Introduction
With the aging population and improvement in
treatment for coronary heart disease, the prevalence and incidence
of chronic heart failure (CHF) has increase steadily (1), particularly in developing countries.
Heart failure is reported to have an annual mortality rate of ~10%
and a five-year mortality rate of ~50%, which results in a heavy
economic burden on families and society (2,3).
It is currently considered that myocardial energy
metabolic disorder may be one of the most important factors
preventing the success of heart failure treatment (4). The heart is arguably the most energy
consuming organ in the body and it has been demonstrated that heart
failure induces a reduction in energy production, promotes
ventricular remodeling and impairs mitochondrial function, which
further reduces energy production, leading to worsening heart
function and subsequently aggravating the heart failure (5,6).
In normal conditions, myocardial energy metabolism
includes three processes: Substrate utilization, oxidative
phosphorylation and adenosine triphosphate (ATP) transfer and
utilization, and changes in either process may affect myocardial
energy metabolism (7). When heart
failure occurs, glucose is the preferred substrate to free fatty
acids, resulting in an overall reduction of oxidative metabolism.
If changes in energy substrate utilization are inhibited at the
early stage of heart failure, ATP production may increase and the
progression of heart failure hindered (8). It is well-known that peroxisome
proliferator-activated receptors (PPARs) upregulate the expression
of β oxidase in myocardial cells, promote free fatty acid oxidation
and increase ATP production (9).
Therefore, activated PPAR inhibits cardiac hypertrophy, and PPAR
activators are able to enhance oxidation and utilization of free
fatty acids, preventing ventricular remodeling-induced
cardiomyopathy or heart failure (10).
PPAR is a ligand-activated nuclear transcription
factor, which belongs to the nuclear receptor superfamily, of which
three subtypes have been described: PPARα, PPARβ and PPARγ. PPARα
can be activated by synthetic ligands, including fibrates, and is
predominantly associated with lipid metabolism, affecting target
genes involved in fatty acid ω-and β-oxidation (11). Fenofibrate (FF), a clofibric acid
derivative lipid regulating drug, is an efficient PPARα ligand with
a long history of clinical use, reducing levels of low density
lipoprotein, cholesterol and triglycerides, and increasing
quantities of high-density lipoprotein (12).
During the process of heart failure, the expression
of uncoupling protein (UCP) in myocardial cells is upregulated
(13). This results in uncoupling
of oxidative phosphorylation and a decrease in ATP production,
further promoting the progression of heart failure. UCP2 is known
to transport protons into the matrix, preventing their use in ATP
synthesis, an effect that promotes free energy consumption of the
electrochemical proton gradient, oxidative phosphorylation
uncoupling and a reduction in ATP production (14). In addition, these events can reduce
calcium excess and mitochondrial membrane potential, thereby
inhibiting the production of reactive oxygen species (ROS)
(15).
Therefore, heart failure may be treated by improving
myocardial energy metabolism. In our previous study, an
isoproterenol (ISO)-induced heart failure rat model was established
(16). The present study aimed to
further investigate the effects of FF on heart failure and energy
metabolism, examine the underlying mechanisms using primary
cultured neonatal rat cardiomyocytes and analyze the associations
between PPARα, UCP2, myocardial energy metabolism and heart
failure. The data aimed to provide a solid basis for understanding
the effect of this drug class in the treatment of heart
failure.
Materials and methods
Rats
A total of 80 male Sprague-Dawley rats (weight,
200–250 g; ~10 weeks old) were provided by the Laboratory Animal
Center of the Medical College of Nanchang University (Jiangxi,
China). The rats were housed at 2–25°C in 40–80% humidity. The
lighting was maintained at 12 h light/dark. Male and female rats
were raised separately. The study procedures were reviewed and
approved by the Animal Ethics Committee of the 2nd Affiliated
Hospital of Nanchang University (Nanchang, China). All animals were
allowed free access to food and water.
Reagents
Horseradish peroxidase (HRP)-conjugated rabbit
anti-rat PPARα (cat. no. sc-130640) and goat anti-rat UCP2 (cat.
no. sc-390189) primary antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). HRP-conjugated goat
anti-rabbit (cat. no. sc-45101) and rabbit anti-goat (cat. no.
sc-358922) IgG secondary antibodies were provided by Beijing
Zhongshan Golden Bridge Biotechnology Co., Ltd. (Beijing,
China).
Establishment of the heart failure animal
model
The healthy inbred male SD rats were randomly
divided into four groups (n=10): CON (control), ISO, FF and FF+ISO
groups. The rats in the CON group were fed a regular diet. In the
ISO group, the rats were subcutaneously injected with 2.5 mg/kg ISO
(Shanghai Harvest Pharmaceutical Co., Ltd., Shanghai, China) for 4
weeks to induce heart failure (HF model group). In the FF group,
the animals were treated with 100 mg/kg/d FF (France Libofuni
pharmaceutical company, Libofuni, France) for 4 weeks. The rats in
FF+ISO group were pretreated with 100 mg/kg/d FF for 4 weeks prior
to the induction of heart failure, as described for the ISO group.
At 4 weeks post-ISO treatment, echocardiography and histopathology
examinations were performed to evaluate the heart failure model and
assess the effects of FF on heart failure. In addition, the heart
weight index was calculated to evaluate heart enlargement. The
serum B-type brain natriuretic peptide (BNP; Kaiji, Nanjing, China)
content in the three groups was also determined.
