Introduction
Non-small cell lung cancer (NSCLC) is one of the
most prevalent malignancies associated with morbidity and mortality
worldwide (1). The main
therapeutic method used to treat NSCLC is surgical resection.
However, when initially diagnosed with NSCLC, >50% of patients
will already be in the advanced stage, and some may have missed the
opportunity for surgery. Furthermore, the patients eligible for
surgery will often also require adjuvant chemotherapy. Therefore,
chemotherapy has become the widest used clinical approach in the
treatment of NSCLC. Unfortunately, traditional chemotherapy has
numerous clinical limitations, due to its poor specificity and
severe side effects. In order to overcome the shortcomings of
traditional chemotherapy, individualized treatment has recently
been extensively used. In recent years, molecular targeted drugs
have been popularized in individualized cancer treatment, due to
their improved specificity and reduced side effects. Epidermal
growth factor receptor tyrosine-kinase inhibitors (EGFR-TKIs), such
as gefitinib and erlotinib, are the most common molecular targeted
drugs used to treat NSCLC. Previous studies have shown that NSCLC
cells which harbor activating EGFR mutations, such as exon 19
deletion and the exon 21 missense mutation (L858R), will be
sensitive to EGFR-TKIs (2–4). Unfortunately, the majority of
patients with NSCLC who are initially sensitive to EGFR-TKIs, will
ultimately develop acquired resistance to the drug. Therefore,
exploring the mechanisms behind the acquired drug resistance of
NSCLC to EGFR-TKI has become an urgent clinical problem.
Some mechanisms regarding the acquired resistance of
NSCLC to EGFR-TKIs, have been reported as follows: Secondary
mutations, such as EGFR T790M exon 20; encoding gene mutations of
Kirsten rat sarcoma viral oncogene homolog (KRAS), v-Raf murine
sarcoma viral oncogene homolog B (BRAF), mitogen-activated protein
kinase and phosphoinositide-3 kinase (PI3K) in the EGFR, downstream
of signal transduction; gene amplification of other signal
transduction pathways, such as MET; and epithelial-mesenchymal
transition (EMT) (5–13). The T790M mutation occurs in ~50% of
NSCLC patients who have developed acquired drug resistance to
EGFR-TKIs. Therefore, the EGFR T790M mutation has been generally
considered as the molecular genetic basis of TKI-acquired drug
resistance. However, it remains unclear how the cells, having
harbored the T790M mutation, develop acquired drug resistance. The
establishment of an acquired gefitinib resistant subline from an
NSCLC cell line, harboring sensitive (exon 21; L858R) and resistant
(exon 20; T790M) mutations of EGFR may be helpful for exploring the
problem of acquired drug resistance to EGFR-TKIs. The NCI-H1975
human NSCLC cell line was established in 1988, prior to the
clinical use of TKIs, and harbors the L858R and T790M double
mutations (6). This cell line
should initially be sensitive and easily develop acquired
resistance to TKIs, following TKI stimulation. Therefore, the
NCI-H1975 cell line is an ideal cell line to use for the study of
TKI-acquired resistance based on the T790M mutation.
It was previously reported that tumor cells with
exon 21 mutations (L858R) or exon 19 deletions in EGFR, showed
higher tyrosine kinase activity (2). EGFR-TKIs can suppress the higher
tyrosine kinase activity due to these mutations, and block signal
transduction from EGFR (14–16).
The T790M mutation substitutes methionine for threonine in the
‘gatekeeper’ region of EGFR, and the bulkier methionine prevents
the EGFR-TKIs from binding the ATP pocket of EGFR tyrosine kinase
(5). Some studies have suggested
that the T790M mutation most likely causes acquired drug resistance
by enhancing the ATP affinity of EGFR L858R, and thus reducing the
efficacy of the ATP-competitive TKIs (17). These studies indicate that the
T790M mutation may cause the cells harboring the EGFR activating
mutations, such as L858R, to maintain the tyrosine kinase activity
of EGFR, and subsequently lose sensitivity to EGFR-TKIs. However,
it remains unclear how the T790M mutation induces tumor cells that
were initially sensitive to EGFR-TKIs, to escape from the
inhibition of the drug. The present study developed an acquired
gefitinib-resistant cell line (NCI-H1975/GR) from the NCI-H1975
cell line. Furthermore, by detecting the protein expression levels
of the EGFR/KRAS/BRAF transduction pathway, and observing the EMT
in the process of acquired gefitinib resistance development, the
possible mechanisms by which the T790M mutation induces NSCLC to
develop acquired resistance to TKIs were investigated.
Materials and methods
Drugs and cell line
Gefitinib powder was purchased from Selleckchem
(Radnor, PA, USA). The NCI-H1975 human NSCLC cell line was
purchased from the Cell Culture Center of the Institute of Basic
Medical Sciences, Chinese Academy of Medical School (Beijing,
China). The cells were cultured in RPMI-1640 medium supplemented
with 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml
streptomycin (Gibco-BRL, Grand Island, NY, USA), and maintained in
a 5% CO2 humidified incubator at 37°C.
Establishment of an acquired
gefitinib-resistant cell line NCI-H1975/GR
To develop the acquired gefitinib-resistant cell
line, the NCI-H1975 cells were initially exposed to 12 μmol/l
gefitinib for 24 h. The surviving cells were washed with RPMI-1640,
and cultured in the drug-free medium until they had reached 80%
confluence. The cells were further exposed to 12 μmol/l gefitinib
for 24 h. This process was repeated with increasing drug
concentrations, until 80 μmol/l. After the cells had been cultured
in drug-free medium for two weeks, the subsequent investigations
were conducted.
