Introduction
Hepatocellular carcinoma (HCC) is the sixth most
prevalent type of cancer and the third most frequent cause of
cancer-related mortality (1). In
2014, the estimated number of novel cases and estimated number of
fatalities of the disease in the United States were 33,190 and
23,000, respectively (2). The most
common risk factors of HCC are chronic hepatitis B (HBV) and C
(HCV) viral infection, chronic alcohol consumption and
aflatoxin-B1-contaminated food (3–6). HCC
often develops during the advanced stages of liver fibrosis and is
the leading cause of mortality among patients with cirrhosis
(7). Patients with cirrhosis are
at the highest risk of developing HCC and should be monitored every
6 months (8). Hepatocarcinogenesis
is a complex multistep process in which numerous signaling cascades
are altered, leading to a heterogeneous molecular profile. The
molecular analysis of human HCC has shown a number of genetic and
epigenetic alterations that result in the deregulation of key
oncogenes and tumor-suppressor genes, including TP53, β-catenin,
ErbB receptor family members, MET and its ligand hepatocyte growth
factor (HGF), p16 (INK4a), E-cadherin and cyclooxygenase 2 (COX2)
(9). Despite this progress, the
neoplastic evolution of HCC remains to be defined.
Mitogen-activated protein kinases (MAPKs) are
signaling components that are important in converting extracellular
stimuli into a wide range of cellular responses. ERK1/2 MAPKs are
preferentially activated in response to growth factors and were
found to be upregulated in human tumors (10). p38 kinases are more responsive to
stress stimuli ranging from osmotic shock and ionizing radiation to
cytokine stimulation and have been found to be involved in
inflammation, cell growth, cell differentiation, the cell cycle,
and cell death (11,12). Moreover, p38 MAPK family members
and their isoforms have been reported as tumor suppressors or
oncoproteins in specific cell types (13,14).
In addition, several negative regulators of p38 MAPK signaling have
been found to be overexpressed in human tumors and cancer cell
lines, including the phosphatases PPM1D and DusP26, and the
inhibitors of the MAP3K apoptosis signal regulating kinase 1
(AsK1), glutathione S-transferase Mu 1 (gsTM1) and gsTM2 (13).
Each family of MAPKs is composed of a set of three
evolutionarily conserved sequentially acting kinases: An MAPK, an
MAPK kinase (MAPKK), and an MAPKK kinase (MAPKKK) (15). As to p38 (MAPK), MKK3 and MKK4 are
two MAPK kinases. A recent study demonstrated its downregulated in
HCC compared with normal liver tissue (16). However, whether it functions as a
tumor suppressor in HCC is unclear. Therefore, in the present
study, the role of MKK3 in HCC cell lines was investigated to
determine whether MKK3 acts as a tumor suppressor in HCC, compared
with normal liver tissue.
Materials and methods
Cell culture, reagents and plasmids
The HepG2 and PLC-PRF-5 hepatocellular carcinoma
cell lines were purchased from the American Type Culture Collection
(Manassas, VA, USA). All hepatocellular carcinoma cell lines were
maintained in Eagle's Minimum Essential Medium (EMEM) supplemented
with 10% fetal bovine serum (FBS). p38 inhibitor, SB203580, was
obtained from Sigma-Aldrich (St. Louis, MO, USA). The MKK3
expression plasmid (plasmid 14671) was obtained from Addgene
(Cambridge, MA, USA). Transient transfection was performed using
the Lipofectamine 2000 reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
For total RNA extraction, samples were processed
using the RNeasy Mini kit (Qiagen, Hilden, Germany) according to
manufacturer's instructions. RNA (1 µg) was
reverse-transcribed using M-MLV Reverse Transcriptase and oligo-dT
primers (Invitrogen Life Technologies). Target genes and controls
were analyzed by RT-qPCR using a StepOnePlus™ Real-Time PCR System
(Invitrogen Life Technologies) and SYBR® Select Master
mix (Invitrogen Life Technologies). β-actin was used as control;
fold changes were calculated using the ΔΔCt method in Microsoft
Excel. Primer sequences were as follows: Forward:
CTTGGTGACCATCTCAGAACTGG and reverse: CTTCTGCTCCTGTGAGTTCACG for
MKK3 and forward: CGTGACATTAAGGAGAAGCTG and reverse:
CTAGAAGCATTTGCGGTGGAC for β-actin.