Echocardiography assessment of cardiac
function in rats
Cardiac function in rats was assessed by measuring
end-diastolic and end-systolic diameters, ejection fraction and
shortening fraction of the left ventricle. Prior to analysis, the
rats were anesthetized by intraperitoneal injection of 0.3% sodium
pentobarbital solution (10 ml/kg) purchased from Sigma–Aldrich
(Beijing, China). Ultrasonic detection of cardiac function
indicators in the rats was performed on a Siemens Acuson Sequoia
512 echocardiographic instrument (Siemens Healthcare, Shanghai,
China) with a 13 MHz frequency wire probe (Siemens Healthcare)
located near the sternum of the rat.
Preparation of myocardial pathological
sections
The rats were sacrificed via an intraperitoneal
injection of 0.3% pento-barbital sodium (10 ml/kg) and fixed on the
plate. The chest was opened and the heart was exposed in full. The
heart was cut open at the root of aorta and washed in cold saline.
The surrounding connective tissues and blood vessels were cut off.
The hearts were fixed with 10% formaldehyde solution (Jiancheng
Bioengineering Institute, Nanjing, China) and embedded in paraffin
(Jiancheng Bioengineering Institute). Using conventional methods,
the heart specimens were dehydrated and embedded in paraffin. From
the specimens, five slices (4 μm in thickness) were cut
along the cross section every other l mm in the long axis of the
left ventricle, submitted to hematoxylin and eosin staining
(Jiancheng Bioengineering Institute), and analyzed using optical
microscopy (BX51; Olympus Corporation, Tokyo, Japan).
Determination of heart weight index
The rats were weighed, as described above, and
fastened. Following sacrifice via intra-peritoneal injection of
0.3% pentobarbital sodium (10 ml/kg), the chest was cut open using
curved and straight scissors (325px; RWD Life Science, Shenzhen,
China). Following thoracotomy, the fully exposed hearts were cut
along aortic roots, and washed with precooled normal saline
solution to remove residual blood. Subsequently, the connective
tissues and blood vessels around the hearts were removed, and water
was filtered out of the organs. The extracted hearts were weighed
and the heart weight index was calculated for each, as a ratio of
heart to body weight.
Determination of serum BNP
Rat serum BNP was determined using an enzyme-linked
immunosorbent assay (ELISA) with a B-type BNP kit (Uscnk Life
Science, Inc., Wuhan, China), according to the manufacturer's
instructions. A 96-well ELISA plate was prepared by sequential
incubation with 50 μl anti-BNP antibodies, 100 μl
sample or standard and 50 μl tracer solution. The mixture
was dried and the plate was rinsed with phosphate-buffered saline
(PBS) five times, followed by sequential incubation with 100 ml
prepared HRP-streptavidin solution (Santa Cruz Biotechnology,
Inc.), 100 μl TMB One-Step Substrate reagent (Fengshou
Technology, Shanghai, China) and 100 μl stop solution.
Absorbance was read for samples and standards at 450 nm using an
enzyme labelling instrument (Synergy HT; BioTek Instruments, Inc.,
Winooski, VT, USA). A standard curve (S-type) was plotted with
known concentrations on the x-axis and measured optical density
values on the y-axis, on a semi-logarithmic scale. The quantities
of BNP in samples were derived from the standard curve.
Determination of free fatty acid content
in myocardial tissue and serum
Free fatty acid content was determined using
solid-phase sandwich ELISA with a Free Fatty Acids kit (Nanjing
Jiancheng Bioengineering Institute, Nanjing, China), according to
the manufacturer's instructions. The samples or standard (50
μl) were added to 96-well plates for 1 h, followed by
incubation with 50 μl biotin-labeled antibodies at 37°C for
1 h. Following washing in PBS, the cells were incubated with 80
μl HRP-streptavidin for 30 min at 37°C. Subsequently, 50
μl substrate A and 50 μl substrate B (mixed gently by
agitation) were added for detection at 37°C in the dark for 10 min.
Finally, 50 μl stop solution was added and the absorbance
was measured at 450 nm, as described above. A standard curve was
plotted to allow quantification of free fatty acids.
Determination of lactic acid and pyruvic
acid contents
The lactic acid and pyruvic acid contents were
determined using a Lactic Acid and Pyruvic Acid kit (Nanjing
Jiancheng Bioengineering Institute), according to the
manufacturer's instructions. Lactic acid and pyruvic acid were
measured at 530 and 505 nm, respectively, and the contents of
lactic acid and pyruvic acid in the samples were calculated as
follows:
Lactic acid content = (absorbance of sample -
absorbance of blank) / (absorbance of standard - absorbance of
blank) x standard concentration (3 μmol/ml).
Pyruvic acid content = (absorbance of sample -
absorbance of blank) / (absorbance of standard - absorbance of
blank) x standard concentration (0.2 μmol/ml).
Isolation, identification and in vitro
culture of myocardial cells
Myocardial tissue was collected from healthy male
Sprague-Dawley rats (1–3 days old), provided by the Laboratory
Animal Center of Jiangxi University of Traditional Chinese Medicine
(Jiangxi, China; 2010-0388). The rats were housed separately at
20–25°C in 60–70% humidity, and were sacrificed using
intraperitoneal injection of 0.3% pentobarbital sodium (10 ml/kg).
