Introduction
Previous studies have demonstrated that an increase
in glomerular capillary permeability is a key step leading to
proteinuria in patients with diabetes and nephropathy (1). Glomerular capillary endothelial cells
(GECs) constitute the first barrier preventing blood
macromolecules, such as proteins, from passing through the
endothelial wall (2–4). Therefore, the structure and
distribution of GECs is closely associated with the permeability of
the capillary. Previous studies have suggested that the
reorganization and redistribution of the cytoskeleton protein
F-actin and the cortical actin binding protein cortactin in
endothelial cells is crucial to the increase in capillary
permeability observed (5–7). In addition, the Ras-related C3
botulinum toxin substrate 1 (Rac1) signaling pathway has been
identified to be involved in mediating the contraction of
endothelial actin-myosin. This induces an alteration in cell
morphology, destroying the cell-cell connections and forming the
gap between cells (8–12), suggesting an association between
the Rac1 signaling pathway and capillary permeability. It has been
reported that advanced glycation end-products (AGEs) are involved
in inducing alterations in the distribution of cytoskeletal
proteins and endothelial cell permeability in diabetic patients
with microvascular complications (13–16).
However, the function of the Rac1 signaling pathway in this process
remains elusive. In the present study, immunofluorescence staining
and confocal microscopic analysis were conducted in order to
determine the effect of AGEs on the structure and distribution of
F-actin and cortactin in GECs. In addition, the levels of Rac1
activity were investigated using a pull-down assay, endothelial
permeability was analyzed using a Transwell assay and the potential
involvement of the Rac1 signaling pathway in this process was
examined. The present study aimed to improve the understanding of
the pathogenesis of nephropathic proteinuria in diabetics and
provide novel therapeutic targets for diabetes and nephropathy.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium, MCDB131, trypsin
and fetal bovine serum (FBS) were purchased from Gibco Life
Technologies (Carlsbad, CA, USA). Vascular endothelial growth
factor was purchased from BD Biosciences (San Jose, CA, USA) and
fluorescein isothiocyanate (FITC)-phalloidin was obtained from
Molecular Probes Life Technologies (Grand Island, NY, USA).
FITC-labeled goat anti-rabbit antibody, human serum albumin (HSA)
and FITC-bovine serum albumin were from Sigma-Aldrich (St. Louis,
MO, USA). DAPI was purchased from Invitrogen Life Technologies
(Carlsbad, CA, USA) and 2′-O-methyladenosine-3′,5′-cyclic
monophosphate (O-Me-cAMP) was purchased from Aladdin Reagents Co.,
Ltd (Shanghai, China). Transwell was purchased from Corning Life
Sciences (Tewksbury, MA, USA) and a Rac1 Activity Detection kit was
purchased from EMD Millipore (Billerica, MA, USA). A Bradford kit
was purchased from Shenneng Bocai Biotechnology Co., Ltd.
(Shanghai, China).
Isolation and culture of GECs
The isolation and culture of primary GECs was
conducted as previously described (17–19).
All studies were approved by the Ethics Committee of Anhui
Provincial Hospital (Hefei, China) for Animal Experiments and
conformed to the Guide for the Care and Use of Laboratory Animals
by the National Institutes of Health. Primary GECs were isolated
from the kidneys of two male Wistar rats (body weight, 80–120 g)
(Animal Department of Anhui Medical University, China; certificate
no. 003). Primary GECs were cultured in MCDB131 medium supplemented
with 10% FBS in a humidified atmosphere with 5% CO2. All
operations were performed under 10% chloral hydrate (Hechang
Chemical Company, Wuhan, China) and all efforts were made to
minimize suffering.
Preparation of AGE-modified HAS
AGE-HSA was prepared by incubating HSA with glucose,
as previously described (20–23).
The reaction system contained 1.5 g HSA and 3.0 g D-glucose (Meilun
Bio, Dalian, China), which were dissolved in 10 ml
phosphate-buffered saline (PBS; 0.2 mol; pH 7.4; Gibco Life
Technologies) and filtered with 0.22 μm microporous
membranes (EMD Millipore). The solution was then maintained in a
container filled with nitrogen, which was sealed, protected from
light and incubated at 37°C for three months. The unbound materials
were removed using a dialysis bag (molecular weight, 10,000;
Corning, Inc., Corning, NY, USA). The same procedure was completed
without the addition of D-glucose for the control. The AGE value of
the samples (1 mg/ml) was detected by fluorescence scanning (BX43;
Olympus Corp., Tokyo, Japan) and samples were stored at −20°C.