Immunoblot assay
Whole-cell extracts were obtained by lysis of cells
in ice-cold radioimmunoprecipitation assay (RIPA) buffer. Cell
lysates were separated on 10% SDS denatured polyacrylamide gel
electrophoresis gels (Beyotime Institute of Biotechnologies,
Haimen, China), transferred to nitrocellulose membranes (EMD
Millipore, Temecula, CA, USA) and blocked in phosphate-buffered
saline/Tween-20 containing 5% non-fat milk. Membranes were
incubated with dilutions of primary antibodies against MKK3 (rabbit
anti-human monoclonal antibody; cat. no. 8355; 1:1,000), Bmi-1
(rabbit anti-human monoclonal antibody; cat. no. .2830, 1:1000),
cyclin D1 (rabbit anti-human monoclonal antibody, cat. no. 2978;
1:1,000), cyclin E (mouse anti-human monoclonal antibody; cat. no.
4129; 1:1,000), p21 Cip1 (rabbit anti-human monoclonal antibody,
Cat. 2947, 1:1,000), p27 Kip1 (Rabbit anti-human monoclonal
antibody; cat. no. 3686, 1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA), p19 INK4D (rabbit anti-human polyclonal
antibody; cat. no. ab80; 1:1,000), p18 INK4C (rabbit anti-human
polyclonal antibody; cat. no. ab192239; 1:1,000), p16 INK4A (rabbit
anti-human polyclonal antibody; cat. no. ab108349; 1:3,000), p15
INK4B (rabbit anti-human polyclonal antibody,; cat. no. ab126625;
1:1,000; Abcam, Cambridge, UK) and tubulin (mouse anti-human
polyclonal antibody; cat. no. T6199; 1:10,000; Sigma-Aldrich),
followed by incubation with horseradish peroxidase-conjugated
secondary antibodies. After extensive washing, the targeted
proteins were visualized by enhanced chemiluminescence and exposure
to film (Fujifilm, Tokyo, Japan).
Cell cycle staging analysis
Cell cycle staging was analyzed by propidium iodide
(PI) staining (BD Biosciences, Franklin Lakes, NJ, USA). Cells were
treated with 1 mg/ml RNase A (BD Biosciences), fixed with 70%
ethanol and then labeled with 20 mg/ml PI solution. The DNA content
of cells was measured by flow cytometry (BD FACS Calibur, BD
Biosciences, Franklin Lakes, NJ, USA). The proportions of cells in
the G1, S, and G2/M phases were analyzed using ModFit Software
(version 4.0; Verity Software House, Topsham, ME, USA).
MTS assay
HCC cells transfected with either MKK3
overexpression plasmid or vector were seeded in 96-well plates. 12
h later, MTS reagent (Promega Corporation, Madison, WI, USA) was
added at a 1:10 dilution. Plates were read at 450 nm using an
ELx800 microplate reader (BioTek Instruments, Inc.) 90 min later.
The absorbance was recorded at day 0. Then at 24, 48 and 72 h,
absorbance was also determined and recorded as day 1, 2 and 3,
respectively.
BrdU incorporation and anaphase
assay
The BrdU incorporation and anaphase assay were
performed as a proliferation indicator. For the BrdU incorporation
assay, a Cell Proliferation ELISA kit (Roche Diagnostics, Manheim,
Germany) was applied and measurements were performed according to
the manufacturer's instructions. For the anaphase assay, the number
of cells, and the number of cells in anaphase were detected using
DAPI and were counted in five visual fields per well.
