accumulation and aggregation of α-synuclein (α-syn) in
susceptible neurons. α-syn is reported to have a central role in
PD. It is known that A53T and A30P, two missense mutations in
α-syn, cause early-onset autosomal dominant PD (1,2) and
transgenic mice with A53T mutant α-syn develop a PD phenotype with
Lewy body-like pathology and locomotor impairment (3,4).
α-syn is degraded by the ubiquitin-proteasome system
(UPS) and autophagy-lysosome pathways (ALP) (5). Impairment to the UPS and primary
non-lysosomal protein degradation pathways have been implicated in
PD (6). Failure of the UPS to
clear unwanted α-syn, eventually leading to the accumulation and
aggregation of α-syn, clearly has a major role in the molecular
pathogenesis of sporadic and familial PD (6–8).
Several loss-of-function studies on the UPS have provided
compelling evidence that UPS impairment is sufficient to cause
neural proteinopathy (9–11). Another pathway relevant to α-syn
clearance is autophagy, a lysosome-mediated degradative pathway,
which mediates the bulk degradation of cytoplasmic proteins or
organelles in the lytic compartment. Autophagy involves the
formation of double-membrane structures, termed autophagosomes,
which fuse with primary lysosomes to become an autophagolysosome.
As a result, the contents of the autophagolysosomes are degraded by
either disposing or recycling back to cells. Autophagy is regulated
by a group of ATG genes. It has been reported that mice that
specifically lacked Atg7 in the central nervous system exhibited
behavioral defects, massive neuronal loss in the cerebral and
cerebellar cortices and accumulation of polyubiquitinated proteins
in autophagy-deficient neurons as inclusion bodies (12). Therefore, the impairment to
autophagy is implicated in the pathogenesis of neurodegenerative
disorders that involve ubiquitin-containing inclusion bodies
(13,14). Associated with PD, the A53T
mutation of α-syn that readily forms aggregates may be more
dependent on autophagy compared with the wild-type protein or A30P
mutation (15).
The UPS and ALP have been viewed as independent
degradation systems. However, several studies have suggested that
they are mechanistically linked (12,16).
For example, accumulation of ubiquitin-positive aggregates was
observed in Atg7-deficient hepatocytes and neurons and autophagy
was induced in response to proteasome inhibition in certain cancer
cells in Drosophila melanogaster (17–20).
In addition, a study in living mouse cortex neurons suggested that
the UPS and ALP may be functionally connected such that impairment
to either one could upregulate the other (21). However, these mechanisms remain to
be clarified and confirmed in the pathogenesis of PD. A PC12 cell
line has been created that stably overexpresses A53T mutant α-syn,
which is considered an ideal alternative to dopaminergic neurons
for PD research. The association between the UPS and ALP in PC12
cells overexpressing A53T mutant α-syn remains to be elucidated. In
the present study, this cell line was treated with the proteasome
inhibitor (PI) MG132 to see whether it could induce autophagy. This
was in order to determine the relevant effects on the degradation
of α-syn and survival of PC12 cells and an attempt to gain insights
into the mechanism and effect of PI-induced autophagy in the
degradation of α-syn associated with the pathogenesis of PD.
Materials and methods
Drugs
MG-132, trehalose and 3-methyladenine (3-MA), which
were all purchased from Sigma (St. Louis, MO, USA), were dissolved
in 100% dimethyl sulfoxide (Sigma) and diluted with Dulbecco’s
modified Eagle’s medium (DMEM; Gibco-BRL, Carlsbad, CA, USA) to the
desired concentration, with a final dimethyl sulfoxide
concentration of 0.1% for in vitro study. Trehalose was
diluted to 1 mol/l with DMEM. 3-MA was dissolved in
dimethylformamide (DMF; Sigma) and diluted with DMEM to the desired
concentration, with a final DMF concentration of 0.2% for in
vitro study. This study was approved by the Ethics Committee of
Changzheng Hospital (Shanghai, China).
Cell culture
A rat PC12 cell line overexpressing human A53T
mutant α-syn was constructed using a pEGFP-SNCA-A53T recombinant
plasmid (kindly provided by Dr Stephanie Cobb, Mayo Clinic, FL,
USA) and the lentiviral gene transfer method. Transfected PC12
cells were further screened with 5 μmol/l blasticidin (Invitrogen
Life Technologies, Carlsbad, CA, USA) and obtained using a limiting
dilution assay. Stably transfected PC12 cells were cultured in DMEM
supplemented with 10% (v/v) heat-inactivated horse serum
(Gibco-BRL), 5% (v/v) fetal bovine serum (Gibco-BRL) and
blasticidin (5 μmol/l). Cells were cultured at 37°C in humidified
air with 5% CO2. All experiments were performed 24–48 h
after cell seeding.