Growth inhibition assay
An MTT assay and the trypan blue dye exclusion
method were used to measure cell sensitivity to gefitinib. The MTT
assay was performed according to the following protocol. The parent
and resistant cells, growing exponentially, were harvested and
seeded into 96-well plates at a density of 5×103
cells/well overnight. The cells were treated with gefitinib at the
indicated doses for 72 h at 37°C, after which 20 μl MTT solution
[Sigma-Aldrich, St Louis, MO, USA; 5 mg/ml in phosphate-buffered
saline (PBS)] was added to each well, and the plates were incubated
for 4 h at 37°C. The plates were then centrifuged at 2,250 × g for
10 min, the medium was aspirated from each well and 150 μl dimethyl
sulfoxide was added to each well, in order to dissolve the formazan
crystals. The optical density was measured at a wavelength of 492
nm using an automatic microplate reader (Thermo Labsystems,
Helsinki, Finland). Absorbance values were expressed as a
percentage of that for untreated cells, and the half maximal
inhibitory concentration of gefitinib (IC50) was
calculated. In addition, the number of viable cells from the parent
and resistant cell lines were determined by the trypan blue
(Spectrum Chemicals & Laboratory Products, Shanghai, China) dye
exclusion method, and the number of viable cells were counted using
a hemocytometer (Shanghai Qiujing Biochemical Reagent and
Instrument Co., Ltd., Shanghai, China) every 24 h for 4 days. Each
assay was performed in triplicate.
Apoptosis assay
The percentage of apoptotic parent and resistant
cells, with or without gefitinib stimulation, were determined using
Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide
(PI) staining (Biouniquer, China). The cells were treated with or
without gefitinib (20 μmol/l) for 24 h, harvested by trypsin
(Sigma-Aldrich) digestion, washed twice with PBS, and then
suspended in 500 μl Annexin V Binding buffer. Thereafter, 5 μl
Annexin V-FITC and 5 μl PI were added to the samples, which were
incubated for 10 min at room temperature in the dark, according to
the manufacturer’s instructions. The apoptotic cells were detected
using a FACSCalibur™ flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA).
Morphological analysis
Changes to the morphology of the parent and
gefitinib-resistant cells, in response to gefitinib treatment,
including size, shape and development of pseudopodia, were directly
observed under an inverted microscope (Olympus Corporation, Tokyo,
Japan).
Cell proliferation assay
The cells, growing exponentially, were harvested and
seeded into 24-well plates at a density of 1.5×104
cells/well. The parent and resistant cells were counted with a
hematocytometer every 24 h for 7 days. The proliferation curves
were charted, and the cell population doubling times were
calculated using the following equation: T = tlg2/(lgNt −
lgN0) (T, population doubling time; t, continuous culture
time; Nt, terminal number of cells; N0,
initial number of cells. Time units in h; lg,
log10).
Cell cycle analysis
The cell cycle distributions of the parent and
resistant cells were analyzed by flow cytometry. The parent and
resistant cells were treated with or without gefitinib (20 μmol/l)
for 24 h. The cells were then harvested by trypsin digestion,
washed twice with ice-cold PBS, fixed in 70% ethanol and then
maintained at 4°C overnight. Following the removal of ethanol by
centrifugation, the cells were washed twice with PBS and stained
with PI/RNase solution for 30 min in a 37°C water bath, according
to the manufacturer’s instructions(Beyotime Institute of
Biotechnology, Haimen, China). Cell cycle distribution was detected
using a FACSCalibur™ flow cytometer, and the data were analyzed
using Cellquest™ (BD Biosciences).
Polymerase chain reaction-high resolution
melting analysis (PCR-HRMA)
Mutation analysis of exons 18–21 of EGFR, exon 2 of
KRAS and exon 15 of BRAF gene was performed using PCR-HRMA
(LightScanner® HRI 96; Biofire Diagnostics, Inc., Salt
Lake City, UT, USA). Genomic DNA was extracted from the parent and
resistant cells using a Genomic DNA Extraction kit (TIANGEN Biotech
Co., Ltd., Beijing, China), and exons 18–21 of EGFR, exon 2 of KRAS
and exon 15 of BRAF were amplified by PCR. The PCR reaction mixture
contained 10X PCR buffer (Takara Biotechnology Co., Ltd., Dalian,
China ), 2.5 mmol/l dNTPs, 25 mmol/l MgCl2, 100 μM
primer, 5 U/μl HotStart Taq (Takara Biotechnology Co., Ltd.,
Dalian, China), 5 ng genomic DNA and 10X LC Green Plus (Biochem,
Salt Lake City, UT, USA). The primer sequences are shown in
Table I. PCR amplification
conditions were set as follows: 1) EGFR exons 18/19: 95°C for 5
min; 45 cycles of 95°C for 15s, 60°C for 1 min; 2) EGFR exon 20/21
and KRAS exon 2: 95°C for 10 min; 45 cycles of 95°C for 30s, 54°C
for 10s and 72°C for 1 min; 3) BRAF exon 15: 95°C for 10 min; 45
cycles of 95°C for 30s, 56°C for 10s and 72°C for 30s. The PCR
products and melting curves were analyzed using the
LightScanner® software Call-IT (version 1.5), according
to the manufacturer’s instructions.
 | Table IPrimers of EGFR, KRAS and BRAF. |
Table I
Primers of EGFR, KRAS and BRAF.
| Primer | Sequence (5′-3′) |
|---|
| Exon 18 of EGFR |
5′-GCTTGTGGAGCCTCTTACA-3′
5′-GCCAGGGACCTTACCTTAT-3′ |
| Exon 19 of EGFR |
5′-TGGATCCCAGAAGGTGAGAA-3′
5′-AGCAGAAACTCACATCGAGGA-3′ |
| Exon 20 of EGFR |
5′-ACTGACGTGCCTCTCCCTC-3′
5′-CCCGTATCTCCCTTCCCTG-3′ |
| Exon 21 of EGFR |
5′-CGCAGCATGTCAAGATCA-3′
5′-CCTCCTTACTTTGCCTCC-3′ |
| Exon 2 of KRAS |
5′-AGGCCTGCTGAAAATGACT-3′
5′-AATGGTCCTGCACCAGTAA-3′ |
| Exon 15 of BRAF |
5′-CTCTTCATAATGCTTGCTCTGATAGG-3′
5′-TAGTAACTCAGCAGCATCTCAGG-3′ |
Western blot analysis
The primary antibodies used for western blot
analysis were as follows: Anti-EGFR (mouse monoclonal antibody,
Thermo Fisher Scientific, Waltham, MA, USA), anti-pEGFR (mouse
monoclonal antibody; Try1068; Cell Signaling Technology, Danvers,
MA, USA), anti-RAS (mouse monoclonal antibody; Abcam, Cambridge,
UK); anti-RAF (mouse monoclonal antibody; Santa Cruz Biotechnology
Inc., Dallas, TX, USA) and anti-β-actin (mouse monoclonal antibody;
Beijing Zhongshan Golden Bridge Biotechnology, Beijing, China).