Statistical analysis
GraphPad Prism 6.0 software (GraphPad Software,
Inc., La Jolla, CA, USA) was used for all statistical analysis. A
two-tailed unpaired Student's t test was used for statistical
evaluation of data. P<0.05 was considered to indicate a
statistically significant difference. Data are expressed as the
mean ± standard deviation.
Results
MKK3 suppresses HepG2 and PLC-PRF-5 cell
proliferation
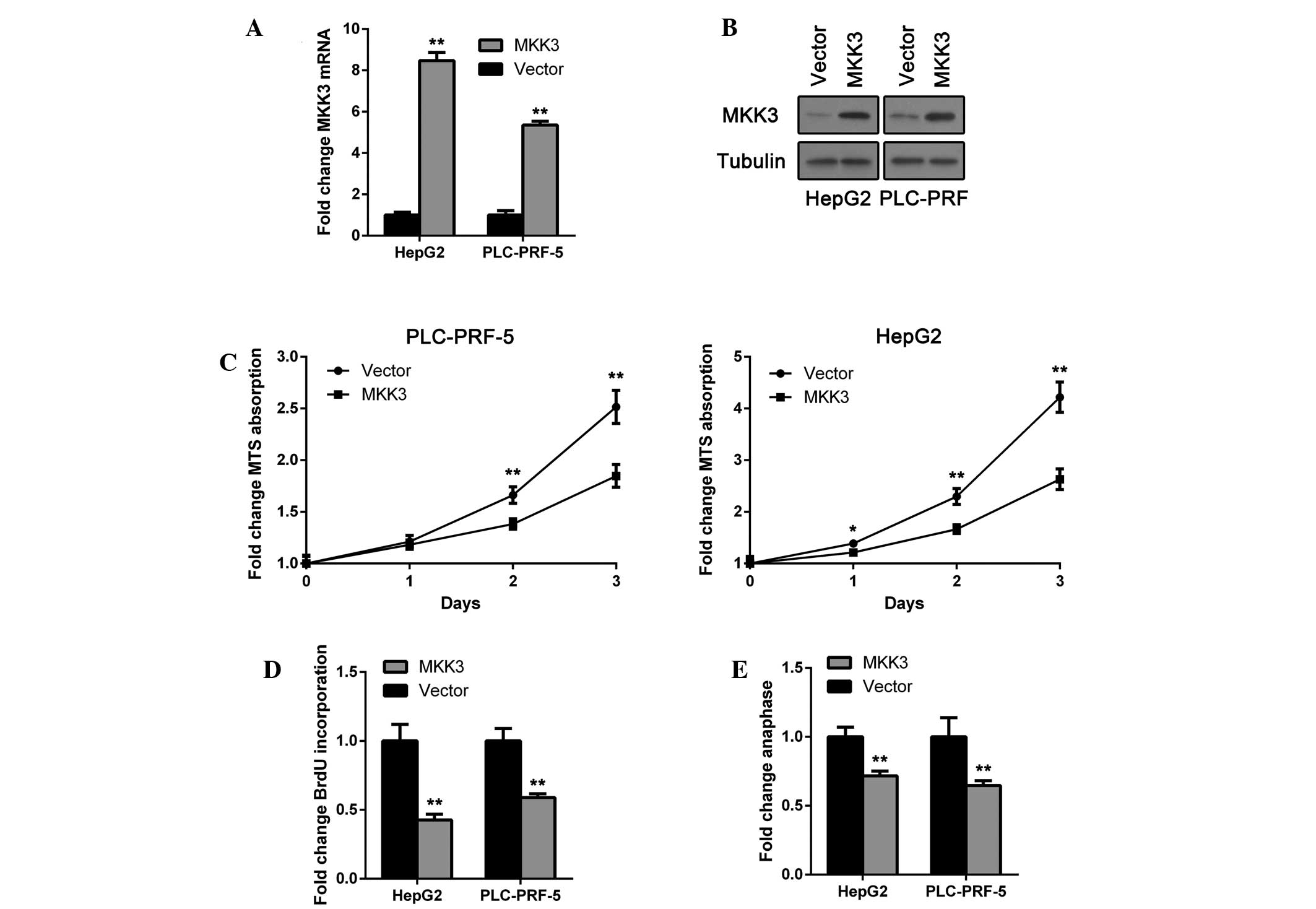
To investigate whether MKK3 acts as a tumor
suppressor in HCC, MKK3 was overexpressed in HepG2 and PLC-PRF-5
HCC cell lines. MKK3 overexpression was confirmed by RT-qPCR and
immunoblotting (Fig. 1A and B). An
MTS proliferation assay was then performed. HepG2 and PLC-PRF-5
cells transfected with the MKK3 expression plasmid exhibited
impaired proliferation (Fig. 1C).
