Introduction
Traumatic brain injury (TBI) is one of the leading
causes of mortality and morbidity in adults and children worldwide
(1). Tissue loss and cell death
following TBI results from both primary injury (direct physical
tissue disruption) and secondary injury (delayed molecular
pathophysiological changes) (2).
Secondary injury occurs following primary injury and may continue
for days or weeks, resulting in progressive neuronal death
(2,3). The mechanisms underlying secondary
injury include inflammation, glutamate excitotoxicity, neuronal
death and neurological dysfunction, all of which result in the
induction of mitochondrial dysfunction as well as the amplification
of biochemical cell death signaling cascades (4–6).
A recent study confirmed that both apoptosis and
autophagy participate in TBI-induced neuronal cell death and
functional loss (7). In addition,
neuronal apoptosis has a significant role in the pathophysiology of
TBI (8). Furthermore, caspase-3
and B cell lymphoma 2 (Bcl-2) are recognized as important apoptotic
regulators, and the expression levels of caspase-3 and Bcl-2
determine the fate of cells (9,10). A
previous study demonstrated that TBI-activated autophagy and
increased microtubule-associated protein 1 light chain 3 (LC3)
immunostaining occurred predominantly in neurons (11). Beclin-1 has also been shown to
participate in the regulation of neuronal autophagy (12). These results indicate that numerous
cell apoptosis mechanisms may contribute to TBI-induced neuronal
cell death. Therefore, identifying neuroprotective agents that
inhibit these numerous cell death mechanisms may provide novel
therapeutic strategies for the treatment of TBI.
Rosiglitazone (RSG) is a peroxisome
proliferator-activated receptor-γ (PPAR-γ) agonist, known for its
anti-inflammatory actions via PPAR-γ activation (13). Previous studies have suggested that
RSG may exert neuroprotective effects in animal models of chronic
brain injuries, such as Alzheimer's disease (14), amyotrophic lateral sclerosis
(15), and Parkinson's disease
(16). The efficacy of RSG has
also been demonstrated in animal models of acute brain injuries,
including focal ischemia (17),
spinal cord injury (18), and TBI
(19). A recent study suggested
that treatment with RSG attenuated TBI-induced excessive neuronal
apoptosis (19); however, whether
RSG treatment is involved in TBI-induced autophagic neuronal death
remains unclear.
In order to determine the potential mechanism
underlying the neuroprotective effects of RSG following TBI, the
present study aimed to investigate the hypothesis that RSG carries
out its neuroprotective effects via the attenuation of neuronal
apoptosis and autophagy following TBI in rats. Furthermore, the
role of RSG in the modulation of inflammatory and glutamate
excitotoxicity, and the impact of RSG on the progression of
functional recovery following TBI were also investigated.
Materials and methods
Animals
Adult female Sprague Dawley rats (weight, 250–300 g;
age, 3 months; Hebei University Animal Center, Baoding, China) were
used for the experiments of the present study. All experiments were
performed in acordance with the institutional guidelines for the
care and use of laboratory animals (Hebei University School of
Medicine). All rats were provided with ad libitum access to
food and water prior to experimentation, and were housed in a 12 h
light/dark environment at 22°C.
Models of TBI
Controlled cortical impact (CCI) injury was carried
out on the rats as previously described (1). The rats were anesthetized by
intraperitoneal injection of 50 mg/kg sodium pentobarbital (Beijing
Solarbio Science & Technology Co., Ltd., Beijing, China), prior
to being placed in a stereotaxic frame. A 5 mm craniotomy was
performed over the left parietal cortex, centered on the coronal
suture and 3 mm laterally to the sagittal suture. Considerable care
was taken to avoid injury to the underlying dura. CCI was performed
using a pneumatic piston with a rounded metal tip (2.5 mm
diameter), angled 22.5° from vertical so that the metal tip was
perpendicular to the brain surface at the center of the craniotomy.
A velocity of 4 m/s and a deformation depth of 2 mm below the dura
were used. The bone flap was immediately replaced and sealed, and
the scalp was closed with sutures (Beijing Solarbio Science &
Technology Co., Ltd.). Body temperature was monitored throughout
the surgery using a rectal probe, and the temperature was
maintained at 37.0±0.5°C using a heated pad. The rats were
subsequently placed in a heated cage in order to maintain constant
body temperature while recovering from anesthesia.
Groups and drug administration
The rats were randomly assigned to a sham-operated
group (sham, n=30); a TBI group, which received 0.9% saline
solution (vehicle, n=60); and a TBI group, which was treated with
RSG (RSG, n=60; Cell Signaling Technology, Inc., Danvers, MA, USA).
RSG was dissolved in 0.9% saline and stored at 4°C. A total of 2
mg/kg RSG was administered via intraperitoneal injection in the RSG
group immediately after TBI. All experiments were carried out as
blind studies, and the animal codes were only revealed at the end
of the behavioral and histologic analyses.
Immunofluorescence
The rats were sacrificed 24 h after TBI by
exsanguination. Prior to exsanguination, the rats were anesthetized
with sodium pentobarbital (i.p.;50 mg/kg). Coronal sections (10
µm) were obtained from the anterior area of the left
hemisphere. The sections were incubated with 10% normal donkey
serum (Beijing Solarbio Science & Technology Co., Ltd.) for 30
min at room temperature in phosphate-buffered saline (PBS)
supplemented with 0.1% Triton X-100 (Beijing Solarbio Science &
Technology Co., Ltd.), prior to being incubated with the
appropriate primary antibodies overnight at 4°C. The following
primary antibodies were used in various combinations:
Anti-neuron-specific nuclear protein (NeuN) (1:200; cat. no.
sc-134481) and anti-caspase 3 (1:50; cat. no. sc-98785), and
anti-LC3 (1:50; cat. no. sc-54237) (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA). Following primary antibody incubation, the
coronal sections were washed four times at room temperature, prior
to being incubated with appropriate fluorescence-labeled secondary
antibodies (1:200) for 1 h at room temperature. A total of 5
µg/ml 4′,6-diamidino-2-phenylindole (Beijing Solarbio
Science & Technology Co., Ltd.) was incubated with the coronal
sections in order to carry out counterstaining of the nucleus. The
sections were then washed with PBS and mounted onto slides using
water-based mounting medium containing anti-fading agents (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). All confocal images
were captured using an Olympus FV1000 laser scanning confocal
microscope, and were analyzed using FV10-ASW 1.5 Viewer digital
imaging software (Olympus Corporation, Tokyo, Japan).