Antibodies were diluted to 1:50, 1:1,000, 1:20, 1:500 and 1:500,
respectively. Whole-cell extracts from the parent and resistant
cells were prepared using a Total Protein Extraction kit (Nanjing
KeyGen Biotech Co., Ltd., Nanjing, China). The protein
concentrations were determined using the Bicinchoninc Acid Protein
Assay kit (Beyotime Institute of Biotechnology, Haimen, China).
Equal amounts of protein (100 μg) were separated by 10% SDS-PAGE
and transferred to polyvinylidene difluoride membranes (EMD
Millipore, Billerica, MA, USA). The membranes were then blocked
with 5% skim milk or 3% bovine serum albumin, and incubated at 4°C
overnight (anti-pEGFR, anti-RAS, anti-RAF and anti-β-actin) or 37°C
for 2 h (anti-EGFR) with the primary antibodies, according to the
manufacturer’s instructions. Subsequent to washing with
Tris-buffered saline with Tween® (TBST) three times, the
membranes were incubated with horseradish peroxidase-conjugated
goat anti-mouse immunoglobulin G secondary antibodies (1:10,000
dilution; Beijing Zhongshan Golden Bridge Biotechnology) for 1 h at
room temperature. The membranes were washed a further three times
with TBST, and the blots were visualized using an Enhanced
Chemiluminescence kit (Beyotime Institute of Biotechnology). The
bands were analyzed using Gel-Pro® Analyzer software
(Media Cybernetics, Rockville, MD, USA).
Immunocytochemistry
Anti-E-cadherin and anti-vimentin were purchased
from Beijing Zhonghshan Golden Bridge Biotechnology. The parent and
resistant cells, having grown to 80% confluence, were fixed in a
chamber slide with cold acetone for 10 min and washed three times
with PBS. The slides were incubated with 3%
H2O2 for 20 min. The cells were then washed a
further three times with PBS and blocked with goat serum for 15
min. The excess serum was poured off of the slides, and the slides
were incubated with the primary antibodies overnight at 4°C,
according to the manufacturer’s instructions. Following the
incubation, the cells were washed three times with PBS and
incubated with biotin-conjugated secondary antibody (SP9002,
monoclonal goat anti-mouse IgG; Beijing Zhonghshan Golden Bridge
Biotechnology) at 37°C for 30 min. The cells were washed again with
PBS three times and incubated with horseradish peroxidase at 37°C
for 30 min. Following another three washes the cells were stained
with diaminobenzidine (Beijing Zhongshan Golden Bridge
Biotechnology). The cell nuclei were stained with hematoxylin
(Beyotime Institute of Biotechnology) for 1–2 min and washed with
water for 10 min, to return to blue. Positive expression was
indicated by a yellow cell membrane or cytoplasm, and the
localization of positive expression was recorded. The positive
expression intensities of the resistant cells were compared to that