The chest was cut open using curved and straight scissors (325px;
RWD Life Science). The tissues were digested with trypsin and
collagenase, filtered and resuspended in Dulbecco's modified
Eagle's medium (DMEM). According to the instructions contained in
the Anti-α Skeletal Muscle Actin immunohistochemistry kit (Wuhan
Boster Biological Technology, Ltd., Wuhan, China), the cells were
labeled with anti-α skeletal muscle actin immunofluorescence at
confluency, in a state of synchronizing pulse, and were used for
myocardial beat detection under an inverted microscope.
Following isolation and purification, the myocardial
cells (1×106/ml) were incubated for 24 h, and randomly
divided into CON, ISO, FF and FF+ISO groups. In the ISO group, the
cells were incubated for 24 h with 100 μmol/l ISO. In the FF
group, the cells were treated with 10 μmol/l FF for 25 h. In
the FF+ISO group, the cells were pretreated with 10 μmol/l
FF for 1 h and cultured in the presence of 100 μmol/l ISO
for 24 h.
Fluorescence microscopy detection of
mitochondrial ROS generation in the in vitro cell culture
Intracellular ROS oxidize non-fluorescent
dichlorofluorescein (DCFH) into fluorescent DCF. Therefore, levels
of intracellular ROS can be determined by assessing DCF
fluorescence. In the present study, myocardial cells
(1×106/ml) were seeded into wells of a 96-well plate
(15,000 cells per well). Following innoculation, the cells were
washed twice with Eagle's solution containing sugar, and were
cultured with 25 μM DCFH-DA (Jiancheng Bioengineering
Institute) at 37°C for 30 mins. Following washes with Eagle's
solution with sugar, the ISO and ISO-FF were administered.
Fluorescence microscopy (excitation, 488 nm; emission, 525 nm; CHC,
Olympus Corporation) was utilized to assess intracellular ROS
levels.
Flow cytometric evaluation of cell
apoptosis and necrosis in vitro
Using an Annexin V-Fluorescein isothiocyanate (FITC)
Apoptosis Detection kit (Nanjing KGI Bio-tech. Co., Ltd., Nanjing,
China), according to the manufacturer's instructions, the
myocardial cells were digested with ethylene diamine tetraacetic
acid-free trypsin and washed twice with PBS. Subsequently,
1−5×105 cells were successively incubated with 500
μl binding buffer, 5 μl Annexin V-FITC and 5
μl propidium iodide, in the dark at room temperature for
5–15 min. Flow cytometry was then performed on a FACSCalibur (BD
Biosciences, San Jose, CA, USA).
Western blot analysis of the expression
levels of PPARα and UCP2
Once the culture medium was aspirated,
5–10×106 cells were collected with trypsin
(Sigma–Aldrich) and washed with PBS. Lysis buffer (0.5 ml;
Sigma–Aldrich), containing 50 mmol/l Tris HCl (pH 7.4), 150 mmol/l
NaCl, 1% Triton X-100, 1 mmol/l Na3VO4 and 1
mmol/l NaF, was added to the cells, and the protein contents were
determined using a Bicinchoninic Acid Protein Assay kit (Thermo
Fisher Scientific, Inc.). Equal quantities of protein (2 μg)
were separated by 5% SDS-PAGE (Gefan Biotechnology, Shanghai,
China) and transferred onto polyvinylidene fluoride membranes
(Sigma–Aldrich) by electroblotting. The membranes were immersed in
blocking buffer [5 g skimmed milk powder with 100 ml Tris-buffered
saline with 20% Tween (TBST)] and shaken for 1 h at room
temperature or remained static at 4°C overnight. The membranes were
then incubated with monoclonal rabbit anti-rat anti-PPARα (1:200;
Santa Cruz Biotechnology, Inc.; cat. no. sc-130640) and monoclonal
goat anti-rat UCP2 (1:500) primary antibodies (Santa Cruz
Biotechnology, Inc.; cat. no. sc-390189) for 2 h at room
temperature. The membranes were washed with TBST (Nanjing Jiancheng
Bioengineering Institute) for 10 mins three times, followed by
incubation with anti-rabbit (Santa Cruz Biotechnology, Inc.; cat.
no. sc-45101) and anti-goat IgG (Santa Cruz Biotechnology, Inc.;
cat. no. sc-358922) secondary antibodies conjugated to HRP
(1:1,000) for 2 h at room temperature. The membranes were then
washed with TBST for 10 mins three times and the target bands were
analyzed using a gel imaging system (Thermo Fisher Scientific,
Inc., Waltham, MA, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Data analysis was performed using the Statistical Package for the
Social Sciences (SPSS) 18.0 software (SPSS, Inc., Chicago, IL,
USA). One way analysis of variance (was used for intergroup
comparisons and P<0.05 was considered to indicate a
statistically significant difference.
Results
FF effectively alleviates ISO-induced
heart failure in rats
Daily subcutaneous injections of ISO for 4 weeks
significantly reduced myocardial systolic function and enlarged
ventricles in the rats. In addition, ISO treatment resulted in
cardiomyocyte hypertrophy, with necrosis and hyperplasia of
myocardial interstitial connective tissue observed. Following 4
weeks of ISO administration, the rats exhibited loss of appetite,
had dry and yellowish fur and exhibited manifestations of heart
failure, including slow weight increase, breathing difficulties and
reduced activity.
Echocardiography data revealed that, compared with
the control animals, left ventricular diameter and systolic
function in the ISO group were significantly enlarged and
decreased, respectively (Table I).