Western blot analysis
Briefly, the rats were anesthetized with sodium
pentobarbital (i.p.; 50 mg/kg) and underwent intra-cardiac
perfusion with 0.1 mol/l PBS (pH 7.4). The cortex region of the
brain was rapidly isolated, homogenized (BestBio Biotechnology,
Beijing, China), and total proteins were extracted using protein
extraction reagent (Bio-Rad Biotechnology, Inc., Shanghai, China).
Protein concentration was determined using a bicinchoninic acid
assay (Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China). The samples were separated by 20% SDS-PAGE, and
were then transferred onto polyvinylidene fluoride membranes (Roche
Diagnostics GmbH, Mannheim, Germany) prior to being blocked with 5%
fat-free dry milk for 1 h at room temperature. The membranes were
subsequently incubated with the following primary antibodies
overnight at 4°C: Rabbit anti-tumor necrosis factor-α (TNF-α)
polyclonal antibody (cat. no. sc-7895), rabbit anti-interleukin
(IL)-6 polyclonal antibody (cat. no. sc-7920), rabbit
anti-glutamate transporter-1 (GLT-1) polyclonal antibody (cat. no.
sc-365634), rabbit anti-capase 3 polyclonal antibody (cat. no.
sc-7148), rabbit anti-Bcl-2 polyclonal antibody (cat. no. sc-783),
rabbit anti-beclin polyclonal antibody (cat. no. sc-292327), rabbit
anti-LC3 polyclonal antibody (cat. no. sc-134226), and mouse
anti-β-actin monoclonal antibody (cat. no. sc-376421) (all 1:500;
Santa Cruz Biotechnology, Inc.). The membranes were then incubated
with horseradish peroxidase-conjugated anti-rabbit immunoglobulin
(Ig)G (cat. no. sc-2027) and anti-mouse IgG (cat. no. sc-2025)
(1:5,000; Cell Signaling Technology, Inc.) for 2 h at room
temperature. The membrane was then visualized with an Enhanced
Chemiluminescence Detection system (BestBio Biotechnology) and the
densitometric signals were quantified using ImageJ 1.41 software
(National Institutes of Health, Bethesda, MD, USA). The
immunoreactive bands of the proteins were normalized to the band
intensity of β-actin. The western blot results were analyzed using
ImageJ 1.41 software (National Institutes of Health, Bethesda, MA,
USA).
Recovery of motor function
The neurobehavioral status of the rats was evaluated
using a set of 10 tasks, collectively termed the neurological
severity score (NSS), which tests the reflexes, alertness,
coordination, and motor abilities of the rats. One point is awarded
for failure to perform a particular task, and thus a score of 10
reflects maximum impairment, whereas a score of 0 is normal. The
NSS was evaluated at 1, 4 and 7 days post-injury. An observer who
was unaware of the treatment the animal had received assessed each
rat. The difference between the initial NSS and the NSS at a later
time point was calculated for each rat (ΔNSS), and this value
reflects the spontaneous or treatment-induced recovery of motor
function.
Statistical analysis
All data were presented as the mean ± standard
error. SPSS 16.0 (SPSS, Inc., Chicago, IL, USA) was used for all
statistical analyses of the data. Statistical analysis was
performed using one-way analysis of variance, followed by
Student-Newman-Keuls post-hoc tests. P<0.05 was considered to
indicate a statistically significant difference.
Results
Treatment with RSG attenuates TBI-induced
motor deficits
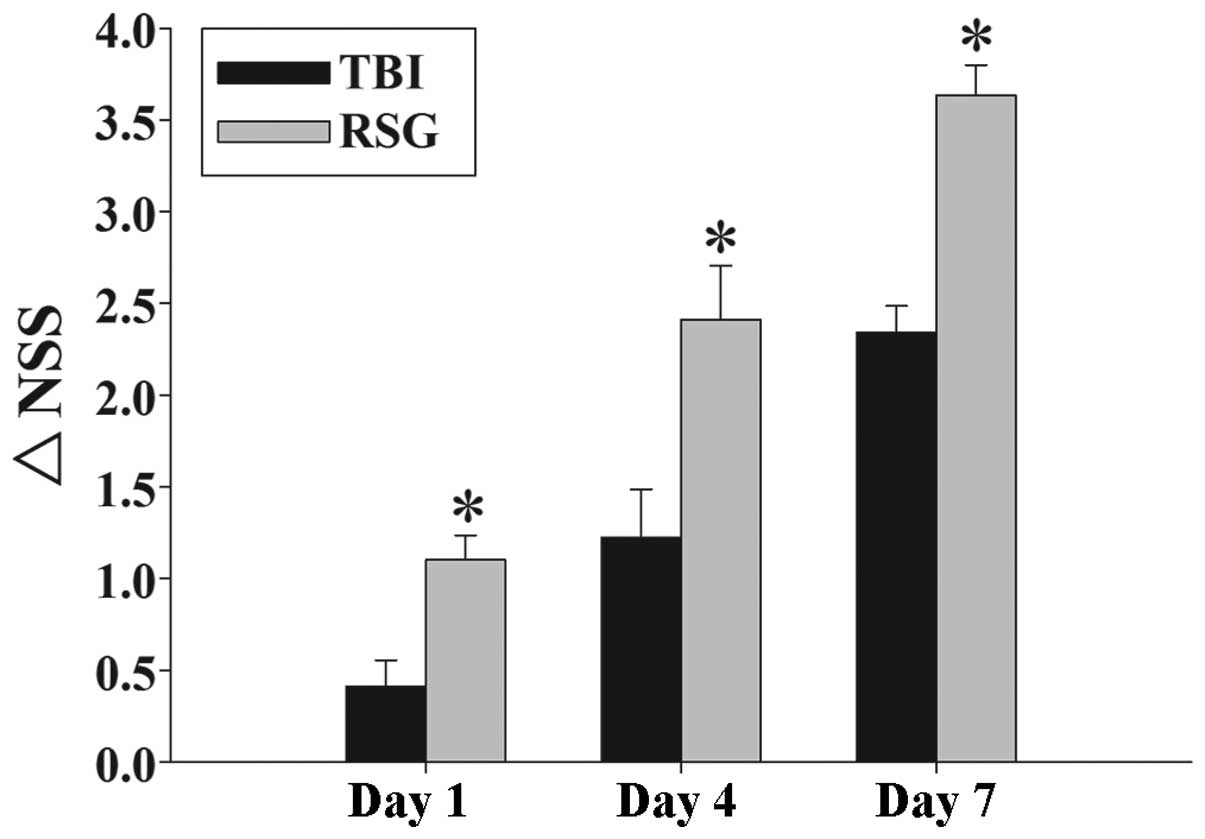
Fig. 1 exhibits the
temporal changes in functional recovery of the TBI rats, expressed
as ΔNSS. Post-injury administration of RSG markedly improved motor
function recovery on days 1, 4, and 7 following TBI.