of the parent cells in the corresponding expression
localizations.
Statistical analysis
All of the assays were repeated ≥3 times. An
independent sample t-test and a one-way analysis of variance were
used to determine the statistical significance of the mean
differences between the groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Growth inhibition assay
Over eight months, the acquired gefitinib-resistant
cell line, generated from the NCI-H1975 NSCLC cell line, was
established and named NCI-H1975/GR. The resistance index of
NCI-H1975/GR was 2.009 and the IC50 was 12.343 μmol/l,
which was markedly higher as compared with the parent NCI-H1975
cells (6.145 μmol/l) (Fig. 1A).
The number of viable NCI-H1975/GR cells treated with gefitinib at
various concentrations decreased on day 1, as did those of the
parent cells. However, the number of viable NCI-H1975GR cells
reached a stable level and did not continuously decrease between
days 2–4 (Fig. 1B). Furthermore,
the number of viable NCI-H1975/GR cells treated with gefitinib at
the indicated concentrations were higher, as compared with the
parent cells.
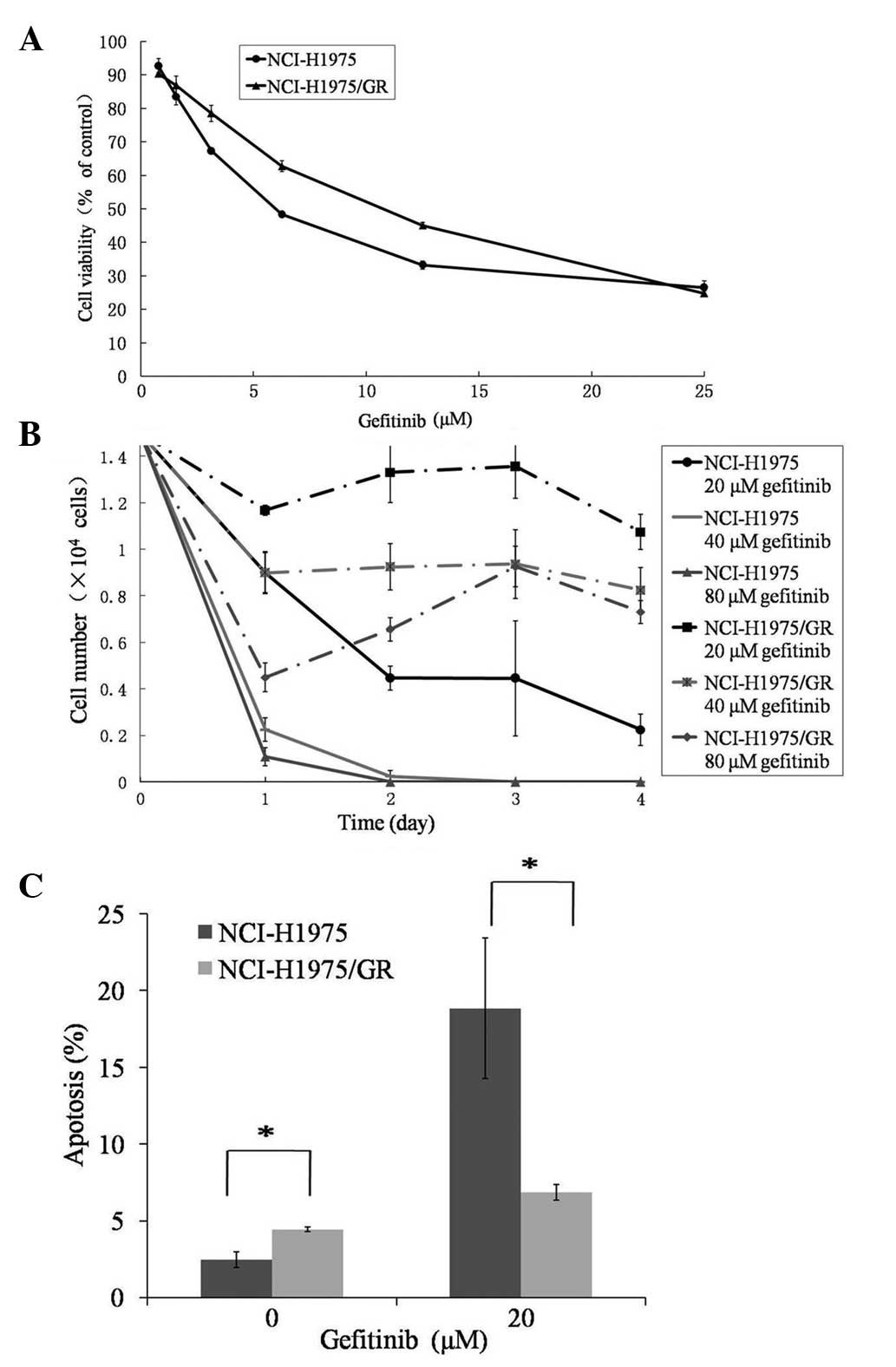 | Figure 1Assessment of the gefitinib resistance
of the NCI-H1975/GR human non-small cell lung cancer cell line. (A)
Cell viability of the parent NCI-H1975 and NCI-H1975/GR cell lines
treated with gefitinib, at the indicated concentrations for 72 h,
as determined by MTT assay. (B) The number of viable NCI-H1975 and
NCI-H1975/GR cells, treated with gefitinib at concentrations of 20,
40 and 80 μmol/l, was determined by trypan blue dye exclusion
method, with the number of cells counted using a hemocytometer
every 24 h for 4 days. (C) The percentage of apoptotic NCI-H1975/GR
and NCI-H1975 cells, with or without gefitinib treatment, were
detected by Annexin V-fluorescein isiothiocyanate and propidium
iodide staining, using flow cytometry. The values represent the
mean ± standard deviation of >3 independent experiments.
*P<0.05. GR, gefitinib-resistant. |
Apoptosis assay
The percentage of apoptotic NCI-H1975GR cells
(4.45±0.14%) was significantly higher, as compared with the
NCI-H1975 cells (2.47±0.51%) (P<0.05), in the absence of
gefitinib (Fig. 1C). Whereas, the
percentage of apoptotic NCI-H1975/GR cells was significantly lower,
as compared with the parent cells, in response to 20 μmol/l
gefitinib treatment (P<0.05).
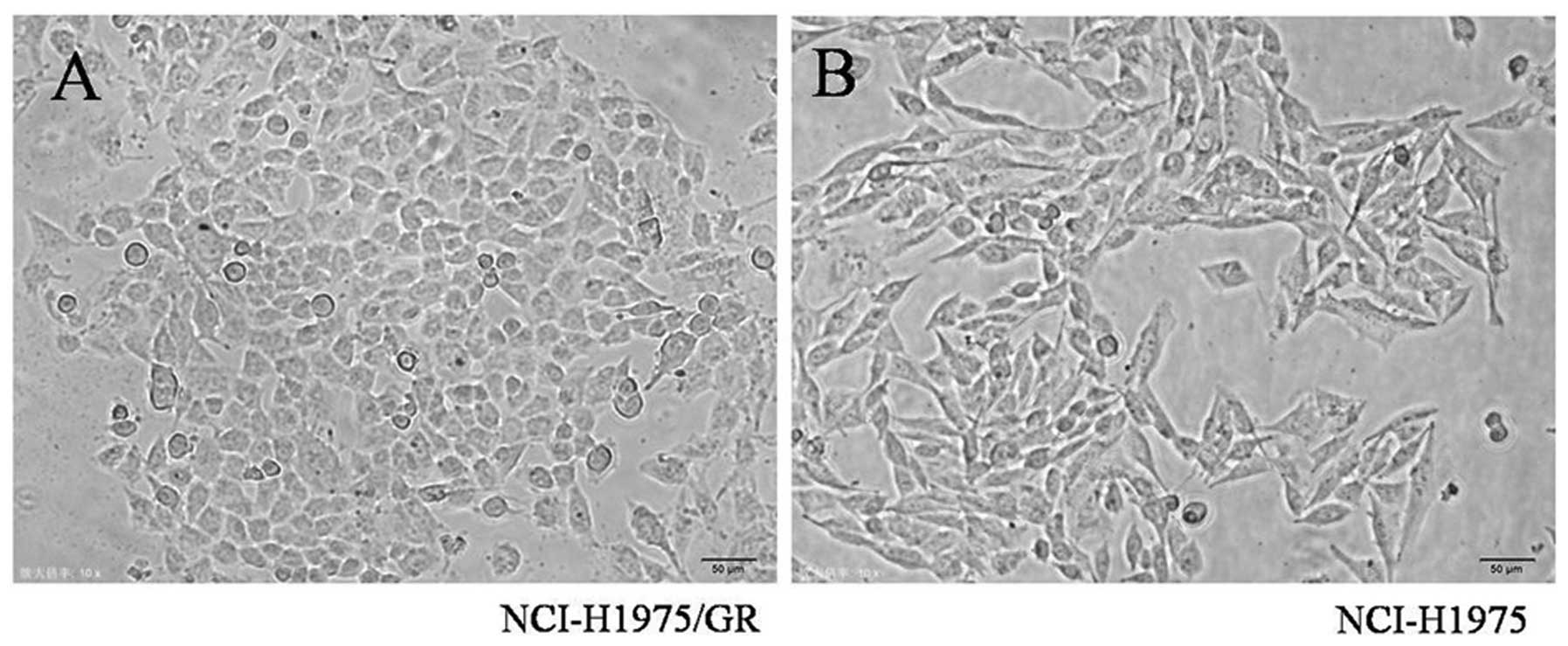
Morphological analysis
Notable morphological differences between the
NCI-H1975/GR and NCI-H1975 cells were observed. The NCI-H1975/GR
cells acquired an oval shape from the long spindle shape of the
parent cells. Furthermore, the resistant cells were smaller, as
compared with the parent cells, and some developed pseudopodia
(Fig. 2).