Experimental cell treatment
To investigate the effect of an autophagy enhancer
or inhibitor on MG132-induced autophagy, the macroautophagy
inhibitor 3-MA was applied at a concentration of 2 mmol/l 3 h prior
to treatment with MG132 and mammalian target of rapamycin
(mTOR)-independent autophagy enhancer trehalose was applied
simultaneously with MG132. The effect of MG132 (500 nmol/l) on PC12
cells overexpressing A53T α-syn was evaluated after 24 h
incubation. PC12 cells that overexpressed A53T α-syn with solely
3-MA or trehalose for 24 h were used as the control.
Western blot analysis for
microtubule-associated protein 1A/1B light chain (LC3) and
α-syn
Total cell lysates of the treated PC12 cells were
prepared in ice-cold extraction buffer consisting of 20 mM Tris-HCl
(pH 7.4), 10 mmol/l potassium acetate (AcK), 1 mmol/l
dithiothreitol, 0.25% NP-40, 1 mmol/l EDTA, 2 mmol/l ethylene
glycol tetraacetic acid, 1 mmol/l phenylmethylsulfonyl fluoride and
a protease inhibitor cocktail (Sigma), containing 104 mM
4-(2-Aminoethyl)benzenesulfonyl fluoride hydrochloride, 80 μM
aprotinin, 4 mM bestatin, 1.4 mM E-64, 2 mM leupeptin and 1.5 mM
pepstatin A. The samples were homogenized and centrifuged at 20,000
× g for 10 min at 4°C and then the protein content was determined
by the BCA protein assay kit (Pierce Biotechnology, Inc., Rockford,
IL, USA). The total quantity of protein (50 μg) was electrophoresed
on a 12% SDS-PAGE, transferred to polyvinylidene difluoride,
blocked and probed overnight at 4°C with the following primary
antibodies: Anti-LC3 antibody (rabbit anti-rat; 1:1,000; Abcam,
Cambridge, MA, USA), anti-α-syn antibody (mouse anti-rat; 1:1,000;
Sigma) and anti-β-actin antibody (mouse anti-rat; 1:5,000; Sigma).
The samples were then incubated with the appropriate secondary
antibodies and developed with enhanced chemiluminescence. The blots
were quantitated by computer-assisted image analysis
software(Quantity One 4.62; Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Immunofluorescence of autophagosomes and
autolysosomes
Cells were seeded on polylysine-coated glass slides
(Sigma) in 6-well plates, treated with the experimental drugs,
incubated as necessary, stained with 70 ng/ml
LysoTracker® Red DND-99 (Invitrogen Life Technologies)
for 30 min, washed with phosphate-buffered saline (PBS) at pH 7.2,
fixed in 4% paraformaldehyde for 30 min, washed with PBS and
permeabilized with 0.1% Triton-X-100 (Sigma) for 15 min, followed
by overnight incubation with the LC3 primary antibody (1:1,000;
MBL, Nagoya, Japan) and incubation with a secondary antibody
(1:100; Kangcheng Biotech, Shanghai, China). A laser-scanning
microscope (Olympus BX60; Olympus, Tokyo, Japan) was used to
capture images.
Transmission electron microscopy
(TEM)
The presence of autophagic vacuoles in TEM is the
gold standard for detecting autophagy. The treated PC12 cells were
harvested using 0.25% trypsin, washed with PBS (pH 7.2), collected
by centrifugation for 10 min at 440 × g, fixed in ice-cold 2.5%
glutaraldehyde in PBS, rinsed with PBS, post-fixed in 1% osmium
tetroxide with 0.1% potassium ferricyanide, dehydrated through a
graded series of ethanol (30–90%), embedded in Epon (Energy Beam
Sciences, Agawam, MA, USA) and sliced into 50–60 nm sections with
an LKB-I ultramicrotome (LKB, Bromma, Sweden). The sections were
stained with 3% uranyl acetate and lead citrate and finally
observed by TEM (Philips CM-120; Philips, Amsterdam, Holland).