Treatment with RSG inhibits caspase 3
expression in the cortex following TBI
Co-localization of NeuN and caspase 3 was assessed
by immunofluorescent staining on day 1. As shown in Fig. 2A, the majority of TBI-induced
apoptosis occurred in the neurons. As demonstrated in Fig. 2B, 1, 4 and
7 days after TBI, the expression
levels of caspase 3 were markedly increased in the TBI group, as
compared with the sham group, and treatment with RSG markedly
attenuated caspase 3 expression, as compared with the TBI group
(Fig. 2C).
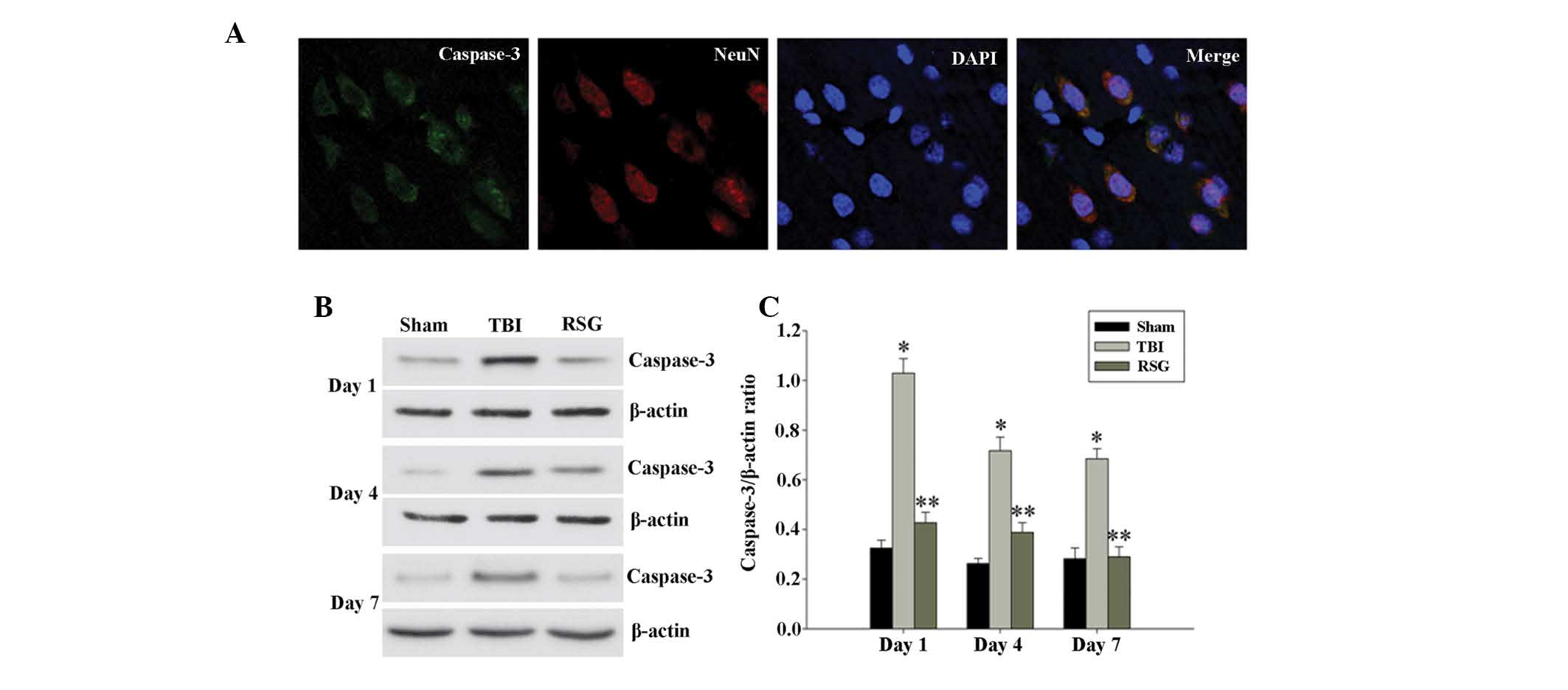 | Figure 2(A) Co-localization of NeuN and
caspase 3 24 h after TBI was determined by immunofluorescent
staining (magnification, ×400), and cell nucleus counterstaining by
4′,6-diamidino-2-phenylindole. (B) Western blot analysis was used
to detect the expression levels of caspase 3 and β-actin in the
cortex at 1, 4 and 7 days after TBI. (C) The expression levels of
caspase 3 were normalized to those of β-actin. The data are
presented as the mean ± standard error (n=5). The expression levels
of caspase 3 were significantly increased in the TBI group 1, 4 and
7 days after TBI (*P<0.01, vs. the sham group), and
treatment with RSG significantly decreased the protein expression
levels of caspase 3 (**P<0.05, vs. the TBI group).