Immunofluorescence staining
Staining was conducted as previously described
(24–27). The slides were washed in PBS twice
and fixed in 4% paraformaldehyde at room temperature for 30 min.
Subsequently, the slides were treated with 0.1% Triton X-100
(Sangon Biotech Co., Ltd, Shanghai, China) for 15 min (F-actin
staining). Subsequent to blocking with 1% bovine serum albumin
(BSA; Sigma-Aldrich) for 1 h at room temperature, the samples were
incubated with FITC-phalloidin for 1 h at room temperature in the
dark (F-actin staining), or with rabbit anti-mouse monoclonal
cortactin antibody (SAB1305513; 1:100) overnight at 4°C. For
cortactin staining, samples were then incubated with DyLight
605-labeled goat anti-rabbit antibody (SAB4600398; 1:500) for 1 h
at room temperature in the dark following washing three times with
PBS for 5 min. Antibodies were obtained from Santa Cruz
Biotechnology, Inc. (Beijing, China) and samples were incubated
with antibodies for 2 h at room temperature. Glycerol (50%; Xilong
Chemical Company, Guangzhou, China) was used to mount the glass
slides with cells, and the images were captured using a confocal
microscope.
Rac1 activity analysis
Racl activity was analyzed using a pull-down assay,
as previously described (9,28).
The protein was extracted using a chemical cleavage method, and the
protein concentration was detected using the Bradford assay.
Samples (50 μg) underwent SDS-PAGE (10%) and were
subsequently transferred to polyvinylidene difluoride membranes
(EMD Millipore). The membranes were incubated with RAC1-GTP
antibody and were then incubated with horseradish
peroxidase-conjugated secondary antibodies (Zhongshan Jinqiao
Company, Beijing, China). The chemiluminescent images were obtained
using a Kodak Image Station 2000R system (Kodak, Rochester, NY,
USA) and the results were analyzed using ImageJ software version
1.44e (National Institutes of Health, Bethesda, MD, USA).
Cell permeability analysis
Using a previously reported method (29), GECs were seeded into the top
compartment of a Transwell chamber with FITC-albumin (100
μl; 1 mg/ml; Sigma-Aldrich). Subsequent to incubation, the
fluorescence intensity of samples was analyzed using a HTS-7000 Bio
Assay Reader (BioAssay Systems, Hayward, CA, USA) with 495 nm
excitation and 520 nm emission filters. The apparent permeability
coefficient [(Pa) = (F/t)(1/A)(v/L)] was used, where F
indicates the fluorescence intensity in the bottom chamber, t
indicates time (sec), A is the membrane area (cm2), v is
the solution volume in the bottom chamber and L indicates the
fluorescence intensity in the top chamber. The results are
expressed as a percentage [Pa% = (experimental
Pa value/control Pa value) × 100%]. The
experiments were repeated a minimum of five times.
Statistical analysis
Statistical analysis was performed using SPSS
software, version 13.0 (SPSS, Inc., Chicago, IL, USA). All data are
presented as the mean ± standard error and were analyzed by a
one-way analysis of variance. P<0.05 was considered to indicate
a statistically significant difference.
Results
Effect of AGE-HSA on F-actin and
cortactin morphologies in GECs
Under normal conditions, endothelial cells appear
smooth and intact (30). F-actin
is filamentous among the long axis and near cell junctions and is
present in reticular, intact and continuous lines, predominantly in
the edges of cells and the inner membranes. A small amount of
reticular nuclear matrix is also present around the nucleus.
Cortactin is predominantly distributed in the cytoplasm and is
occasionally also present in the cell membrane. With an increase in
AGE-HSA concentration or treatment time, the edge of the F-actin
peripheral dense band was observed to become rough and irregular,
with a jagged appearance. F-actin was observed to be diffusely
distributed in cells and the number of stress fibers, composed of a
single row of non-polar actin filaments, increased. The
distribution of cytoplasmic cortactin became disorganized and it
was unclear in the cortex and membrane. Cells became round and
retracted when treated with AGE-HSA for 8 h at a concentration of
100 μg/ml; however, HSA alone did not produce these effects
(Figs. 1 and 2).