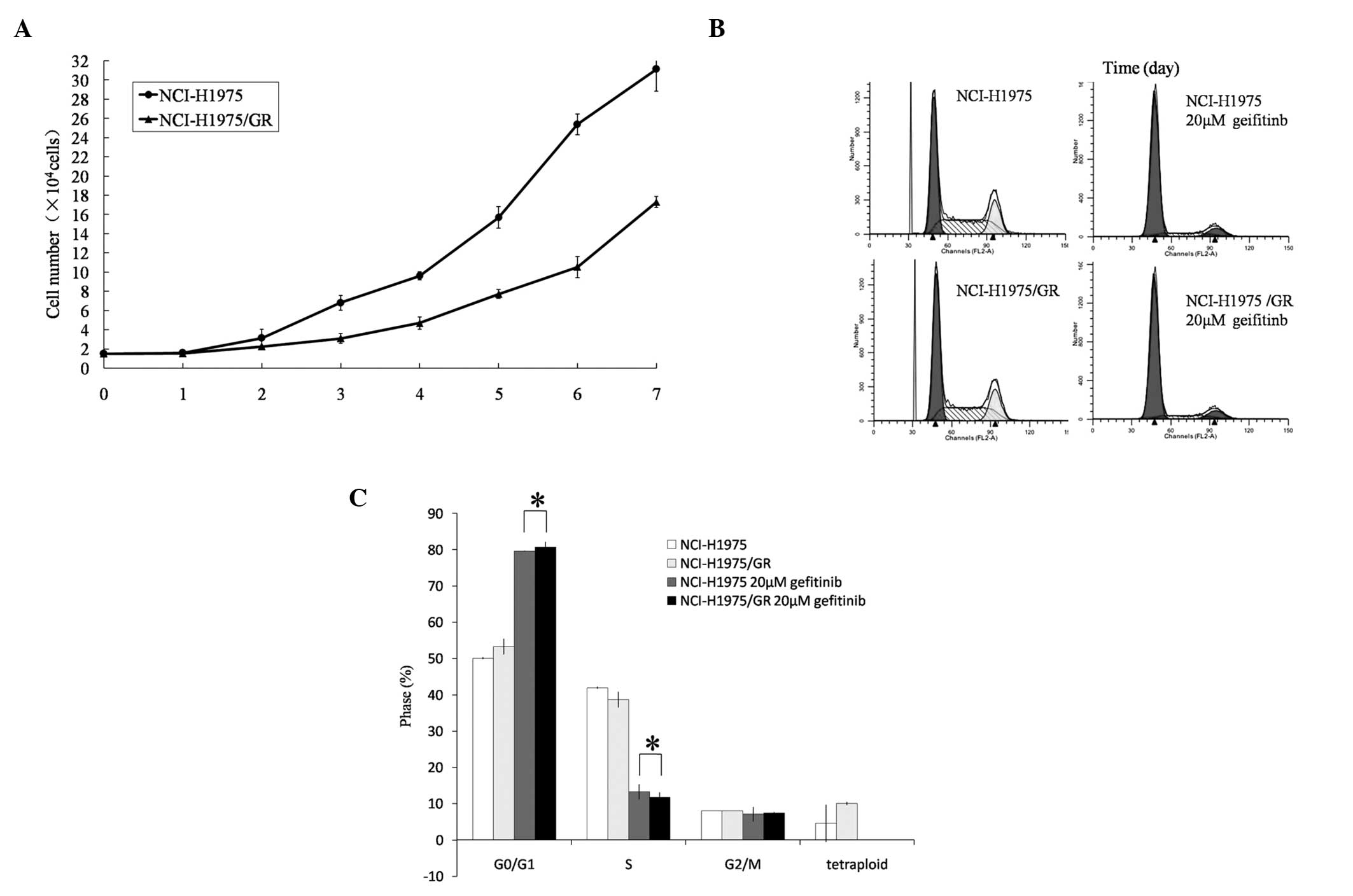
Cell proliferation assay
Cell proliferation curves were charted for the
parent and gefitinib-resistant cells. The cell population doubling
time of the NCI-H1975/GR cells was 46.535±0.428 h, which was
16.004±1.426 h longer as compared with the NCI-H1975 cells
(30.531±1.823 h) (P<0.05; Fig.
3A).
Cell cycle analysis
The proportion of NCI-H1975/GR cells within the
G0/G1 phase was slightly higher, as compared
with the NCI-H1975 cells, and the proportion of tetraploid cells
was also slightly higher (P>0.05). Following treatment with
gefitinib (20 μmol/l) for 24 h, the proportion of NCI-H1975/GR
cells within the G0/G1 phase was markedly
increased and within the S phase decreased (Fig. 3B and C). Furthermore, the
tetraploid cells disappeared following gefitinib treatment.
Mutation analysis of EGFR, KRAS and
BRAF
With the exception of T790M and L858R, no novel
mutations were observed in the EGFR, KRAS and BRAF genes of the
NCI-H1975/GR cell line, by PCR-HRMA.