Furthermore, a BrdU incorporation assay was also performed to
examine proliferation alterations. In accordance with the results,
MKK3 suppressed BrdU incorporation in the two cell lines (Fig. 1D). Moreover, the number of
detectable anaphase cells, assessed in parallel as an indicator of
active proliferation, was selectively reduced in
MKK3-overexpressing HepG2 and PLC-PRF-5 cells (Fig. 1E). The results indicate that MKK3
may function as a tumor suppressor via inhibiting
proliferation.
MKK3 induces cell cycle arrest in HepG2
and PLC-PRF-5 cells
Cell cycle deregulation is a common feature of human
cancer, which leads to unscheduled proliferation, genomic
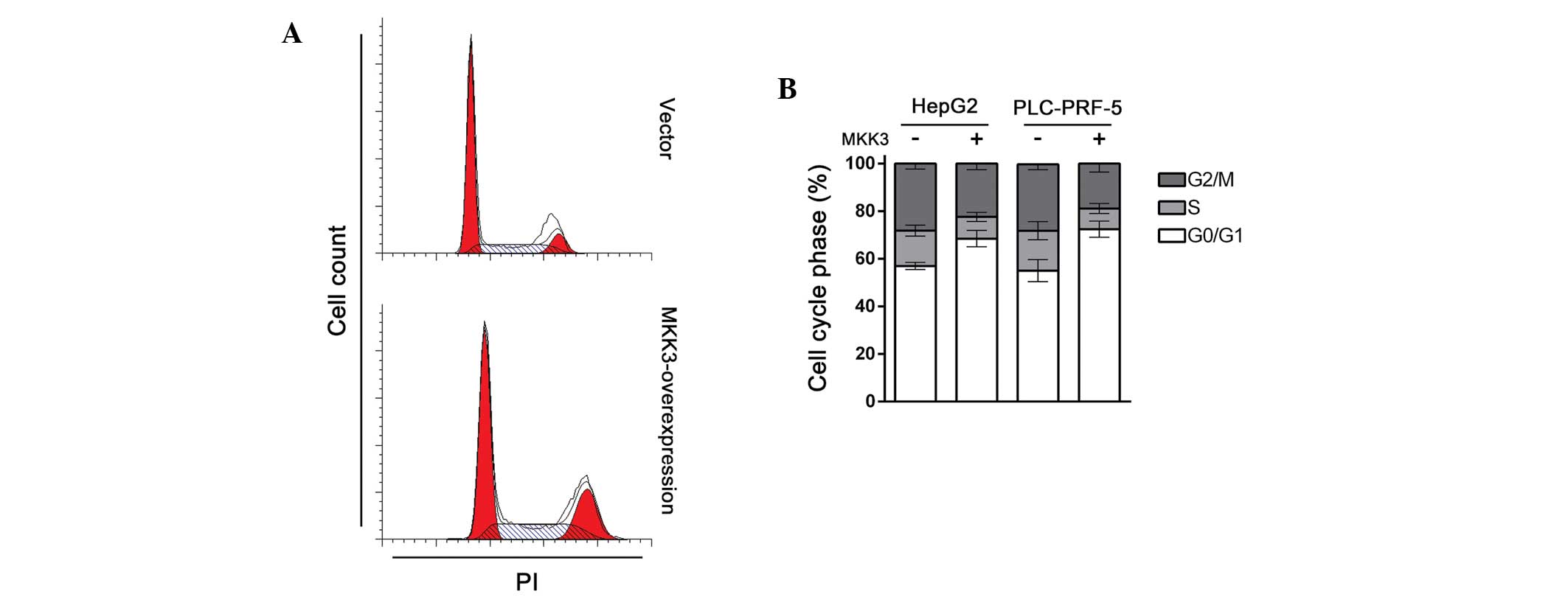
instability and chromosomal instability (17). Given that MKK3 suppresses HCC cell
proliferation, it was speculated that MKK3 may participate in cell
cycle regulation. To test this hypothesis, cell cycle distribution
in HepG2 and PLC-PRF-5 cells transfected with either MKK3