Activation of Rac1 inhibits
AGE-HSA-induced morphological alterations to F-actin and cortactin
in GECs
The reconfiguration of the structure of F-actin,
formation of central stress fibers and retraction in GECs induced
by AGE-HSA was markedly inhibited by pre-treatment with O-Me-cAMP.
In addition, similar inhibitory effects were observed in
AGE-HSA-induced cortactin disorganization and cell retraction with
O-Me-cAMP-pre-treatment. However, HSA alone did not produce this
inhibitory effect (Fig. 3). These
observations further suggested an involvement of the Rac1 signaling
pathway in the development of AGE-induced morphological and
structural alterations in GECs.
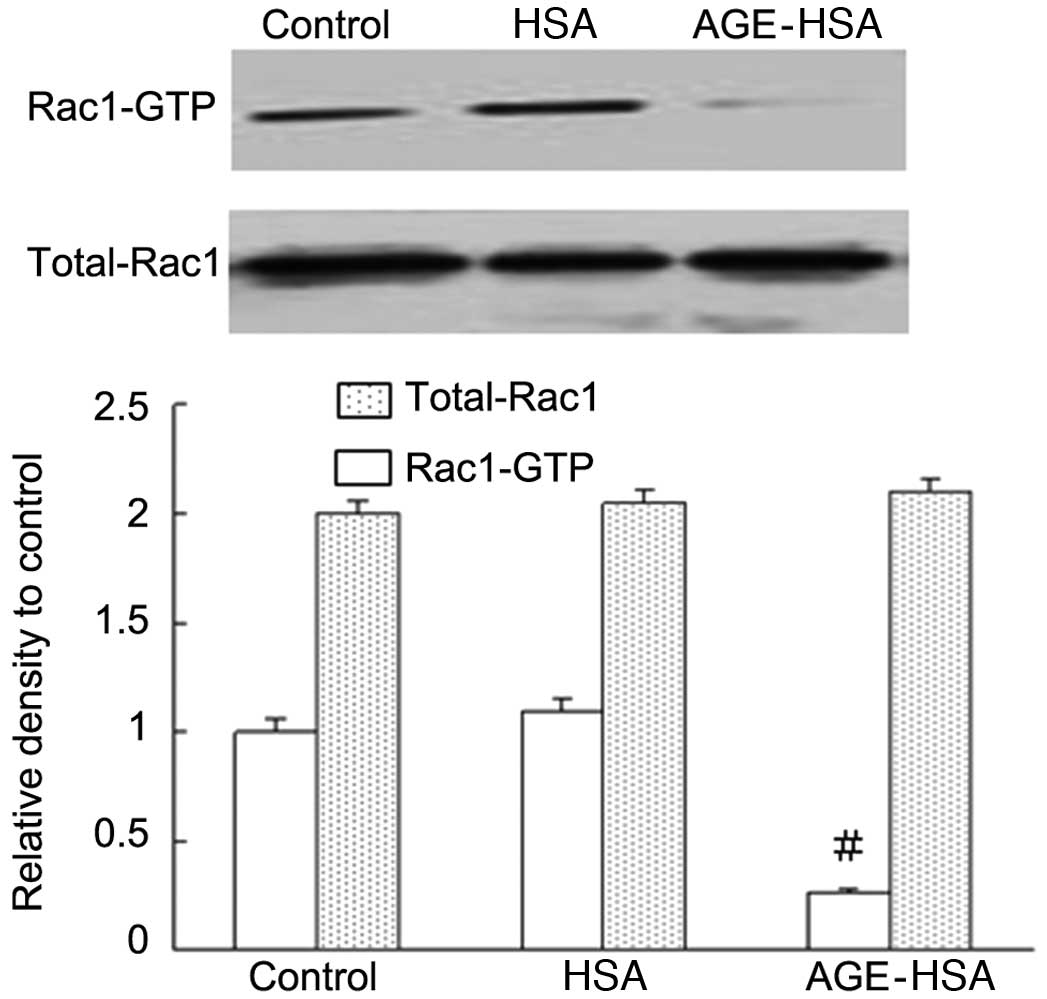
Effect of AGE-HSA on the levels of Rac1
activity in GECs
The levels of Rac1 activity (but not total Rac1
levels) in the AGE-HSA (100 μg/ml) treatment group were
significantly reduced compared with those in the control group
(P=0.002), whereas HSA alone did not produce this effect (Fig. 4). These results suggested that the
Rac1 signaling pathway is important in the mediation of AGE-induced
functional alterations in endothelial cells.