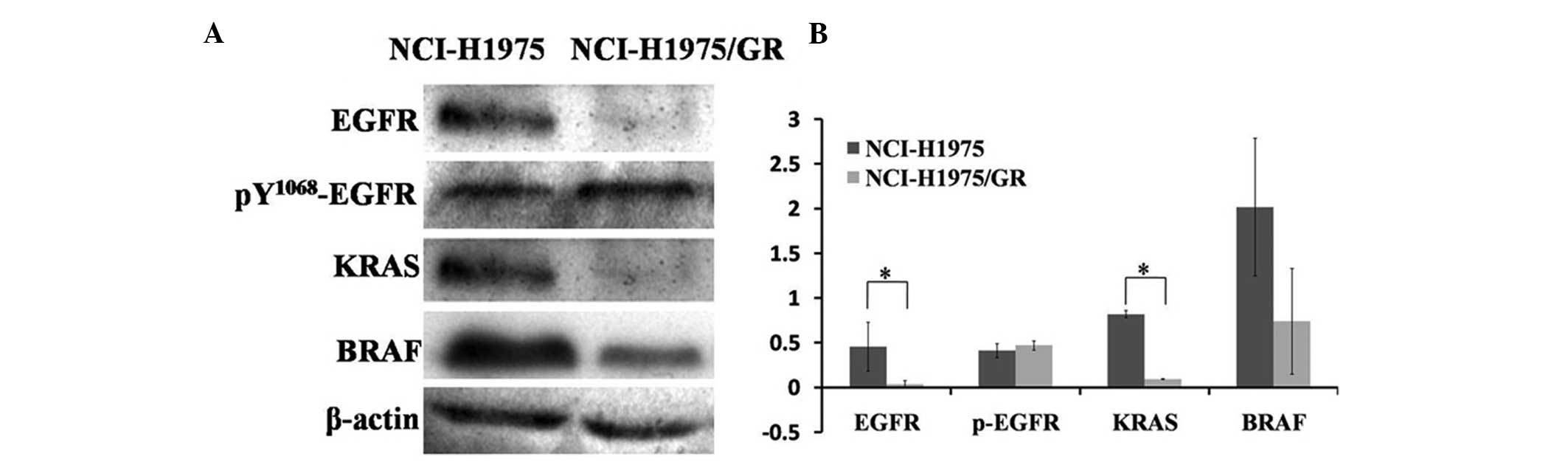
Protein expression levels of the
EGFR/KRAS/BRAF transduction pathway
The protein expressions in the EGFR/KRAS/BRAF
transduction pathway were detected by western blotting. The protein
expression levels in the NCI-H1975/GR cells were lower, as compared
with the NCI-H1975 cells; however, the expression levels of
pY1068-EGFR protein were slightly higher in the NCI-H1975/GR cells,
as compared with the NCI-H1975 cells (Fig. 4). These results indicate that the
EGFR/KRAS/BRAF pathway was not re-activated in the development of
resistance to EGFR-TKIs.
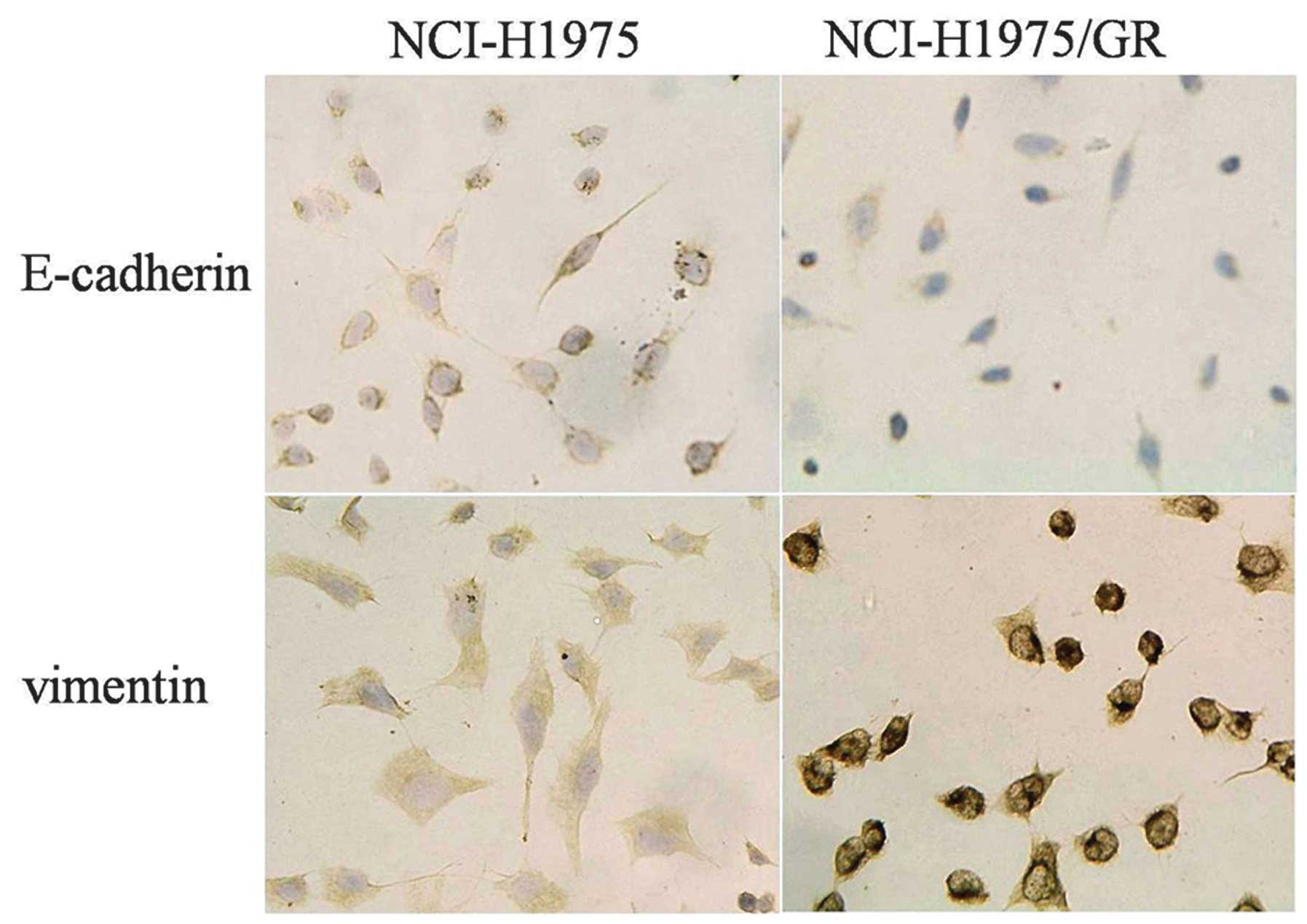
EMT of NCI-H1975/GR cells, as detected by
immunocytochemistry
The expression of E-cadherin was lower, whereas the
expression of vimentin was higher in the NCI-H1975/GR cells, as
compared with the NCI-H1975 cells (Fig. 5).