Cell viability assay
Cell viability was measured using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Cells were plated at a density of 2×104 cells per
well in 96-well plates. Following treatment with 500 nmol/l MG132
for 24 h, MTT solution (5 mg/ml) was added to each well and the
plates were incubated for another 4 h. Subsequently, 150 μl
dimethyl sulfoxide was added to each well to resuspend and dissolve
the MTT metabolic product. Finally, the optical density was read at
570 nm, with the subtraction of background at 670 nm using a
Multiskan MK3 microplate reader (Thermo Electron Corporation,
Marietta, OH, USA).
Statistical analysis
All experiments were repeated at least three times
and the data are expressed as the mean ± standard error of the
mean. Differences between groups were analyzed by one-way analysis
of variance or Student’s t-test using SPSS 12.0 software (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
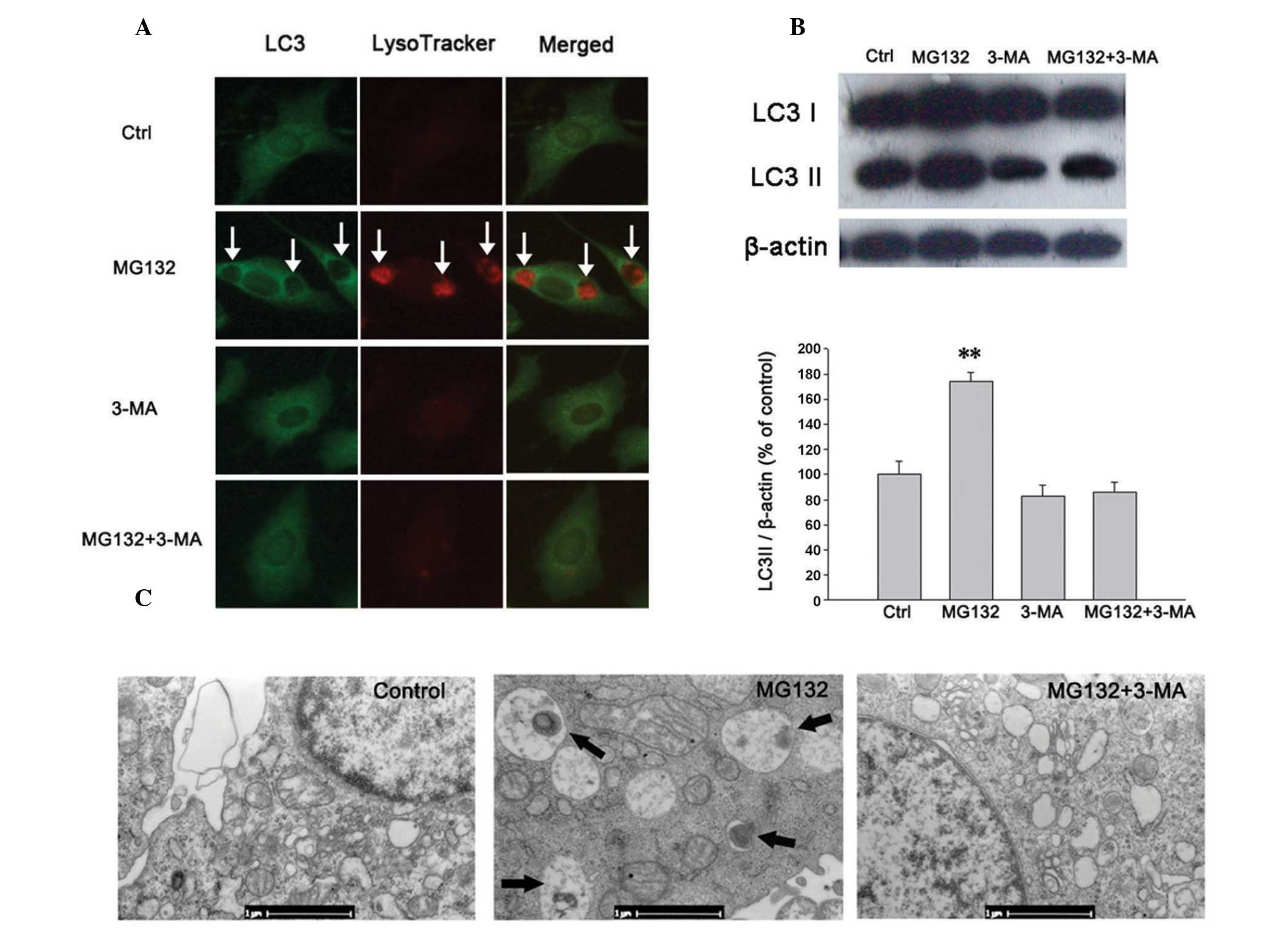
Mg132 induces macroautophagy in PC12
cells overexpressing A53T mutant α-syn
The induction of autophagy was assessed by detecting
an increase in the autophagosomal membrane form of LC3, which is a
mammalian homolog of the autophagy-related gene 8 in yeast. It is
recruited to the autophagosomal membrane during autophagy, making
it a specific marker of autophagy. Since the quantity of LC3
protein, particularly LC3-II, has been previously demonstrated to
correlate with the extent of autophagy (22–24),
the ratio of LC3II/β-actin was determined in the present study to
estimate autophagy. LysoTracker® and LysoSensor™ probes
are also commonly used to investigate the acidification of
lysosomes and the alterations of lysosomal function or trafficking
that occurs in live cells (5,25).