Rac1 agonist inhibits the AGE-HSA-induced
increase in GEC permeability
The permeability of GECs to FITC-BSA was
significantly increased with AGE-HSA-treatment (P<0.05), but not
with HSA alone (P>0.05). This effect was inhibited by O-Me-cAMP,
as demonstrated by reduction in Pa from 189.32±6.16 to
128.52±3.53% subsequent to treatment with O-Me-cAMP (Fig. 5).
Discussion
The initial step in the development of diabetes and
nephropathy is the structural and functional impairment of GECs,
which induces glomerular capillary lesions and leads to disease
progression (31,32). The increase in glomerular capillary
permeability is considered to indicate the development of diabetes
and nephropathy and leads to the development of pathological
proteinuria (33). The proximal
damage among cells is considered to be the basis for the increase
in endothelial cell gap formation and vascular permeability
(2,34–36).
A number of studies have demonstrated that the alterations in
cellular morphology and the distribution of cytoskeletal proteins
is closely associated with the integrity of the endothelial
cell-cell connections (37,38).
The AGE content in diabetic patients has been observed to be
significantly increased, which may induce damage to GEC structure
and function (39–41). Additional studies have demonstrated
that AGE increases the permeability of umbilical vein endothelial
cells and induces alterations in the distribution and morphology of
cytoskeletal proteins (8,11,12,14).
To mimic the pathological process of glomerular capillary
endothelial damage in diabetes and nephropathic patients, primary
GECs were used in the present study. The effects of AGE on the
distribution and morphology of F-actin and cortactin in GECs were
investigated and it was determined whether or not these processes
were mediated by the Rac1 signaling pathway. Rac1 is a member of
the Ras protein superfamily and belongs to the Rho family of
guanosine triphosphatases (42).
Rac1 has multiple functions, including controlling cellular
morphology, actin movement, transcriptional activation and
apoptotic signals (43–46). Rac1 has two active forms, including
an active form bound to guanosine triphosphate (GTP) on the cell
membrane and an inactive form bound to GTP in the cytoplasm. The
two forms can change as a result of upstream stimuli, Rac 1
combining with GTP on the cell membrane is activated, whereas on
the cytoplasm it is inactivated, which in turn regulate the
functions of downstream effectors.
It has been demonstrated that Rac1 can be activated
by specific extracellular signals, which in turn induce actin
cytoskeleton-directed assembly, resulting in characteristic
morphological alterations, including cell stretch (47), an increase in cortical actin
polymerization (48) and an
enhancement of connections between cells (10). Therefore, the Rac1 signaling
pathway is suggested to be necessary for maintaining the stability
of vascular endothelial cell connections. Previous studies have
suggested that activation of the Rac1 signaling pathway can promote
cortactin translocation to the plasma membrane and cortex, thus
inhibiting cell collapse and gap-formation and strengthening
cell-cell connections (49–51).
The mechanism for strengthening these connections is frequently
associated with the increase in cortical actin polymerization,
cortical cortactin and the enhancement of binding to the cortical
actin cytoskeleton. Thus, cortical actin assembly is suggested to
be closely associated with cortactin.
In the present study, the observations suggested
that the AGE-induced increase in GEC permeability was closely
associated with inhibition of Rac1 activity. It was observed that
the F-actin peripheral dense band became thicker and disorganized,
the number of central stress fibers increased, the expression
levels of cortactin in cell membranes were reduced, the boundaries
of cells were unclear and cells retracted and deformed upon
treatment with AGE in a time- and dose-dependent manner (Figs. 1 and 2). These alterations are associated with
the damage to endothelial cell integrity and the increase in
permeability. The pull-down assay was used to investigate Rac1
activity upon treatment with different concentrations of AGE-HSA,
and it was observed that AGE-HSA was able to significantly reduce
Rac1 activity (P<0.05; Fig. 4).