Discussion
The majority of cases of NSCLC which harbor
activating EGFR mutations, such as exon 19 deletion and exon 21
mutation (L858R substitution), are initially sensitive to
EGFR-TKIs; however, the vast majority of them ultimately acquire
resistance to the drug (18–20).
Notably, the tumors of ~50% of patients who develop acquired
resistance to EGFR-TKIs harbor the exon 20 T790M EGFR mutation. The
T790M mutation may develop during the process of acquired
resistance to TKIs, but could also be primary, since a small number
of patients with NSCLC already harbor the T790M mutation, prior to
EGFR-TKI exposure (21–24). The T790M mutation is considered to
be the basis by which NSCLC develops acquired resistance to TKIs.
Therefore, it is necessary to explore the mechanisms through which
the T790M mutation results in the development of resistance to
TKIs, for the increasing benefits of patients with NSCLC. The
present study developed an acquired gefitinib-resistant cell line
from the NCI-H1975 NSCLC cell line, which was considered to be
sensitive (harboring L858R mutation) and have the potential to
develop resistance to TKIs easily (harboring T790M mutation).
Until now, numerous acquired resistance cell lines
have been established. The representative PC-9/ZD was established
in 2005 and was the first human NSCLC cell line resistant to
gefitinib. It was generated from the PC-9 cell line with an exon 19
deletion of EGFR. PC-9/ZD cells are 182-fold more resistant to
gefitinib, as compared with their parent cells. However, there were
no significant differences observed between the PC-9/ZD and PC-9
parent cells regarding cell proliferation, microscopic morphology
and the DNA sequence of EGFR (25). The H3255 GR was established in 2006
and is another representative NSCLC cell line resistant to
EGFR-TKIs. It was developed by prolonged exposure of the
gefitinib-sensitive H3255 cell line, with EGFR L858R, to gefitinib.
The H3255 GR cells are 100-fold more resistant to gefitinib, as
compared with their parental cells. It was demonstrated that the
H3255 GR cell line acquired a T790M mutation in a small fraction of
the amplified alleles, which was detected by a highly sensitive
high performance liquid chromatography-based technique, but not by
common direct DNA sequencing (26). The HCC827 GR was established in
2007 and is another NSCLC cell line resistant to EGFR-TKIs, which
was also developed by exposure of gefitinib-sensitive HCC827 cells,
with exon 19 deletion of EGFR, to increasing concentrations of
gefitinib. The HCC827 GR cells are 100-fold more resistant to
gefitinib, as compared with their parental cells. The HCC827 GR
cells showed amplification of MET, which caused gefitinib
resistance by driving human epidermal growth factor receptor
3-dependent activation of PI3K (27).
The present study used the NCI-H1975 cell line,
which is genetically different from the cell lines mentioned above.
The NCI-H1975 cell line harbors not only the L858R mutation but
also the T790M mutation. In order to develop a gefitinib-resistant
cell line, the NCI-H1975 cells were exposed to increasing
concentrations of gefitinib. The established gefitinib-resistant
cell line, NCI-H1975/GR, was 2.009-fold more resistant to
gefitinib, as compared with their parental cells. The percentage of
apoptotic NCI-H1975/GR cells decreased, in response to treatment
with gefitinib (20 μmol/l), as compared with the NCI-H1975 cells.
In addition, the speed of growth of the NCI-H1975/GR cells slowed
down (doubling time, 46.535±0.428 h), as compared with the
NCI-H1975 cells (30.531±1.823 h). These results indicate that the
NCI-H1975/GR cell line has low resistance to gefitinib, and that it
may be used to explore the mechanisms of TKI-acquired resistance,
based on the T790M mutation.
The T790M mutation in the NCI-H1975/GR cells was
initially confirmed using PCR-HRMA. The results of the present
study indicate that the development of resistance to TKIs was not
directly associated with the presence of the T790M mutation. It is
well known that NSCLC tumor cells harboring activating mutations in
exons 18, 19 and 21 of the EGFR, are sensitive to EGFR-TKIs. These
exons were detected by PCR-HRMA in the present study; however,
these mutations were not identified, nor the disappearance of the
L858R mutation, which had presented in the NCI-H1975 parent cells.
The activating mutations of KRAS and BRAF genes have been shown to
correlate with primary resistance of NSCLC to TKIs (28). However, these activating and new
mutations of exon 2 of KRAS and exon 15 of BRAF were not observed
in the present study. These results suggest that the common
mutations, which are known to associated with resistance, could not
directly result in the development of acquired resistance to TKIs.
Generally, the development of acquired resistance to TKIs, based on
the T790M mutation, has been attributed to EGFR reactivation, due
to drug-binding deficiency (5,6).
However, in the present study, the protein expression levels of
EGFR, as well as those of KRAS and BRAF, in the NCI-H1975/GR cells
were decreased, as compared with the parental cells. These results
suggest that the EGFR transduction pathway was not reactivated in
the process of acquired resistance in NCI-H1975 cells. Therefore,
other mechanisms of resistance to TKIs should be considered.
In 2011, Suda et al (29) established an acquired
erlotinib-resistant HCC4006ER NSCLC cell line. The parental cell
line harbored the activating mutation of EGFR (exon 19 deletion).
In the resistant cell line, no novel mutations of EGFR were
detected, whereas some morphological changes associated with EMT
were observed, such as loss of intercellular connection and
polarity. These findings indicate that EMT was associated with the
acquired resistance to TKIs of NSCLC cells harboring a mutation in
the EGFR gene. In addition, Rho et al (30) established an acquired
gefitinib-resistant A549/GR NSCLC cell line. In this cell line, EMT
was also observed. In the present study, some mesenchymal
morphologies were detected in the NCI-H1975/GR cells. Furthermore,
the EMT in the NCI-H1975/GR cells was examined by detection of the
epithelial marker E-cadherin and the mesenchymal marker vimentin.