TBI, traumatic brain injury; RSG, rosiglitazone; NeuN,
neuron-specific nuclear protein. |
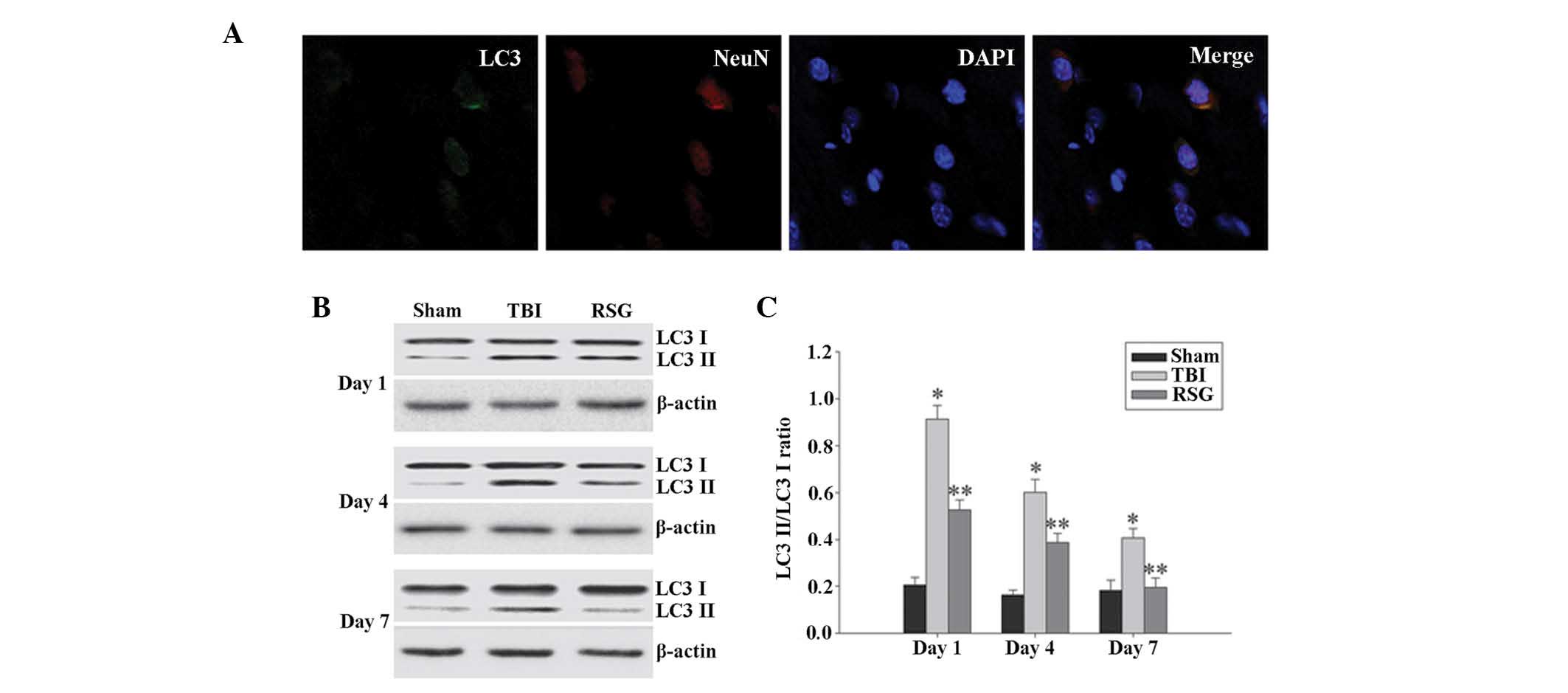 | Figure 4(A) Co-localization of NeuN and LC3 24
h following TBI as determined by immunofluorescent staining
(magnification, ×400), and cell nucleus counterstaining by
4′,6-diamidino-2-phenylindole. (B) Western blot analysis was used
to detect the expression levels of LC3-II/LC3-I in the cortex 1, 4
and 7 days after TBI. (C) The expression levels of LC3-II were
normalized to those of LC3-I. The data are presented as the mean ±
standard error (n=5). The expression levels of LC3-II/LC3-I were
significantly increased in the TBI group 1, 4 and 7 days after TBI
(*P<0.01, vs. sham group), and treatment with RSG
significantly decreased the expression levels of LC3-II/LC3-I
(**P<0.05, vs. TBI group). TBI, traumatic brain
injury; RSG, rosiglitazone; LC3, microtubule-associated protein 1
light chain 3; NeuN, neuron-specific nuclear protein. |
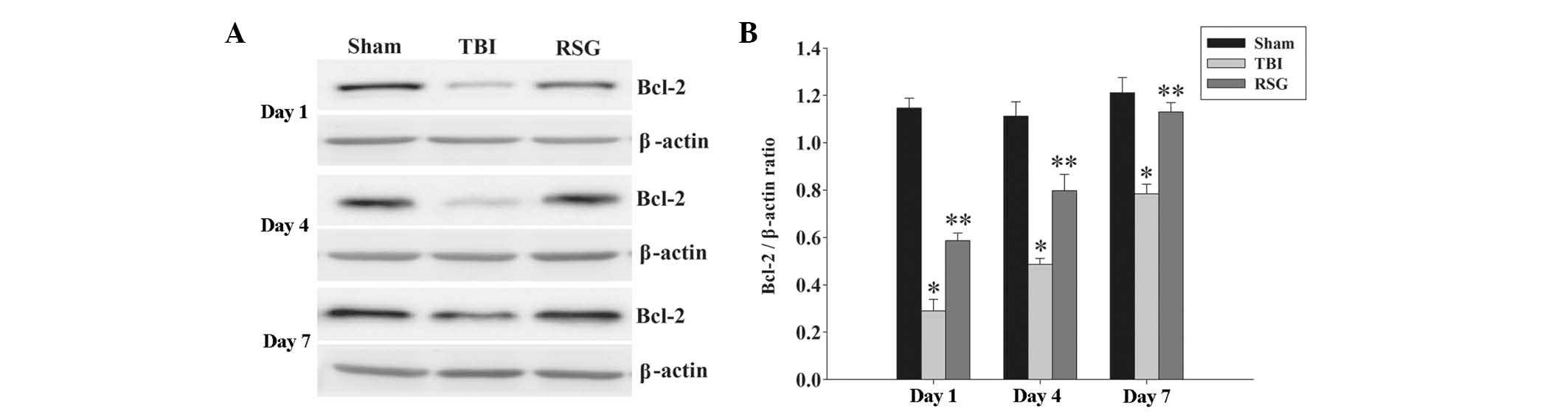
RSG treatment increases the expression
levels of Bcl-2 in the cortex following TBI
The protein expression levels of Bcl-2 in the cortex
were analyzed by western blotting 1, 4 and 7 days after TBI
(Fig. 3). As shown in Fig. 3, the expression levels of Bcl-2
were significantly downregulated in the TBI group, as compared with
the sham group 1, 4 and 7 days after TBI. Treatment with RSG
markedly increased the expression levels of Bcl-2, as compared with
the TBI group.