This suggested an important involvement for Rac1 in the mediation
of functional alterations in GECs induced by AGE. In addition, the
present study demonstrated that with O-Me-cAMP pre-treatment,
AGE-induced alterations in cell morphology and stress
fiber-formation (Fig. 3), in
addition to increased GEC permeability, were significantly
inhibited (P<0.05; Fig. 5).
In conclusion, the results of the present study
suggested that Rac1 signaling is important in mediating AGE-induced
morphological and functional alterations in GECs. This may aid in
the development of novel therapeutic targets for microvascular
complications in patients with advanced diabetes. Furthermore, it
was observed that O-Me-cAMP was not able to completely reverse
AGE-induced alterations, including the disorganization of F-actin
and cortactin distribution and morphology, and the increase in cell
permeability. This suggested that in addition to the Rac1 signaling
pathway, other pathways may be involved in the pathological
processes induced by AGE, which require further investigation.
References
|
1
|
Ying WZ, Lin SY and Qiu CL: Study of
permeability of the glomerular capillary wall in human membranous
nephropathy. Zhonghua Nei Ke Za Zhi. 25:227–231. 2541986.In
Chinese.
|
|
2
|
Satchell SC and Braet F: Glomerular
endothelial cell fenestrations: an integral component of the
glomerular filtration barrier. Am J Physiol Renal Physiol.
296:F947–F956. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Myers BD: Pathophysiology of proteinuria
in diabetic glomerular disease. J Hypertens Suppl. 8:S41–S46. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Remuzzi G and Bertani T: Is
glomerulosclerosis a consequence of altered glomerular permeability
to macromolecules? Kidney Int. 38:384–394. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schnoor M, Lai FP, Zarbock A, et al:
Cortactin deficiency is associated with reduced neutrophil
recruitment but increased vascular permeability in vivo. J Exp Med.
208:1721–1735. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Romero IA, Radewicz K, Jubin E, Michel CC,
Greenwood J, Couraud PO and Adamson P: Changes in cytoskeletal and
tight junctional proteins correlate with decreased permeability
induced by dexamethasone in cultured rat brain endothelial cells.
Neurosci Lett. 344:112–116. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dudek SM and Garcia JG: Cytoskeletal
regulation of pulmonary vascular permeability. J Appl Physiol
(1985). 91:1487–1500. 2001.
|
|
8
|
Spindler V, Peter D, Harms GS, Asan E and
Waschke J: Ultrastructural analysis reveals cAMP-dependent
enhancement of microvascular endothelial barrier functions via
Rac1-mediated reorganization of intercellular junctions. Am J
Pathol. 178:2424–2436. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peng H, Wang C, Ye ZC, et al: How
increased VEGF induces glomerular hyperpermeability: a potential
signaling pathway of Rac1 activation. Acta Diabetol. 47(Suppl 1):
57–63. 2010. View Article : Google Scholar
|
|
10
|
Spindler V, Schlegel N and Waschke J: Role
of GTPases in control of microvascular permeability. Cardiovasc
Res. 87:243–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Semina EV, Rubina KA, Rutkevich PN,
Voyno-Yasenetskaya TA, Parfyonova YV and Tkachuk VA: T-cadherin
activates Rac1 and Cdc42 and changes endothelial permeability.
Biochemistry (Mosc). 74:362–370. 2009. View Article : Google Scholar
|
|
12
|
Mehta D and Malik AB: Signaling mechanisms
regulating endothelial permeability. Physiol Rev. 86:279–367. 2006.
View Article : Google Scholar
|
|
13
|
Guo X, Wang L, Chen B, et al: ERM protein
moesin is phosphorylated by advanced glycation end products and
modulates endothelial permeability. Am J Physiol Heart Circ
Physiol. 297:H238–H246. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo XH, Huang QB, Chen B, et al: Advanced
glycation end products induce actin rearrangement and subsequent
hyperpermeability of endothelial cells. APMIS. 114:874–883. 2006.