End-diastolic and end-systolic diameters, ejection fraction and
shortening fraction of the left ventricle were all significantly
improved in the FF+ISO group, compared with the ISO group. No
statistical significant differences were observed in the values
obtained for the FF group, compared with the control animals
(Table I).
 | Table IEchocardiographic data of rats
following treatment. Indicators of cardiac function of rats in the
CON, FF, ISO and FF+ISO groups were detected by
echocardiography. |
Table I
Echocardiographic data of rats
following treatment. Indicators of cardiac function of rats in the
CON, FF, ISO and FF+ISO groups were detected by
echocardiography.
| Group | n | DD (mm) | SD (mm) | EF (%) | FS (%) |
|---|
| CON | 10 | 4.64±0.38 | 2.68±0.30 | 81.40±4.98 | 47.00±5.00 |
| FF | 10 | 4.69±0.33 | 2.71±0.29 | 79.84±5.76 | 47.43±4.26 |
| ISO | 10 | 6.46±0.58a | 4.74±0.29a | 54.20±5.40a | 28.60±3.05a |
| FF+ISO | 10 |
5.44±0.36b,d |
3.66±0.43a,c |
71.60±2.70a,c |
35.20±3.49a,d |
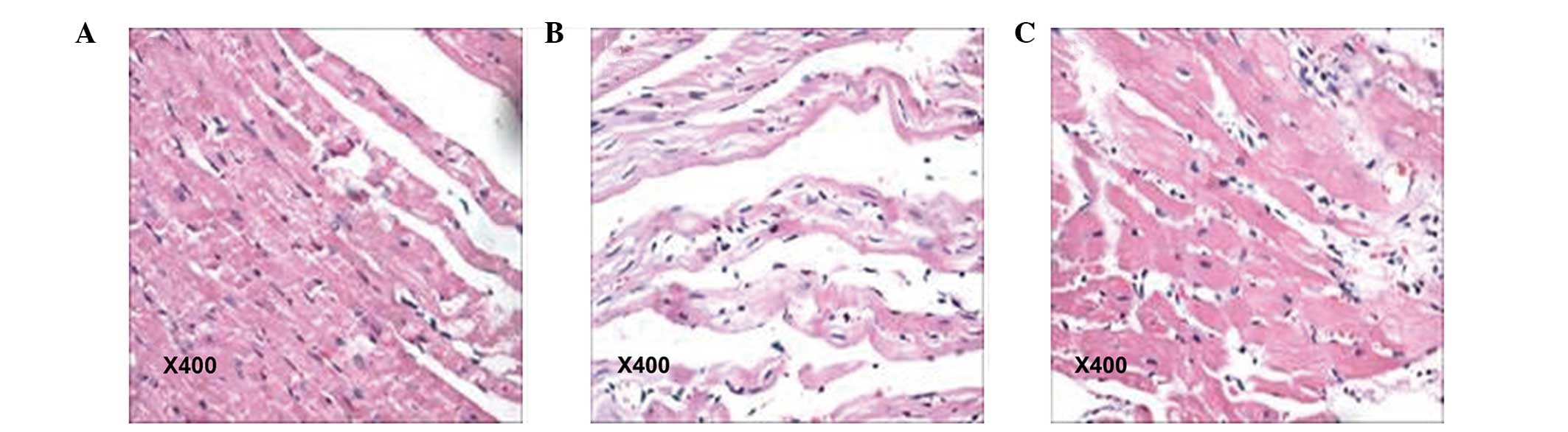
Gross examination revealed a dark purple heart
surface in the rats of the ISO group, whereas the hearts of the
control animals was ruddy. Following analysis under microscopy,
cardiac sections from the ISO group exhibited overt hypertrophy,
acidophilic degeneration or necrosis of a number of myocardial
cells. In addition, focal or diffuse lesions, predominantly
distributed in the left ventricular wall, were observed in these
animals, with certain rats exhibiting cord-like hyperplasia of
myocardial interstitial connective tissue. The pathological damage
observed in the myocardial tissues was significantly reduced in the
FF+ISO group, compared with animals treated with ISO only. As
expected, the myocardial cells were of normal size and neatly
arranged in the control group (Fig.
1). These data demonstrated that FF reversed the ISO-induced
myocardial damage in rats.
The heart weight index, defined as the ratio of
heart to body weight, was higher in the ISO and FF+ISO groups than
in the control group. However, the heart weight index was lower in
FF+ISO group than the ISO group, indicating a relief of the
ISO-induced heart enlargement by FF. A similar trend was observed
in the serum levels of BNP, which were significantly higher in the
ISO and FF+ISO groups, compared with the control, but significantly
reduced in the FF+ISO group, compared with the ISO group (Table II). Of note, similar values were
obtained in the CON and FF groups for heart weight index and serum
levels of B-type BNP (Table
II).
| Table IIHWI and serum levels of BNP in rats
following treatment. |
Table II
HWI and serum levels of BNP in rats
following treatment.