The expression of E-cadherin was lower, whereas the expression of
vimentin was higher in the NCI-H1975/GR cells, as compared with the
NCI-H1975 cells. These results indicate that EMT may have a role in
the development of acquired resistance to EGFR-TKIs in NSCLC cells
harboring activating and resistant mutations of EGFR. How EMT
promotes the development of acquired resistance in NSCLC cells
requires further study.
In conclusion, the present study established an
acquired gefitinib-resistant cell line NCI-H1975/GR from the
NCI-H1975 cell line, harboring the L858/T790M double mutation.
Reactivation of the EGFR/KRAS/BRAF transduction pathway was not
observed in the gefitinib-resistant NCI-H1975/GR cells. The results
suggested that the EMT may have an important role in the
development of acquired resistance to EGFR-TKIs in NSCLC cells with
mutations of sensitivity and resistance.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81071805).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Paez JG, Jänne PA, Lee JC, et al: EGFR
mutations in lung cancer: correlation with clinical response to
gefitinib therapy. Science. 304:1497–1500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pao W, Miller V, Zakowski M, et al: EGF
receptor gene mutations are common in lung cancers from
‘neversmokers’ and are associated with sensitivity of tumors to
gefitinib and erlotinib. Proc Natl Acad Sci USA. 101:13306–13311.
2004. View Article : Google Scholar
|
|
5
|
Kobayashi S, Boggon TJ, Dayaram T, et al:
EGFR mutation and resistance of non-small cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pao W, Miller VA, Politi KA, et al:
Acquired resistance of lung adenocarcinomas to gefitinib or
erlotinib is associated with a second mutation in the EGFR kinase
domain. PLoS Med. 2:e732005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ohashi K, Sequist LV, Arcila ME, et al:
Lung cancers with acquired resistance to EGFR inhibitors
occasionally harbor BRAF gene mutations but lack mutations in KRAS,
NRAS, or MEK1. Proc Natl Acad Sci USA. 109:E2127–E2133. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ercan D, Xu C, Yanagita M, et al:
Reactivation of ERK signaling causes resistance to EGFR kinase
inhibitors. Cancer Discov. 2:934–947. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Faber AC, Corcoran RB, Ebi H, et al: BIM
expression in treatment-naïve cancers predicts responsiveness to
kinase inhibitors. Cancer Discov. 1:352–365. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bean J, Brennan C, Shih JY, et al: MET
amplification occurs with or without T790M mutations in EGFR mutant
lung tumors with acquired resistance to gefitinib or erlotinib.
Proc Natl Acad Sci USA. 104:20932–20937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yauch RL, Januario T, Eberhard DA, et al:
Epithelial versus mesenchymal phenotype determines in vitro
sensitivity and predicts clinical activity of erlotinib in lung
cancer patients. Clin Cancer Res. 11:8686–8698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Suda K, Tomizawa K, Fujii M, et al:
Epithelial to mesenchymal transition in an epidermal growth factor
receptor-mutant lung cancer cell line with acquired resistance to
erlotinib. J Thorac Oncol. 6:1152–1161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mok TS, Wu YL, Thongprasert S, et al:
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N
Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mitsudomi T, Morita S, Yatabe Y, et al:
Gefitinib versus cisplatin plus docetaxel in patients with
non-small-cell lung cancer harboring mutations of the epidermal
growth factor receptor (WJTOG3405): an open label, randomized phase
3 trial. Lancet Oncol. 11:121–128. 2010. View Article : Google Scholar
|
|
16
|
Rosell R, Moran T, Queralt C, et al:
Screening for epidermal growth factor receptor mutations in lung
cancer. N Engl J Med. 361:958–967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yun CH, Mengwasser KE, Toms AV, et al: The
T790M mutation in EGFR kinase causes drug resistance by increasing
the affinity for ATP. Proc Natl Acad Sci USA. 105:2070–2075. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Costa DB, Kobayashi S, Tenen DG and
Huberman MS: Pooled analysis of the prospective trials of gefitinib
monotherapy for EGFR-mutant non-small cell lung cancers. Lung
Cancer. 58:95–103. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nguyen KS, Kobayashi S and Costa DB:
Acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in non-small-cell lung cancers dependent on the
epidermal growth factor receptor pathway. Clin Lung Cancer.
10:281–289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sequist LV, Bell DW, Lynch TJ and Haber
DA: Molecular predictors of response to epidermal growth factor
receptor antagonists in non-small-cell lung cancer. J Clin Oncol.
25:587–595. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Inukai M, Toyooka S, Ito S, et al:
Presence of epidermal growth factor receptor gene T790M mutation as
a minor clone in non-small cell lung cancer. Cancer Res.
66:7854–7858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sequist LV, Martins RG, Spigel D, et al:
First-line gefitinib in patients with advanced non-small-cell lung
cancer harboring somatic EGFR mutations. J Clin Oncol.
26:2442–2449. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Toyooka S, Kiura K and Mitsudomi T: EGFR
mutation and response of lung cancer to gefitinib. N Engl J Med.
352:21362005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bell DW, Gore I, Okimoto RA, et al:
Inherited susceptibility to lung cancer may be associated with the
T790M drug resistance mutation in EGFR. Nat Genet. 37:1315–1316.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Koizumi F, Shimoyama T, Taguchi F, Saijo N
and Nishio K: Establishment of a human non-small cell lung cancer
cell line resistant to gefitinib. Int J Cance. 116:36–44. 2005.
View Article : Google Scholar
|
|
26
|
Engelman JA, Mukohara T, Zejnullahu K, et
al: Allelic dilution obscures detection of a biologically
significant resistance mutation in EGFR-amplified lung cancer. J
Clin Invest. 116:2695–2706. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Adjei AA: K-ras as a target for lung
cancer therapy. J Thorac Oncol. 3:S160–S163. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Suda K, Tomizawa K, Fujii M, et al:
Epithelial to mesenchymal transition in an epidermal growth factor
receptor-mutant lung cancer cell line with acquired resistance to
erlotinib. J Thorac Oncol. 6:1152–1161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rho JK, Choi YJ, Lee JK, et al: Epithelial
to mesenchymal transition derived from repeated exposure to
gefitinib determines the sensitivity to EGFR inhibitors in A549, a
non-small cell lung cancer cell line. Lung Cancer. 63:219–226.
2009. View Article : Google Scholar
|