View Article : Google Scholar
|
|
15
|
Guo XH, Huang QB, Chen B, Wang SY, Hou FF
and Fu N: Mechanism of advanced glycation end products-induced
hyperpermeability in endothelial cells. Sheng Li Xue Bao.
57:205–210. 2005.In Chinese. PubMed/NCBI
|
|
16
|
Sheikpranbabu S, Haribalaganesh R, Lee KJ
and Gurunathan S: Pigment epithelium-derived factor inhibits
advanced glycation end products-induced retinal vascular
permeability. Biochimie. 92:1040–1051. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Akis N and Madaio MP: Isolation, culture,
and characterization of endothelial cells from mouse glomeruli.
Kidney Int. 65:2223–2227. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rops AL, van der Vlag J, Jacobs CW, et al:
Isolation and characterization of conditionally immortalized mouse
glomerular endothelial cell lines. Kidney Int. 66:2193–2201. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Laulajainen T, Julkunen I, Haltia A,
Knuutila S, Miettinen A and Holthöfer H: Establishment and
characterization of a rat glomerular endothelial cell line. Lab
Invest. 69:183–192. 1993.PubMed/NCBI
|
|
20
|
Glomb MA and Monnier VM: Mechanism of
protein modification by glyoxal and glycolaldehyde, reactive
intermediates of the Maillard reaction. J Biol Chem.
270:10017–10026. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nagai R, Unno Y, Hayashi MC, Masuda S,
Hayase F, Kinae N and Horiuchi S: Peroxynitrite induces formation
of N(epsilon)-(carboxymethyl) lysine by the cleavage of Amadori
product and generation of glucosone and glyoxal from glucose: novel
pathways for protein modification by peroxynitrite. Diabetes.
51:2833–2839. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hou FF, Boyce J, Chertow GM, Kay J and
Owen WF Jr: Aminoguanidine inhibits advanced glycation end products
formation on beta2-microglobulin. J Am Soc Nephrol. 9:277–283.
1998.PubMed/NCBI
|
|
23
|
Hou FF, Miyata T, Boyce J, et al:
beta(2)-Microglobulin modified with advanced glycation end products
delays monocyte apoptosis. Kidney Int. 59:990–1002. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eiselein L, Wilson DW, Lamé MW and
Rutledge JC: Lipolysis products from triglyceride-rich lipoproteins
increase endothelial permeability, perturb zonula occludens-1 and
F-actin, and induce apoptosis. Am J Physiol Heart Circ Physiol.
292:H2745–H2753. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zheng HZ, Zhao KS, Zhou BY and Huang QB:
Role of Rho kinase and actin filament in the increased vascular
permeability of skin venules in rats after scalding. Burns.
29:820–827. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu H and Parsons JT: Cortactin, an
80/85-kilodalton pp60src substrate, is a filamentous actin-binding
protein enriched in the cell cortex. J Cell Biol. 120:1417–1426.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Paradis H, Islam T, Tucker S, Tao L, Koubi
S and Gendron RL: Tubedown associates with cortactin and controls
permeability of retinal endothelial cells to albumin. J Cell Sci.
121:1965–1972. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ando K, Ishibashi T, Ohkawara H, et al:
Crucial role of membrane type 1 matrix metalloproteinase (MT1- MMP)
in RhoA/Rac1-dependent signaling pathways in thrombin- stimulated
endothelial cells. J Atheroscler Thromb. 18:762–773. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boutoille D, Marechal X, Pichenot M,
Chemani C, Guery B and Faure K: FITC-albumin as a marker for
assessment of endothelial permeability in mice: comparison with
125I-albumin. Exp Lung Res. 35:263–271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kajiya M, Komatsuzawa H, Papantonakis A,
et al: Aggregatibacter actinomycetemcomitans Omp29 is associated
with bacterial entry to gingival epithelial cells by F-actin
rearrangement. PLoS One. 6:e182872011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Swärd P and Rippe B: Acute and sustained
actions of hyperglycaemia on endothelial and glomerular barrier
permeability. Acta Physiol (Oxf). 204:294–307. 2012. View Article : Google Scholar
|
|
32
|
Balakumar P, Chakkarwar VA, Krishan P and
Singh M: Vascular endothelial dysfunction: a tug of war in diabetic
nephropathy? Biomed Pharmacother. 63:171–179. 2009. View Article : Google Scholar
|
|
33
|
Thomas MC: Pathogenesis and progression of
proteinuria. Contrib Nephrol. 170:48–56. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dane MJ, van den Berg BM, Avramut MC, et
al: Glomerular endothelial surface layer acts as a barrier against
albumin filtration. Am J Pathol. 82:1532–1540. 2013. View Article : Google Scholar
|
|
35
|
Stewart RJ and Marsden PA: Vascular
endothelial cell activation in models of vascular and glomerular
injury. Kidney Int Suppl. 45:S37–S44. 1994.PubMed/NCBI
|
|
36
|
Koike K, Aiboshi J, Shinozawa Y, Sekine K,
Endo T and Yamamoto Y: Correlation of glomerular permeability,
endothelial injury, and postoperative multiple organ dysfunction.