| Group | n | HWI (mg/g) | BNP (ng/ml) |
|---|
| CON | 10 | 2.13±0.15 | 0.39±0.03 |
| FF | 10 | 2.11±0.13 | 0.40±0.05 |
| ISO | 10 | 3.58±0.08a | 4.86±0.12a |
| FF+ISO | 10 |
3.16±0.12b,c |
1.79±0.09a,c |
FF effectively alleviates ISO-induced
metabolic abnormalities in a rat model of heart failure
Free fatty acid levels in the serum and myocardial
tissue, and lactate acid and pyruvic acid contents in the
myocardial tissue were determined to assess fatty acid and
carbohydrate metabolism in rats following treatment. Free fatty
acids reflect the efficiency of fatty acid metabolism, with high
contents indicating low metabolic efficiency. In the present study,
free fatty acid contents in the serum and myocardial tissue were
higher in the ISO group, compared with the FF+ISO group, with
control animals exhibiting the lowest values. No statistically
significant differences were observed between the CON and FF groups
(Table III). These findings were
in agreement with the histological data, and provided evidence
supporting fatty acid metabolic dysfunction in ISO-induced heart
failure rats, as well as effective improvement in fatty acid
metabolism following FF treatment.
| Table IIIFFA content in the serum and
myocardial tissues of the rats. |
Table III
FFA content in the serum and
myocardial tissues of the rats.
| Group | n | FFA in serum
(μmol/l) | FFA in myocardial
tissue (μmol/gprot) |
|---|
| CON | 10 | 480.32±44.71 | 493.74±17.72 |
| FF | 10 | 483.97±45.81 | 497.64±20.55 |
| ISO | 10 |
782.76±65.98a |
650.42±54.38a |
| FF+ISO | 10 |
596.62±112.18b,c |
565.70±57.55b,d |
The levels of lactic acid and pyruvic acid, glucose
metabolism intermediates, were detected using spectrophotometry.
The data revealed that lactic acid and pyruvic acid were higher in
content in the myocardial tissues obtained from the ISO group,
compared with those from the FF+ISO group, with the control group
animals exhibiting the lowest levels of these metabolites (Table IV). These findings indicated an
increase in anaerobic glycolysis in the ISO-induced heart failure
rats, which was altered by FF treatment. As shown in Table IV, animals in FF and CON groups
exhibited similar values for all the parameters investigated.
| Table IVLactate and pyruvic acid contents in
myocardial tissue of rats and in supernatants collected from in
vitro cultured cardiac cells. |
Table IV
Lactate and pyruvic acid contents in
myocardial tissue of rats and in supernatants collected from in
vitro cultured cardiac cells.
| Group | n | In vivo
myocardial tissue
| In vivo
cultured cardiac cells
|
|---|
| Lactic acid
(mmol/gprot) | Pyruvic acid
(μmol/gprot) | Lactic acid
(μmol/l) | Pyruvic acid
(nmol/l) |
|---|
| CON | 10 | 0.22±0.34 | 0.26±0.01 | 4.90±0.05 | 212.25±0.86 |
| FF | 10 | 0.43±0.25 | 0.27±0.03 | 4.89±0.14 | 216.40±6.11 |
| ISO | 10 | 0.66±0.65a | 1.17±0.09a | 10.57±1.66a | 549.63±2.41a |
| FF+ISO | 10 |
0.37±0.52a,c |
0.62±0.01a,b |
6.88±0.56a,b |
335.49±1.82a,b |
FF inhibits ISO-induced increases of
anaerobic glycolysis, ROS levels and cell necrosis in vitro
In order to examine the mechanisms underlying the
effects of FF on heart failure, the present study isolated,
purified and identified neonatal rat myocardial cells for in
vitro investigations. Significantly reduced lactic acid and
pyruvic acid contents were found in the FF+ISO group, compared with
the ISO group (P<0.05), with the control animals exhibiting the
lowest values (Table IV). The
high lactic acid and pyruvic acid contents indicated that ISO
induced abnormal glucose metabolism and anaerobic glycolysis in the
in vitro myocardial cell culture, the effects of which were
inhibited by treatment with FF. The cells treated with FF alone
exhibited similar lactic acid and pyruvic acid contents as detected
in the control cells (Table
IV).
To investigate whether abnormal cellular energy
metabolism results in oxidative phosphorylation in mitochondria and
causes abnormal ATP production, the present study subsequently
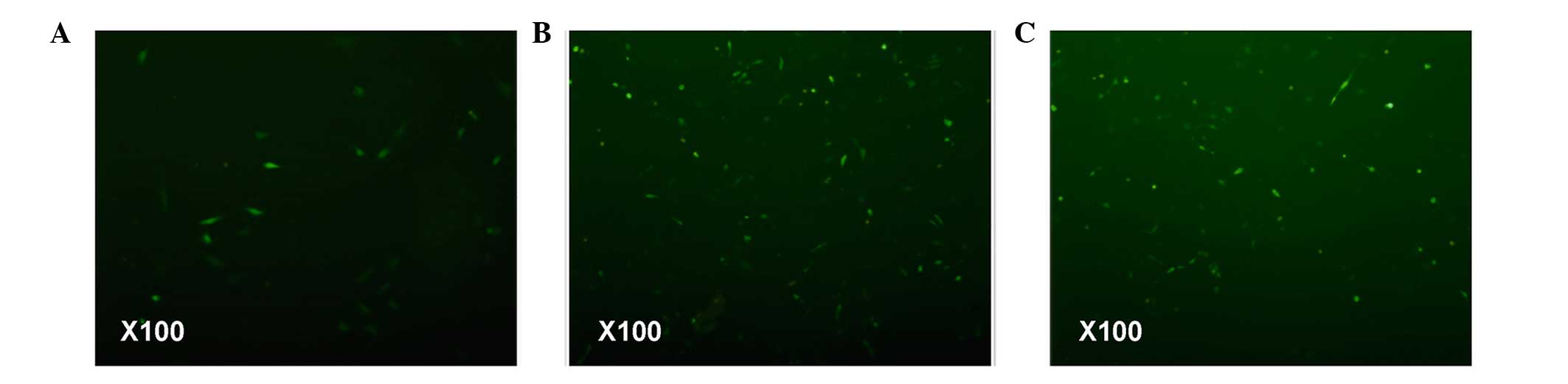
examined mitochondrial ROS. The accumulation of ROS indicates
decreased oxidative phosphorylation, which promotes the enhancement
of mitochondrial membrane potential. The fluorescence microscopy
data revealed that the ISO-treated cardiomyocytes were more
fluorescent, indicating high ROS levels in this group (Fig. 2). Notably, FF pretreatment
decreased myocardial cell fluorescence. However, the fluorescence
intensity observed in the myocardial cells of the FF+ISO group
remained higher, compared with hat of the control cells. These
findings suggested that FF reversed the ISO-induced decrease of
oxidative phosphorylation.