Treatment with RSG decreases the
expression levels of LC3-II/LC3-I in the cortex following TBI
A recent study demonstrated that the expression
levels of autophagy marker protein LC3 were significantly increased
1 day following TBI (7);
therefore, the present study investigated the co-localization of
NeuN and LC3 using immunofluorescent staining 1 day after TBI. As
shown in Fig. 4A, the majority of
TBI-induced autophagy occurred in the neurons. As demonstrated in
Fig. 4B and C, 1, 4 and 7 days
following TBI, the expression levels of LC3-II/LC3-I were
significantly increased in the TBI group, as compared with the sham
group, and treatment with RSG significantly decreased the
expression levels of LC3-II/LC3-I in the rat cortex, as compared
with the TBI group.
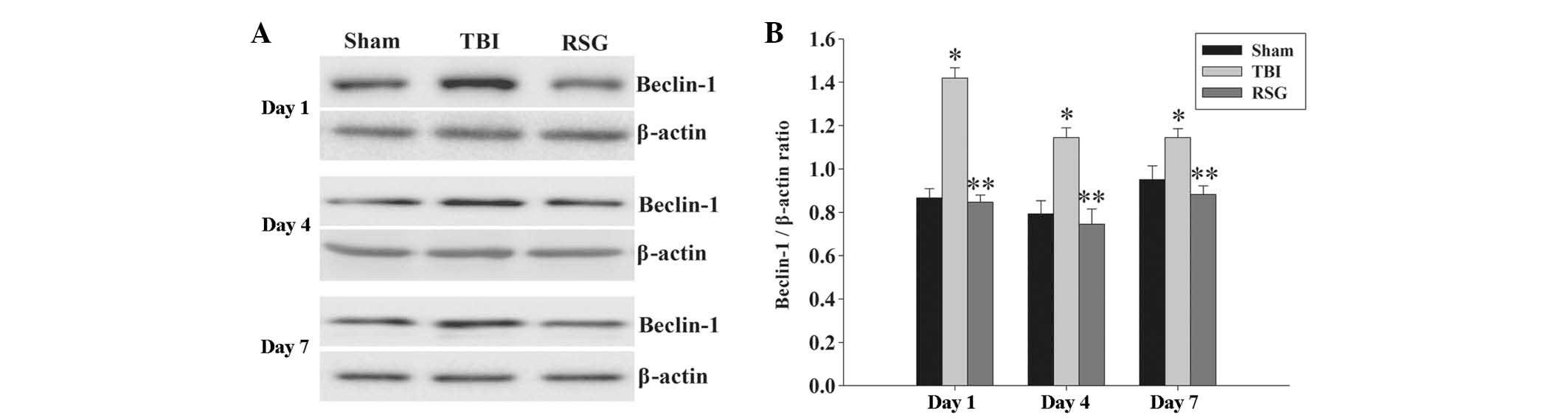
Treatment with RSG decreases the
expression levels of Beclin-1 in the cortex following TBI
The expression levels of Beclin-1 in the cortex were
measured by western blot analysis 1, 4 and 7 days after TBI
(Fig. 5). As shown in Fig. 5, the expression levels of Beclin-1
were significantly increased at the various time points in the TBI
group, as compared with the sham group. Conversely, treatment with
RSG significantly reduced the expression levels of Beclin-1, as
compared with the TBI group.
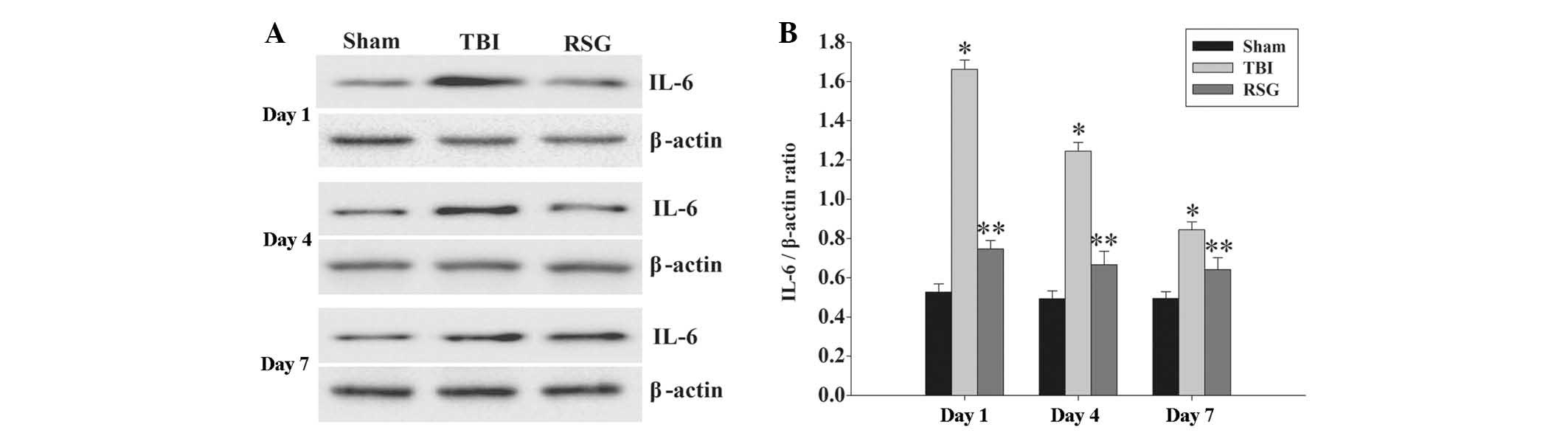
Treatment with RSG decreases the
expression levels of IL-6 in the cortex following TBI
The expression levels of IL-6 in the cortex were
measured by western blot analysis 1, 4 and 7 days after TBI
(Fig. 6). The expression levels of
IL-6 were significantly increased at the various time points in the
TBI group, as compared with the sham group. Conversely, treatment
with RSG significantly reduced the expression levels of IL-6, as
compared with the TBI group.