Surg Today. 34:811–816. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bates DO: Vascular endothelial growth
factors and vascular permeability. Cardiovasc Res. 87:262–271.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bogatcheva NV and Verin AD: The role of
cytoskeleton in the regulation of vascular endothelial barrier
function. Microvasc Res. 76:202–207. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Soulis T, Thallas V, Youssef S, et al:
Advanced glycation end products and their receptors co-localise in
rat organs susceptible to diabetic microvascular injury.
Diabetologia. 40:619–628. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hori O, Yan SD, Ogawa S, Kuwabara K,
Matsumoto M, Stern D and Schmidt AM: The receptor for advanced
glycation end-products has a central role in mediating the effects
of advanced glycation end-products on the development of vascular
disease in diabetes mellitus. Nephrol Dial Transplant. 11(Suppl 5):
13–16. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yamagishi S: Role of advanced glycation
end products (AGEs) and receptor for AGEs (RAGE) in vascular damage
in diabetes. Exp Gerontol. 46:217–224. 2011. View Article : Google Scholar
|
|
42
|
Bond M, Wu YJ, Sala-Newby GB and Newby AC:
Rho GTPase, Rac1, regulates Skp2 levels, vascular smooth muscle
cell proliferation, and intima formation in vitro and in vivo.
Cardiovasc Res. 80:290–298. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Szczepanowska J: Involvement of
Rac/Cdc42/PAK pathway in cytoskeletal rearrangements. Acta Biochim
Pol. 56:225–234. 2009.PubMed/NCBI
|
|
44
|
Pai SY, Kim C and Williams DA: Rac GTPases
in human diseases. Dis Markers. 29:177–187. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Williams LM, Lali F, Willetts K, et al:
Rac mediates TNF-induced cytokine production via modulation of
NF-kappaB. Mol Immunol. 45:2446–2454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mondal S, Ghosh-Roy S, Loison F, et al:
PTEN negatively regulates engulfment of apoptotic cells by
modulating activation of Rac GTPase. J Immunol. 187:5783–5794.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chang F, Lemmon C, Lietha D, Eck M and
Romer L: Tyrosine phosphorylation of Rac1: a role in regulation of
cell spreading. PLoS One. 6:e285872011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jacobson JR, Dudek SM, Singleton PA,
Kolosova IA, Verin AD and Garcia JG: Endothelial cell barrier
enhancement by ATP is mediated by the small GTPase Rac and
cortactin. Am J Physiol Lung Cell Mol Physiol. 291:L289–L295. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Weed SA, Du Y and Parsons JT:
Translocation of cortactin to the cell periphery is mediated by the
small GTPase Rac1. J Cell Sci. 111:2433–2443. 1998.PubMed/NCBI
|
|
50
|
Adamson RH, Sarai RK, Altangerel A,
Thirkill TL, Clark JF and Curry FR: Sphingosine-1-phosphate
modulation of basal permeability and acute inflammatory responses
in rat venular microvessels. Cardiovasc Res. 88:344–351. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Maharjan S, Kim K, Agrawal V, et al:
Sac-1004, a novel vascular leakage blocker, enhances endothelial
barrier through the cAMP/Rac/cortactin pathway. Biochem Biophys Res
Commun. 435:420–427. 2013. View Article : Google Scholar : PubMed/NCBI
|