ROS accumulation and mitochondrial activation can
cause cell apoptosis or necrosis. The flow cytometry data
demonstrated that treatment with ISO resulted in marked cell death,
and this effect was markedly reduced following FF pretreatment.
These findings suggested that ISO-induced abnormal energy
metabolism ultimately resulted in cell necrosis, and that FF
treatment restored energy metabolism and reduced cell necrosis
(Fig. 3).
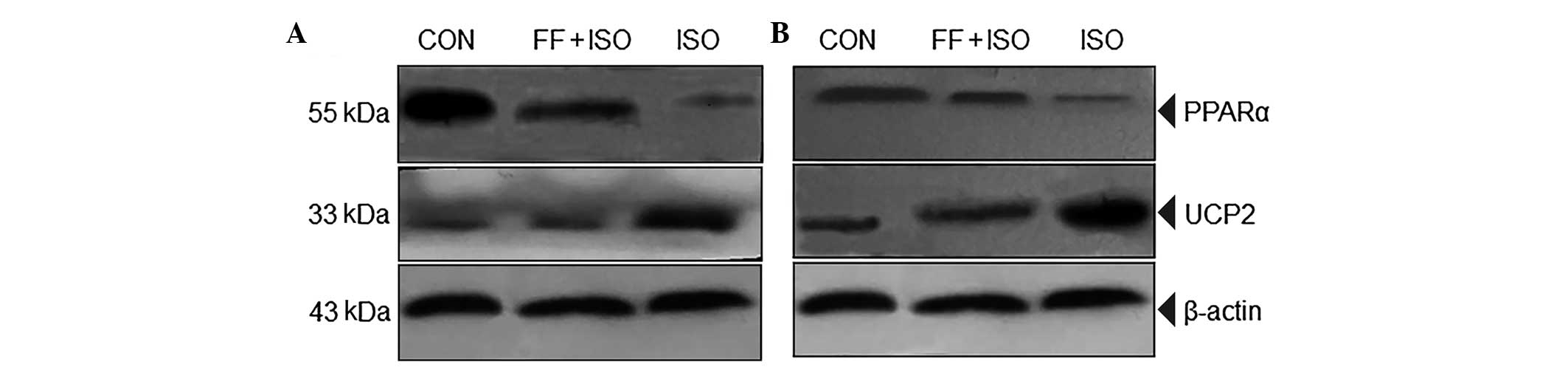
ISO and FF regulate the expression levels
of PPARα and UCP2
To understand the mechanisms by which FF alleviates
ISO-induced cell energy metabolic disorder, necrosis and heart
failure in rats, the present study assessed the expression levels
of PPARα and UCP2 using western blot analysis. The expression of
PPARα was significantly increased and that of UCP2 was
significantly reduced in the myocardial cells of the FF+ISO group,
compared with the ISO groups, in vitro (Fig. 4A) and in vivo (Fig. 4B). These data demonstrated that FF,
a PPARα ligand, may promote the oxidation of fatty acids by
β-oxidase by PPARα activation, therefore providing energy for
myocardial cells. In addition, FF may downregulate the ISO-induced
expression of UCP2, inhibit the uncoupling of oxidative
phosphorylation caused by ISO and provide more ATP or energy in
myocardial cells.
Discussion
Heart failure is a major cause of disability and
mortality, with a five-year mortality rate as high as 50% (17). Its predominant etiologic mechanism
includes myocardial energy metabolic disorder (16). Even for a healthy heart, energy
substrates can only provide ~25% energy, far less than the body
demands. Thus, a small variation in the efficiency of energy
production and utilization can significantly affect myocardial
energy levels (18).
In order to understand the occurrence of heart
failure and the mechanisms underlying the therapeutic effects of FF
in the treatment of heart failure, the present study established
and investigated a heart failure rat model, in vivo and
in vitro, for energy utilization. It is suggested that heart
failure may be accompanied by excessive activation of the
sympathetic nervous system and increased relative myocardial
ischemia and hypoxia, which is possibly associated with
catecholamine-induced lipid peroxidation, membrane permeability
changes and intracellular calcium overload (19). Large doses of ISO in rats results
in diffuse or focal myocardial necrosis, cardiac enlargement,
calcium accumulation in myocardial tissue and enhancement of lipid
peroxidation (20). In this study,
daily subcutaneous 2.5 mg/kg doses of ISO were injected each day
into rats for 4 weeks. At the end of treatment, the survival rate
was high and the majority of the rats exhibited heart failure
symptoms, including yellowish hair, appetite loss, slow weight
increase, breathing accompanied by wheezing and reduced activity.