To investigate the effect of PIs on autophagy, alterations in
LC3+ autophagosomes and lysotracker-positive
autolysosomes were detected by immunofluorescence. The expression
of LC3-II was assayed by western blot analysis and changes in the
morphology of PC12 cells were examined by TEM. It was found that in
the PC12 cells overexpressing A53T α-syn, MG132 (500 nmol/l)
significantly increased the LC3+ autophagosome and
lysotracker-positive autolysosome levels (Figs. 1A and 2A), upregulated the expression of LC3-II
(Figs. 1B and 2B) and promoted the formation of
autophagosomes as shown by TEM (Fig.
1C).
PI-induced autophagy can be inhibited by
3-MA
The class III phosphoinositide 3-kinase (PI3K)
inhibitor 3-MA is known to convert LC3-I to LC3-II and is
implicated in the modulation of autophagy. To investigate whether
the autophagy induced by MG132 in these experiments was mediated by
the PI3K pathway, PC12 cells were pretreated with 2 mmol/l 3-MA for
3 h. It was revealed that 3-MA reduced the formation of autophagic
vacuoles and the expression of LC3-II protein induced by 500 nmol/l
MG132, as shown by immunofluorescence (Fig. 1A), western blot analysis (Fig. 1B) and TEM (Fig. 1C). In summary, it was demonstrated
that the presence of MG132-activated macroautophagy in the PI3K
pathway can be inhibited by 3-MA.
Effects of mTOR-independent autophagy
enhancer trehalose on PI-induced autophagy
Trehalose is an mTOR-independent autophagy enhancer
and can induce autophagy and promote the clearance of A53T α-syn