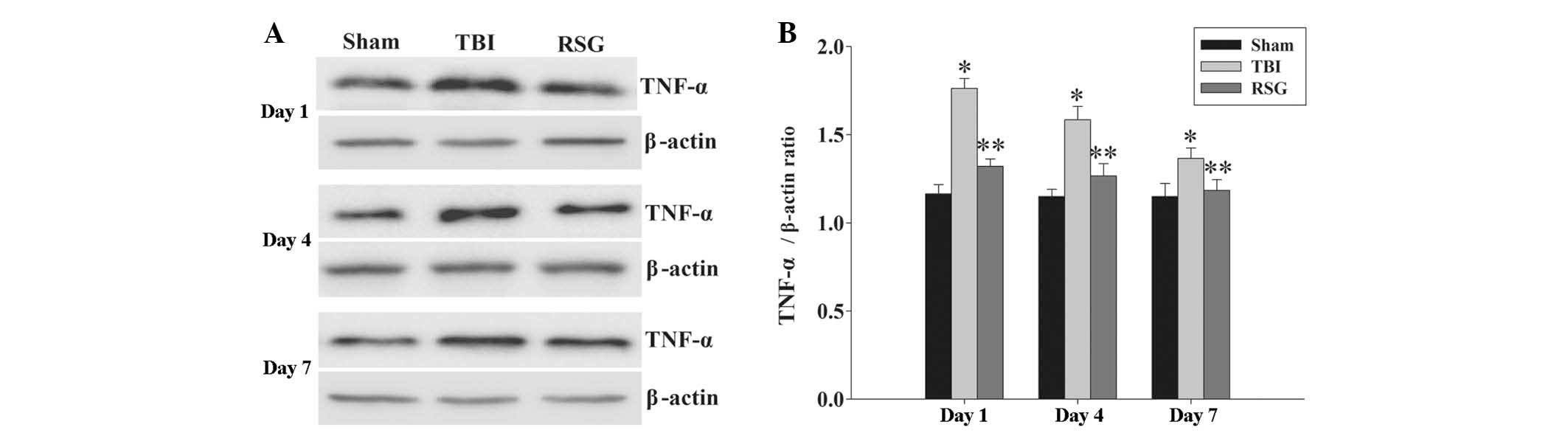
Treatment with RSG attenuates the
expression levels of TNF-α in the cortex following TBI
The expression levels of TNF-α in the cortex were
measured by western blot analysis 1, 4 and 7 days after TBI
(Fig. 7). The expression levels of
TNF-α were significantly increased at the various time points in
the TBI group, as compared with the sham group. Conversely,
treatment with RSG significantly reduced the TBI-induced
upregulation of TNF-α expression.
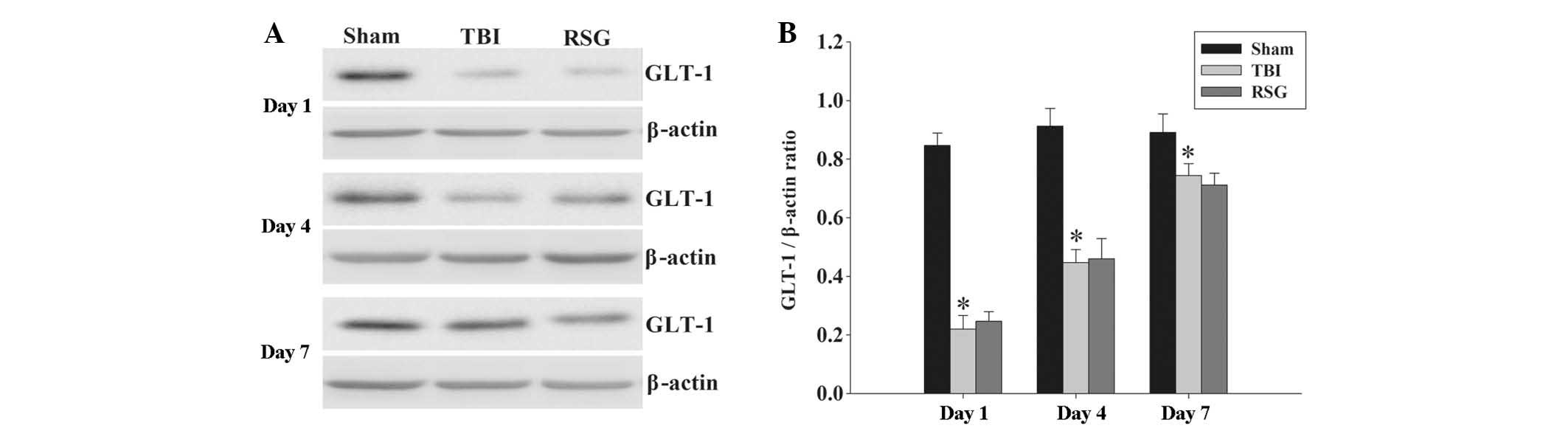
No significant changes were observed in the protein
expression levels of GLT-1 in the cortex following RSG treatment.