Cardiac function, heart weight index and serum levels of BNP in
rats were significantly altered, suggesting a successful rat model
of heart failure.
During the occurrence and development of CHF,
substrates for myocardial energy metabolism change from fatty acids
to glucose, resulting in increased consumption of ATP. However, the
decrease in fatty acid oxidation cannot be fully compensated by the
enhanced glucose oxidation. This results in insufficient energy
production and further aggravates heart failure (8). Therefore, CHF mechanisms may include
changes in mitochondrial energy source, depending on the expression
of enzymes in the fatty acid oxidation pathway (β-oxidation is
reduced). It has been demonstrated that the reduction of fatty acid
utilization in myocardial cells with heart failure may be
associated with decreased expression of β-oxidase (21,22).
β-oxidase promotes β-oxidation of fatty acids in the mitochondria
of myocardial cells, resulting in increased ATP, and activated
PPARα regulates myocardial fatty acid β-oxidase and promotes fatty
acid uptake, activation and metabolism in myocardial cells
(23). However, during heart
failure, the expression of PPARα is also decreased in the
mitochondria of myocardial cells, (24). which may be a major reason why the
myocardium changes its preferred energy source from fatty acid to
glucose.
The present study revealed decreased expression
levels of PPARα in the ISO and FF+ISO groups, compared with the
normal myocardial tissue. Of note, the ISO group exhibited the
lowest expression of this enzyme. In contrast, fatty acid contents
in the serum and myocardial tissue were highest in the ISO group
and lowest in the control group, with the FF+ISO group between.
These data suggested that the decreased expression of PPARα in
myocardial tissue during heart failure in rats of the ISO group
reduced PPARα activity. Notably, the data revealed significantly
improved cardiac function, as assessed by echocardiography,
myocardial biopsy and serum BNP content in the FF+ISO group. These
findings indicated that FF, as a highly selective PPARα agonist,
may increase fatty acid β-oxidation enzymes by activating PPARα,
promoting fatty acid oxidation in the mitochondria, regulating
myocardial energy metabolism, and improving ventricular
remodeling.
Uncoupling proteins (UCPs) are constituents of the
mitochondrial inner membrane, of which they can lower the
electrochemical gradient by promoting H+ permeability
between the mitochondrial inner membrane and substrate, and
therefore, decrease ATP production. The UCP family includes UCP1-5,
UCP2 and UCP3, which are predominantly expressed in the
cardiovascular system (25), and
the expression of UCP2 has been observed to increase when heart
failure occurs. Noma et al (26) reported increased expression of UCP2
and decreased ATP production with the progression of heart failure
in a mouse model of aortic regurgitation, indicating that
UCP2-induced ATP deficiency is a predominant cause of heart
failure.
In addition, previous studies have demonstrated that
the myocardial protein expression of UCP2 is positively correlated
with serum free fatty acid content, and it is known that high
plasma free fatty acid content can promote the myocardial
expression of UCP2 (27).
The data of the present study demonstrated that
serum free fatty acid content was reduced and myocardial UCP2
protein was downregulated in the FF+ISO group, compared with the
ISO group, however, ventricular remodeling and cardiac function
were significantly improved in the FF+ISO group in comparison with
the ISO group. Therefore, in agreement with our previous work
(28,29), this data indicated that the
regulation of the expression of UCP2 may be involved in the
mechanism by which lipid-lowering drugs relieve heart failure.
In the present study, only one dose of FF was
examined in the in vivo and in vitro experiments, and
achieved partial inhibition of the effects of ISO. The hemodynamic
effects of FF are not clear, as data regarding the blood pressure
and heart rates of the rats are limited. Future investigations are
required to assess higher, but not toxic doses, also in other
animal models.
Overall, the in vivo and in vitro
results of the present study demonstrated that FF efficiently
alleviated ISO-induced HF in rats, which was possibly due to the
promotion of fatty acid oxidative metabolism. In addition,
UCP2-induced uncoupling of oxidative phosphorylation may be
involved in the FF effect.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (grant no, 1140019), the
Natural Science Foundation of JiangXi Province (grant no.
20122BAB205007) and the Science and Technology Support Project of
Nanchang (grant no. 49-10).
References
|
1
|
Stewart S, MacIntyre K, Capewell S and
McMurray JJ: Heart failure and the aging population: An increasing
burden in the 21st century? Heart. 89:49–53. 2003. View Article : Google Scholar
|
|
2
|
Bettari L, Fiuzat M, Felker GM and
O'Connor CM: Significance of hyponatremia in heart failure. Heart
Fail Rev. 17:17–26. 2012. View Article : Google Scholar
|
|
3
|
Dayer M and Cowie MR: Heart failure:
Diagnosis and healthcare burden. Clin Med. 4:13–18. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sinatra ST: Metabolic cardiology: The
missing link in cardiovascular disease. Altern Ther Health Med.
15:48–50. 2009.PubMed/NCBI
|
|
5
|
Herrmann G and G D: The chemical nature of
heart failure. Ann Intern Med. 12:1233–1244. 1939. View Article : Google Scholar
|
|
6
|
Neubauer S: The failing heart-an engine
out of fuel. N Engl J Med. 356:1140–1151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
J I: ATP and the heart [M]. Kluwer
academic publishers USA; pp. 197–216. 2002
|
|
8
|
Nagoshi T, Yoshimura M, Rosano GMC,
Lopaschuk GD and Mochizuki S: Optimization of cardiac metabolism in
heart failure. Curr Pharm Des. 17:3846–3853. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim T and Yang Q:
Peroxisome-proliferator-activated receptors regulate redox
signaling in the cardiovascular system. World J Cardiol. 5:164–174.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Planavila A, Rodriguez-Calvo R, Jové M,
Michalik L, Wahli W, Laguna JC and Vázquez-Carrera M: Peroxisome
proliferator-activated receptor beta/delta activation inhibits
hypertrophy in neonatal rat cardiomyocytes. Cardiovasc Res.