(26). As a positive control,
trehalose increased LC3− and Lysotracker RED-positive
autolysosome levels following using lysotracker and LC3 staining
(Fig. 2A). It also enhanced the
expression of LC3-II as shown by western blot analysis (Fig. 2B). In addition, trehalose (50
mmol/l) markedly increased the number of LC3+
autophagosomes and lysotracker-positive autolysosomes (Fig. 2A) and the expression of LC3-II
induced by MG132 (Fig. 2B).
Effects of autophagy enhancers or
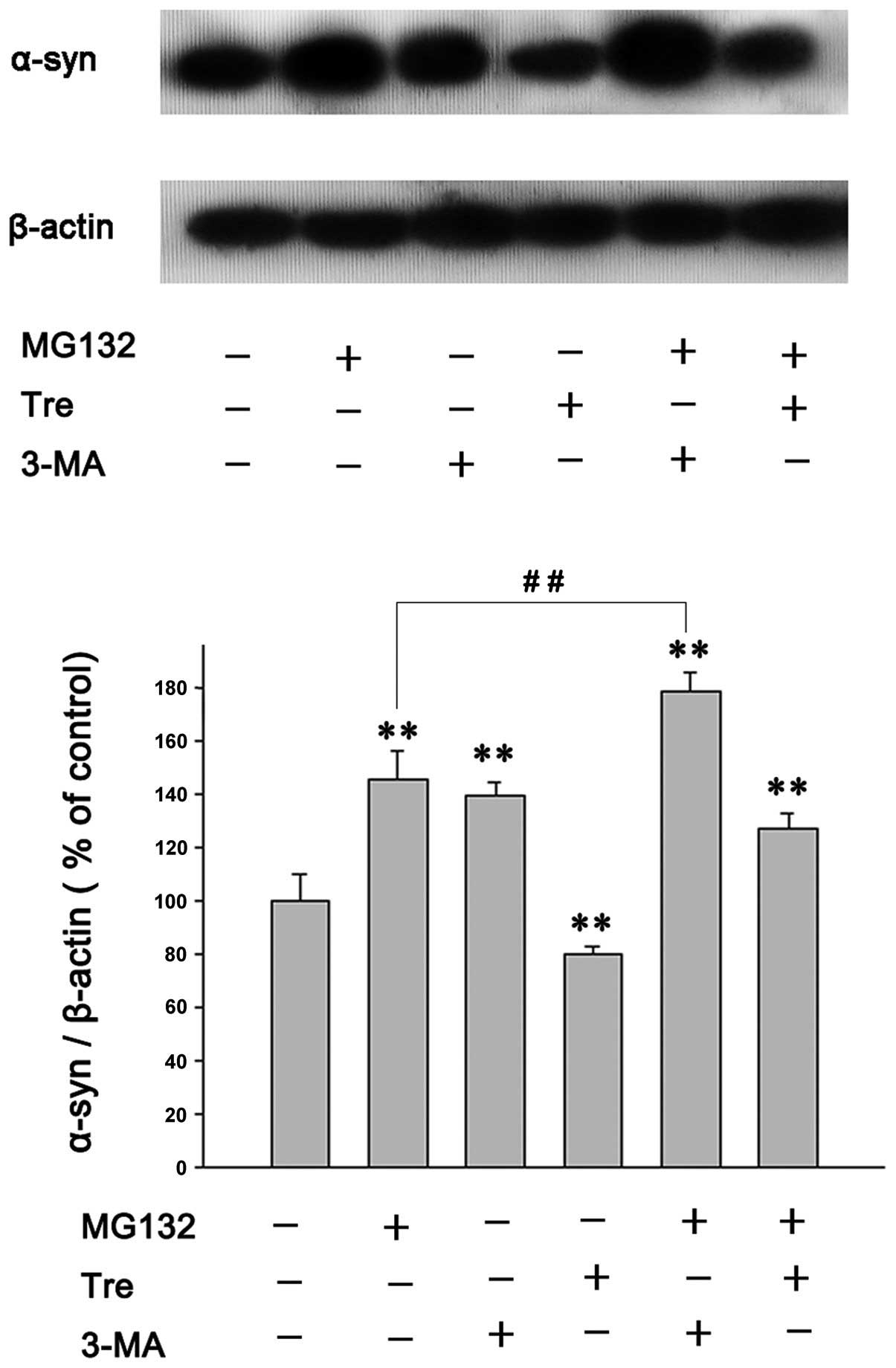
inhibitors on MG132-induced α-syn accumulation
α-syn can be degraded by the UPS and ALP. To
identify the effects of autophagy inducers or inhibitors on A53T
α-syn clearance in PC12 cells treated with the PI, A53T
α-syn-overexpressing PC12 cells were treated with MG132, with or
without trehalose or 3-MA, for 24 h. The results were as follows:
MG132 (500 nmol/l) increased the level of A53T α-syn by inhibiting
its degradation (Fig. 3) and
trehalose (50 mmol/l) attenuated the increase of A53T α-syn induced
by MG132 (Fig. 3). Additionally,
pretreatment with 3-MA (2 mmol/l) clearly increased the
accumulation of A53T α-syn induced by MG132 (Fig. 3), a phenomenon that was similarly
observed in previous experiments (27). However, although the inhibition of
proteasomes or autophagy, or both, significantly increased the
accumulation of A53T α-syn in the present study, immunofluorescence
did not detect the inclusion bodies containing aggregated α-syn in
PC12 cells (data not shown).