The protein expression levels of GLT-1 in the cortex were analyzed
by western blotting at 1, 4 and 7 days after TBI (Fig. 8). The expression levels of GLT-1
were significantly downregulated in the TBI group, as compared with
the sham group 1, 4 and 7 days after TBI; however, treatment with
RSG induced no significant changes in the expression levels of
GLT-1, as compared with the TBI group.
Discussion
The present study investigated the effectiveness of
RSG, a PPAR-γ agonist, as a therapeutic option for the treatment of
TBI. The results indicated that a single injection of RSG
immediately following TBI significantly reduced neuronal apoptosis
and autophagy, and increased functional recovery. These effects
correlate with a decrease in the protein expression levels of TNF-α
and IL-6 in the brain cortex. However, no significant changes were
observed in the protein expression levels of GLT-1 in the rats
treated with RSG. Previous studies have demonstrated that RSG
exerts neuroprotective effects in numerous acute brain injury
models, including focal ischemia, spinal cord injury, and TBI
(14–19). Using the CCI model of TBI, the
present study confirmed these previous results, and extended these
observations by providing the first demonstration, to the best of
our knowledge, that post-TBI treatment with RSG exerts
neuroprotective effects via the attenuation of neuronal apoptosis
and autophagy in the cortex, and these neuroprotective effects were
not mediated by GLT-1.
A previous study demonstrated that TBI initiates
physiopathological cascades of cell death signals and induces
numerous cell death pathways (20). Apoptosis is an important type of
programmed cell death that occurs following TBI (8). Caspase 3, which is regarded as an
effector caspase, may be activated via the amplification of
extrinsic or intrinsic apoptotic signals (9). Conversely, Bcl-2 is regarded as an
anti-apoptotic member of the Bcl-2 protein family, which has an
important role in the regulation of caspase-dependent and
caspase-independent apoptosis (10). The results of the present study
demonstrated that treatment with RSG resulted in decreased protein
expression levels of activated caspase 3, but increased protein
expression levels of Bcl-2 in the cortex following TBI. Previous
studies have suggested that treatment with RSG decreased the number
of apoptotic neurons following TBI (19), results which are confirmed by the
findings of the present study, which demonstrated that RSG is able
to exert neuroprotective effects via attenuation of TBI-induced
neuronal apoptosis.
A previous study demonstrated that autophagy is
activated in damaged brain tissue samples of numerous and distinct
animal brain injury models (21).
Erlich et al (22)
evaluated the effects of treatment with an autophagy agonist in a
closed head injury model. The results indicated that treatment with
rapamycin resulted in improved neurobehavioral function and
increased neuronal survival in the injured region (22). Conversely, numerous studies have
demonstrated that attenuation of TBI-induced neuronal autophagy
improved cognitive performance and reduced histological damage
(7,23). Therefore, whether the role of
autophagy is detrimental or beneficial following TBI remains
uncertain and controversial. Notably, in the present study,
treatment with RSG attenuated the TBI-induced elevated expression
levels of LC3 II and Beclin-1 in the brain cortex. It is therefore
conceivable to hypothesize that TBI overactivates neuronal
autophagy, which causes neuronal self-digestion and induces
neuronal cell death, and the neuroprotection of RSG may be
associated with the attenuation of TBI-induced over-activated
neuronal autophagy.
TNF-α and IL-6 are crucial proinflammatory cytokines
involved in TBI-induced inflammatory responses (24). A previous study demonstrated that
activated TNF-α and IL-6 expression in the initial post-injury
period is harmful, and attenuation of these cytokines may exert
neuroprotective effects following TBI (25). The results of the present study
demonstrated that RSG is able to downregulate the expression levels
of inflammatory cytokines TNF-α and IL-6 in the cortex following
TBI. These results are concordant with those of previous studies,
leading to the hypothesis that apoptotic and autophagic pathways
may be influenced by the downregulation of inflammatory cytokines.
In addition, numerous studies have confirmed the important role of
glutamate-mediated excitotoxicity in the pathophysiology of TBI
(26,27). In the central nervous system, the
activation of glutamate transporter systems, which promote
glutamate uptake, is the principle mechanism by which extracellular
glutamate concentrations are maintained below the level of
excitotoxicity (27). Among these
transporter mechanisms, GLT-1 is responsible for ~90% of all
glutamate transport in adult brain tissue (28). Therefore, the pharmacological
modulation of GLT-1 may provide novel therapeutic applications in
TBI. However, in the present study, no significant changes were
observed in the protein expression levels of GLT-1 in the rats
treated with RSG. The results of the present study suggested that
the neuroprotective effects of RSG are not mediated by the
modulation of GLT-1 expression in a rat model of TBI.
In conclusion, the present study demonstrated that
treatment with RSG reduced the levels of neuronal apoptosis and
autophagy, and increased the functional recovery in a rat model of
TBI. Furthermore, RSG may also decrease the protein expression
levels of TNF-α and IL-6, but no significant changes were observed
in the protein expression levels of GLT-1 in the cortex. These
results suggest that RSG may exert neuroprotective effects via the
reduction of neuronal apoptosis and autophagy following
experimental TBI in rats, and the mechanism underlying these
neuroprotective effects may be associated with the
anti-inflammatory effects of RSG.
Acknowledgments
The present study was supported by a grant from the
Natural Science Foundation of Hebei Province (grant no.
H2013201283)
References
|
1
|
Liu Y, Yi XC, Guo G, Long QF, Wang XA,
Zhong J, Liu WP, Fei Z, Wang DM and Liu J: Basic fibroblast growth
factor increases the transplantation-mediated therapeutic effect of
bone mesenchymal stem cells following traumatic brain injury. Mol
Med Rep. 9:333–339. 2014.
|
|
2
|
Greve MW and Zink BJ: Pathophysiology of
traumatic brain injury. Mt Sinai J Med. 76:97–104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gaetz M: The neurophysiology of brain
injury. Clin Neurophysiol. 115:4–18. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Headrick JP, Bendall MR, Faden AI and Vink
R: Dissociation of adenosine levels from bioenergetic state in
experimental brain trauma: Potential role in secondary injury. J
Cereb Blood Flow Metab. 14:853–861. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khan M, Im YB, Shunmugavel A, Gilg AG,
Dhindsa RK, Singh AK and Singh I: Administration of
S-nitrosoglutathione after traumatic brain injury protects the
neurovascular unit and reduces secondary injury in a rat model of
controlled cortical impact. J Neuroinflammation. 6:322009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cui C, Cui Y, Gao J, Sun L, Wang Y, Wang
K, Li R, Tian Y, Song S and Cui J: Neuroprotective effect of
ceftriaxone in a rat model of traumatic brain injury. Neurol Sci.