65:832–841. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Umemoto T and Fujiki Y: Ligand-dependent
nucleo-cytoplasmic shuttling of peroxisome proliferator-activated
receptors, PPAR α and PPAR γ. Genes Cells. 17:576–596. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Scott R, O'Brien R, Fulcher G, Pardy C,
D'Emden M, Tse D, Taskinen MR, Ehnholm C and Keech A; Fenofibrate
Intervention Event Lowering in Diabetes (FIELD) Study
Investigators: Effects of fenofibrate treatment on cardiovascular
disease risk in 9,795 individuals with type 2 diabetes and various
components of the metabolic syndrome: The fenofibrate intervention
and event lowering in diabetes (FIELD) study. Diabetes Care.
32:493–498. 2009. View Article : Google Scholar :
|
|
13
|
Teshima Y, Akao M, Jones SP and Marbán E:
Uncoupling protein-2 overexpression inhibits mitochondrial death
pathway in cardiomyocytes. Circ Res. 93:192–200. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Echtay KS: Mitochondrial uncoupling
proteins - what is their physiological role? Free Radic Biol Med.
43:1351–1371. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Deng S, Yang Y, Han Y, Li X, Wang X, Zhang
Z and Wang Y: UCP2 inhibits ROS-mediated apoptosis in A549 under
hypoxic conditions. PLoS One. 7:e307142012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ingwall JS: Energy metabolism in heart
failure and remodelling. Cardiovasc Res. 81:412–419. 2009.
View Article : Google Scholar :
|
|
17
|
Askoxylakis V, Thieke C, Pleger ST, Most
P, Tanner J, Lindel K, Katus HA, Debus J and Bischof M: Long-term
survival of cancer patients compared to heart failure and stroke: A
systematic review. BMC Cancer. 10(105)2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Joiner ML, Koval OM, Li J, He BJ,
Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, et al:
CaMKII determines mitochondrial stress responses in heart. Nature.
491:269–273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Andalib S, Shayanfar A, Khorrami A,
Maleki-Dijazi N and Garjani A: Atorvastatin reduces the myocardial
content of coenzyme Q10 in isoproterenol-induced heart failure in
rats. Drug Res (Stuttg). 64:246–250. 2014.
|
|
20
|
Heymes C, Bendall JK, Ratajczak P, Cave
AC, Samuel JL, Hasenfuss G and Shah AM: Increased myocardial NADPH
oxidase activity in human heart failure. J Am Coll Cardiol.
41:2164–2171. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Burkart EM, Sambandam N, Han X, Gross RW,
Courtois M, Gierasch CM, Shoghi K, Welch MJ and Kelly DP: Nuclear
receptors PPARbeta/delta and PPARalpha direct distinct metabolic
regulatory programs in the mouse heart. J Clin Invest.
117:3930–3939. 2007.PubMed/NCBI
|
|
22
|
Wolins NE, Quaynor BK, Skinner JR, Tzekov
A, Croce MA, Gropler MC, Varma V, Yao-Borengasser A, Rasouli N,
Kern PA, et al: OXPAT/PAT-1 is a PPAR-induced lipid droplet protein
that promotes fatty acid utilization. Diabetes. 55:3418–3428. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sack MN, Rader TA, Park S, Bastin J,
McCune SA and Kelly DP: Fatty acid oxidation enzyme gene expression
is downregulated in the failing heart. Circulation. 94:2837–2842.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garnier A, Fortin D, Deloménie C, Momken
I, Veksler V and Ventura-Clapier R: Depressed mitochondrial
transcription factors and oxidative capacity in rat failing cardiac
and skeletal muscles. J Physiol. 551:491–501. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Murray AJ, Anderson RE, Watson GC, Radda
GK and Clarke K: Uncoupling proteins in human heart. Lancet.
364:1786–1788. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Noma T, Nishiyama A, Mizushige K, Murakami
K, Tsuji T, Kohno M, Rahman M, Fukui T, Abe Y and Kimura S:
Possible role of uncoupling protein in regulation of myocardial
energy metabolism in aortic regurgitation model rats. FASEB J.
15:1206–1208. 2001.PubMed/NCBI
|
|
27
|
Young ME, Patil S, Ying J, Depre C, Ahuja
HS, Shipley GL, Stepkowski SM, Davies PJ and Taegtmeyer H:
Uncoupling protein 3 transcription is regulated by peroxisome
proliferator-activated receptor (alpha) in the adult rodent heart.
FASEB J. 15:833–845. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li P, Yin K, Luo SK and Cheng XS: Effect
of uncoupling protein-2 on angiotensin II induced reactive oxygen
species generation in mitochondria. Chinese Journal of
Hypertension. 18:940–945. 2010.In Chinese.
|
|
29
|
Luo SK, Li P and Cheng XS: Establishment
of model of isoprenaline-induced chronic heart failure in rats.
Chongqing Yixue. 41:352–354. 2012.
|