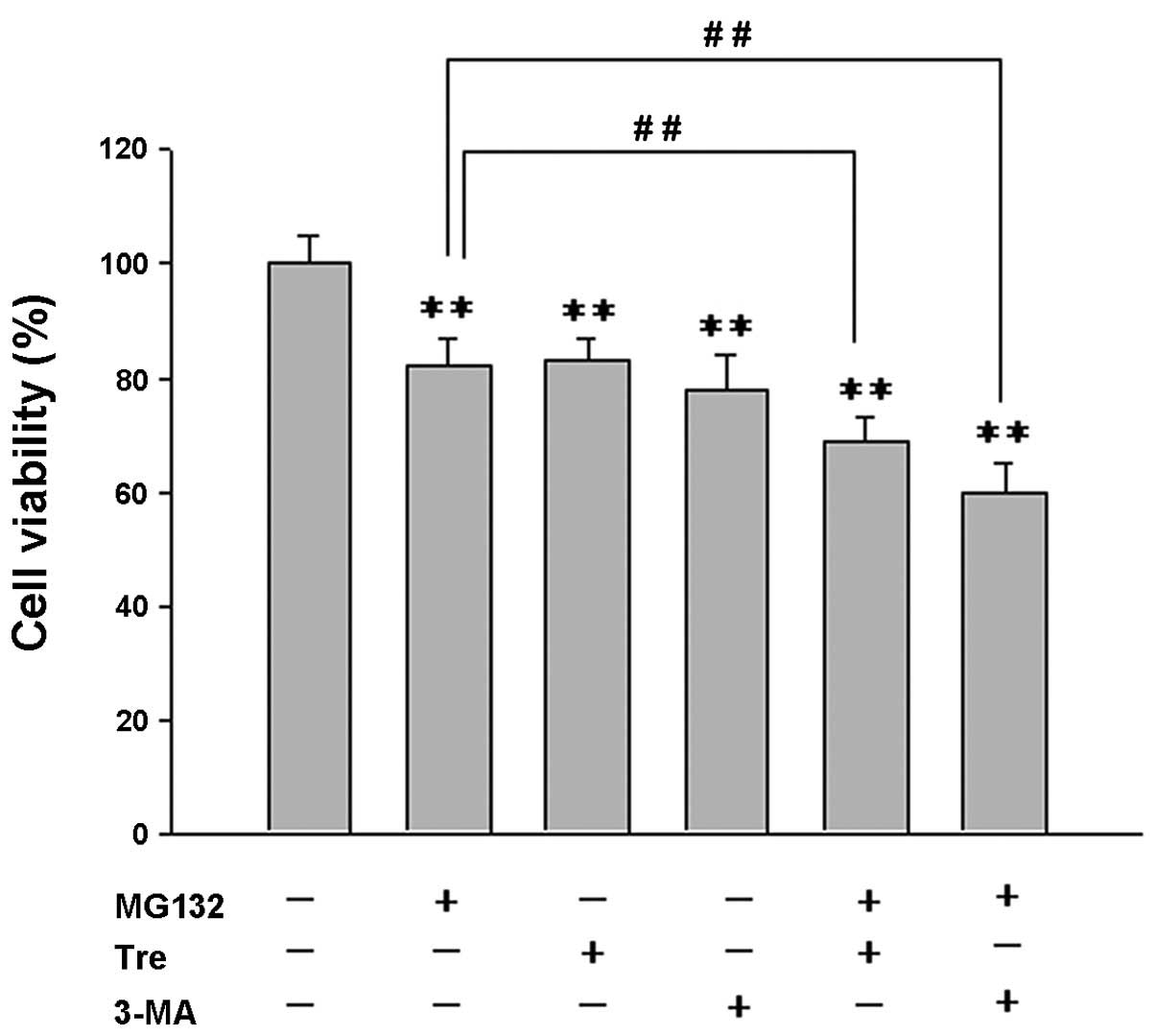
Effects of autophagy enhancers or
inhibitors on PI-induced cell death
To examine the role of PI-induced autophagy in cell
survival in A53T α-syn-overexpressing PC12 cells, an MTT assay was
used to assess the effect of a combined autophagy
enhancer/inhibitor with MG132 on cell viability (Fig. 4). It was found that the combination
of MG132 (500 nmol/l) with trehalose (50 mmol/l) or 3-MA (2 mmol/l)
markedly decreased the cell viability compared with treatment using
either single agent (Fig. 4).
Discussion
In the present study, the effects of the PI MG132 on
PC12 cells overexpressing A53T mutant α-syn, an ideal alternative
to dopaminergic neurons, were examined and it was found that MG132
significantly increased the number of autophagosomes and
autolysosomes. MG132 also upregulated the expression of LC3-II and
promoted the formation of autophagosomes as shown by TEM, thus
confirming the presence of PI-induced autophagy in PC12 cells
overexpressing A53T mutant α-syn. When the UPS was impaired, it was
identified that autophagy operated via a compensatory degradation
system. In addition, PI-induced autophagy could be completely
inhibited by the selective class III PI3K inhibitor 3-MA,
indicating that PI-induced autophagy is mediated by the
upregulation of the macroautophagy class III PI3K pathway.
Trehalose, an autophagy enhancer, not only induced autophagy in
PC12 cells overexpressing A53T mutant α-syn (27), but also significantly improved
autophagy induced by MG132. These results indicate that a
combination of PI and other autophagy enhancers could produce a
synergistic effect in inducing autophagy.