35:695–700. 2014. View Article : Google Scholar
|
|
7
|
Wang YQ, Wang L, Zhang MY, Wang T, Bao HJ,
Liu WL, Dai DK, Zhang L, Chang P, Dong WW, et al: Necrostatin-1
suppresses autophagy and apoptosis in mice traumatic brain injury
model. Neurochem Res. 37:1849–1858. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rink A, Fung KM, Trojanowski JQ, Lee VM,
Neugebauer E and McIntosh TK: Evidence of apoptotic cell death
after experimental traumatic brain injury in the rat. Am J Pathol.
147:1575–1583. 1995.PubMed/NCBI
|
|
9
|
Clark RS, Kochanek PM, Watkins SC, Chen M,
Dixon CE, Seidberg NA, Melick J, Loeffert JE, Nathaniel PD, Jin KL
and Graham SH: Caspase-3 mediated neuronal death after traumatic
brain injury in rats. J Neurochem. 74:740–753. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Graham SH, Chen J and Clark RS: Bcl-2
family gene products in cerebral ischemia and traumatic brain
injury. J Neurotrauma. 17:831–841. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Clark RS, Bayir H, Chu CT, Alber SM,
Kochanek PM and Watkins SC: Autophagy is increased in mice after
traumatic brain injury and is detectable in human brain after
trauma and critical illness. Autophagy. 4:88–90. 2008. View Article : Google Scholar
|
|
12
|
Cao Y and Klionsky DJ: Physiological
functions of Atg6/Beclin 1: A unique autophagy-related protein.
Cell Res. 17:839–849. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mohanty P, Aljada A, Ghanim H, Hofmeyer D,
Tripathy D, Syed T, Al-Haddad W, Dhindsa S and Dandona P: Evidence
for a potent antiinflammatory effect of rosiglitazone. J Clin
Endocrinol Metab. 89:2728–2735. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Risner ME, Saunders AM, Altman JF, Ormandy
GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA and Roses AD;
Rosiglitazone in Alzheimer's Disease Study Group: Efficacy of
rosiglitazone in a genetically defined population with
mild-to-moderate Alzheimer's disease. Pharmacogenomics J.
6:246–254. 2006.PubMed/NCBI
|
|
15
|
Schütz B, Reimann J, Dumitrescu-Ozimek L,
Kappes-Horn K, Landreth GE, Schürmann B, Zimmer A and Heneka MT:
The oral antidiabetic pioglitazone protects from neurodegeneration
and amyotrophic lateral sclerosis-like symptoms in superoxide
dismutase-G93A transgenic mice. J Neurosci. 25:7805–7812. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chaturvedi RK and Beal MF: PPAR: A
therapeutic target in Parkinson's disease. J Neurochem.
106:506–518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luo Y, Yin W, Signore AP, Zhang F, Hong Z,
Wang S, Graham SH and Chen J: Neuroprotection against focal
ischemic brain injury by the peroxisome proliferator-activated
receptor-gamma agonist rosiglitazone. J Neurochem. 97:435–448.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Q, Hu W, Meng B and Tang T: PPARγ
agonist rosiglitazone is neuroprotective after traumatic spinal
cord injury via anti-inflammatory in adult rats. Neurol Res.
32:852–859. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yi JH, Park SW, Brooks N, Lang BT and
Vemuganti R: PPARgamma agonist rosiglitazone is neuroprotective
after traumatic brain injury via anti-inflammatory and
anti-oxidative mechanisms. Brain Res. 1244:164–172. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stoica BA and Faden AI: Cell death
mechanisms and modulation in traumatic brain injury.
Neurotherapeutics. 7:3–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu M and Zhang HL: Death and survival of
neuronal and astrocytic cells in ischemic brain injury: A role of
autophagy. Acta Pharmacol Sin. 32:1089–1099. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Erlich S, Alexandrovich A, Shohami E and
Pinkas-Kramarski R: Rapamycin is a neuroprotective treatment for
traumatic brain injury. Neurobiol Dis. 26:86–93. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lai Y, Hickey RW, Chen Y, Bayir H,
Sullivan ML, Chu CT, Kochanek PM, Dixon CE, Jenkins LW, Graham SH,
et al: Autophagy is increased after traumatic brain injury in mice
and is partially inhibited by the antioxidant
gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab.
28:540–550. 2008. View Article : Google Scholar
|
|
24
|
Csuka E, Morganti-Kossmann MC, Lenzlinger
PM, Joller H, Trentz O and Kossmann T: IL-10 levels in
cerebrospinal fluid and serum of patients with severe traumatic
brain injury: Relationship to IL-6, TNF-alpha, TGF-beta 1 and
blood-brain barrier function. J Neuroimmunol. 101:211–221. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He J, Evans CO, Hoffman SW, Oyesiku NM and
Stein DG: Progesterone and allopregnanolone reduce inflammatory
cytokines after traumatic brain injury. Exp Neurol. 189:404–412.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Palmer AM, Marion DW, Botscheller ML,
Swedlow PE, Styren SD and DeKosky ST: Traumatic brain
injury-induced excitotoxicity assessed in a controlled cortical
impact model. J Neurochem. 61:2015–2024. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yi JH and Hazell AS: Excitotoxic
mechanisms and the role of astrocytic glutamate transporters in
traumatic brain injury. Neurochem Int. 48:394–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rao VL, Başkaya MK, Doğan A, Rothstein JD
and Dempsey RJ: Traumatic brain injury down-regulates glial
glutamate transporter (GLT-1 and GLAST) proteins in rat brain. J
Neurochem. 70:2020–2027. 1998.PubMed/NCBI
|