It was also revealed that MG132 increased the
expression of A53T α-syn and trehalose counteracted the increase of
A53T α-syn induced by MG132. These results are consistent with the
previous finding that trehalose accelerated the clearance of mutant
α-syn by enhancing autophagy (26). Combined inhibition of autophagy and
the proteasome significantly increased the accumulation of A53T
α-syn compared with treatment using either single agent. These
results suggest that A53T α-syn could be degraded by the UPS and
ALP and that PI-induced autophagy may act as a compensatory
degradation system for A53T α-syn degradation when the UPS is
impaired.
Although treatment with MG132 and/or 3-MA inhibited
A53T α-syn degradation and significantly increased its
accumulation, immunofluorescence failed to detect inclusion bodies
containing aggregated α-syn in PC12 cells in the present study,
which may suggest that treatment with PIs is not a reliable model
for replicating all the neuropathological abnormalities of PD.
Ample evidence supports the hypothesis that
autophagy is a survival pathway required for cellular viability
(28). In the present study, it
was also observed that there is increased cell death in PC12 cells
following treatment with 3-MA alone. In addition, the effect of
PI-induced autophagy on the survival rate of PC12 cells
overexpressing A53T α-syn was examined and it was revealed that
pre-treatment with 3-MA potentiated PI-induced cell death in
vitro, as determined by measuring cell viability with an MTT
assay. These findings indicate that autophagy activation may
directly contribute to the survival of PC12 cells treated with
proteasome inhibitors.
Several lines of evidence have suggested that
autophagy enhancers can promote the clearance of aggregate-prone
proteins and decrease PI-induced cell death (29,30).
However, in the present study combined treatment with MG132 and
trehalose resulted in a marked decrease in cell viability in A53T
α-syn-overexpressing PC12 cells compared with treatment by MG132
alone. It was also supported by a previous study, which may be
explained by the change of osmolality and overactivated autophagy
induced by trehalose (27).
In conclusion, in the present study it has been
demonstrated that PI can induce macroautophagy in PC12 cells
overexpressing A53T α-syn. The UPS and ALP are two reciprocally
regulated, not strictly parallel degradation systems that are
important in the cellular mechanisms of A53T α-syn degradation.
PI-induced autophagy can be completely inhibited by the autophagy
inhibitor 3-MA, suggesting that such action depends on class III
PI3K. The combination of PI and trehalose synergistically promotes
autophagy. Trehalose can offset IP-induced accumulation of A53T
α-syn. The inhibition of autophagy can significantly increase the
accumulation of A53T α-syn and proteasome inhibitor-induced cell
death, indicating that PI-induced autophagy may have a
cytoprotective role in the degradation of A53T α-syn and in the
survival of PC12 cells overexpressing A53T α-syn. These results may
prove useful in illuminating the mechanism of PI-induced autophagy
in the pathogenesis of PD.
Acknowledgements
This study was supported by grants from the Natural
Science Foundation of China (nos. 30600663 and 81070070) and the
Ministry of Science and Technology Plan Fund Major Projects (no.
2011ZXJ09202-015).
References
|
1
|
Polymeropoulos MH, Lavedan C, Leroy E, et
al: Mutation in the alpha-synuclein gene identified in families
with Parkinson’s disease. Science. 276:2045–2047. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kruger R, Kuhn W, Muller T, et al:
Ala30Pro mutation in the gene encoding alpha-synuclein in
Parkinson’s disease. Nat Genet. 18:106–108. 1998. View Article : Google Scholar
|
|
3
|
Masliah E, Rockenstein E, Veinbergs I, et
al: Dopaminergic loss and inclusion body formation in
alpha-synuclein mice: implications for neurodegenerative disorders.
Science. 287:1265–1269. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Feany MB and Bender WW: A Drosophila model
of Parkinson’s disease. Nature. 404:394–398. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Webb JL, Ravikumar B, Atkins J, Skepper JN
and Rubinsztein DC: Alpha-Synuclein is degraded by both autophagy
and the proteasome. J Biol Chem. 278:25009–25013. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McNaught KS and Jenner P: Proteasomal
function is impaired in substantia nigra in Parkinson’s disease.
Neurosci Lett. 297:191–194. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moore DJ, Dawson VL and Dawson TM: Role
for the ubiquitin-proteasome system in Parkinson’s disease and
other neurodegenerative brain amyloidoses. Neuromolecular Med.
4:95–108. 2003. View Article : Google Scholar
|
|
8
|
McNaught KS, Belizaire R, Isacson O,
Jenner P and Olanow CW: Altered proteasomal function in sporadic
Parkinson’s disease. Exp Neurol. 179:38–46. 2003. View Article : Google Scholar
|
|
9
|
McNaught KS, Bjorklund LM, Belizaire R,
Isacson O, Jenner P and Olanow CW: Proteasome inhibition causes
nigral degeneration with inclusion bodies in rats. Neuroreport.
13:1437–1441. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McNaught KS, Perl DP, Brownell AL and
Olanow CW: Systemic exposure to proteasome inhibitors causes a
progressive model of Parkinson’s disease. Ann Neurol. 56:149–162.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McNaught KS and Olanow CW: Proteasome
inhibitor-induced model of Parkinson’s disease. Ann Neurol.
60:243–247. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Komatsu M, Waguri S, Chiba T, et al: Loss
of autophagy in the central nervous system causes neurodegeneration
in mice. Nature. 441:880–884. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alvarez-Erviti L, Rodriguez-Oroz MC,
Cooper JM, et al: Chaperone-mediated autophagy markers in Parkinson
disease brains. Arch Neurol. 67:1464–1472. 2010.PubMed/NCBI
|
|
14
|
Yang Q and Mao Z: Parkinson disease: a
role for autophagy? Neuroscientist. 16:335–341. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Conway KA, Harper JD and Lansbury PT:
Accelerated in vitro fibril formation by a mutant alpha-synuclein
linked to early-onset Parkinson disease. Nat Med. 4:1318–1320.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Komatsu M, Waguri S, Ueno T, et al:
Impairment of starvation-induced and constitutive autophagy in
Atg7-deficient mice. J Cell Biol. 169:425–434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ding WX, Ni HM, Gao W, et al: Linking of
autophagy to ubiquitin-proteasome system is important for the
regulation of endoplasmic reticulum stress and cell viability. Am J
Pathol. 171:513–524. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fels DR, Ye J, Segan AT, et al:
Preferential cytotoxicity of bortezomib toward hypoxic tumor cells
via overactivation of endoplasmic reticulum stress pathways. Cancer
Res. 68:9323–9330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pandey UB, Nie Z, Batlevi Y, et al: HDAC6
rescues neurodegeneration and provides an essential link between
autophagy and the UPS. Nature. 447:859–863. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu K, Dunner K Jr and McConkey DJ:
Proteasome inhibitors activate autophagy as a cytoprotective
response in human prostate cancer cells. Oncogene. 29:451–462.
2010. View Article : Google Scholar :
|
|
21
|
Ebrahimi-Fakhari D, Cantuti-Castelvetri I,
Fan Z, et al: Distinct roles in vivo for the ubiquitin-proteasome
system and the autophagy-lysosomal pathway in the degradation of
alpha-synuclein. J Neurosci. 31:14508–14520. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kadowaki M and Karim MR: Cytosolic LC3
ratio as a quantitative index of macroautophagy. Methods Enzymol.
452:199–213. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tanida I, Ueno T and Kominami E: LC3 and
autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dingle JT and Barrett AJ: Uptake of
biologically active substances by lysosomes. Proc R Soc Lond B Biol
Sci. 173:85–93. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sarkar S, Davies JE, Huang Z, Tunnacliffe
A and Rubinsztein DC: Trehalose, a novel mTOR-independent autophagy
enhancer, accelerates the clearance of mutant huntingtin and
alpha-synuclein. J Biol Chem. 282:5641–5652. 2007. View Article : Google Scholar
|
|
27
|
Lan DM, Liu FT, Zhao J, et al: Effect of
trehalose on PC12 cells overexpressing wild-type or A53T mutant
alpha-synuclein. Neurochem Res. 37:2025–2032
|
|
28
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Casarejos MJ, Solano RM, Gomez A, Perucho
J, de Yebenes JG and Mena MA: The accumulation of neurotoxic
proteins, induced by proteasome inhibition, is reverted by
trehalose, an enhancer of autophagy, in human neuroblastoma cells.
Neurochem Int. 58:512–520. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pan T, Kondo S, Zhu W, Xie W, Jankovic J
and Le W: Neuroprotection of rapamycin in lactacystin-induced
neurodegeneration via autophagy enhancement. Neurobiol Dis.
32:16–25. 2008. View Article : Google Scholar : PubMed/NCBI
|