expression plasmid or vector was examined. As expected, compared
with control, MKK3 overexpressing cells showed significant cell
cycle arrest in the G1 phase (Fig. 2A
and B). Together with the results of the proliferation study,
these results indicate that MKK3 regulates the tumor cell cycle
and, thus, exhibits a tumor suppressive role in HCC.
MKK3 upregulates INK4A and INK4B in HepG2
and PLC-PRF-5 cells
The cell cycle is tightly controlled by
cyclin-dependent kinases (CDKs) (18). CDK activity requires binding of
regulatory subunits termed cyclins. In addition, There are two
families of CDK inhibitors (CKIs), INK4 proteins and the Cip and
Kip family (17,19,20).
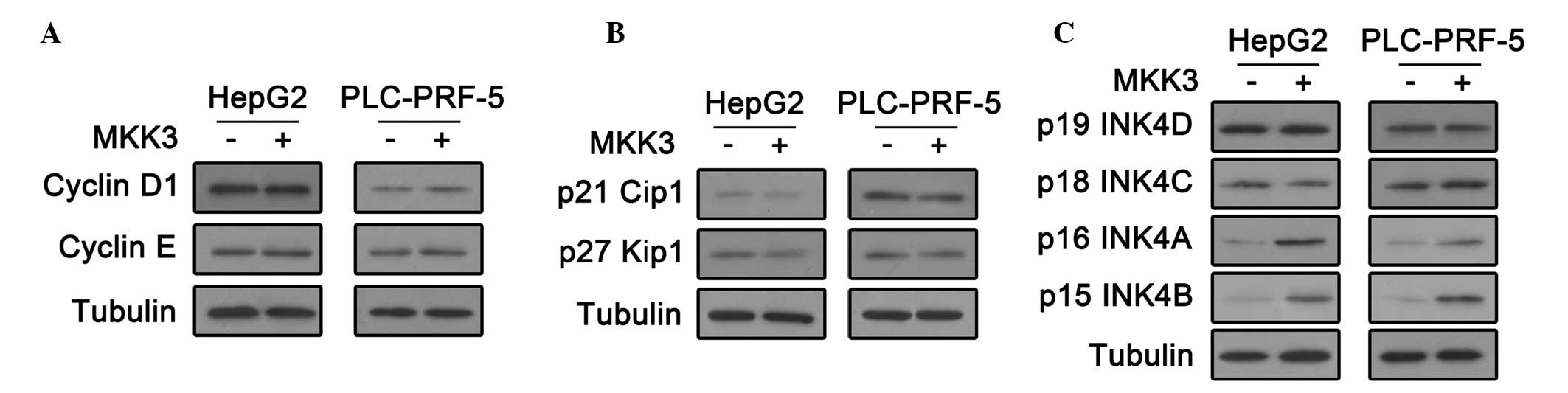
To investigate how MKK3 influences HCC cell cycle regulation, these
cell cycle regulators were investigated. First, cyclin D1 and
cyclin E expression was analyzed by an immunoblot assay. As shown
in Fig. 3A, there was no change in
these levels following MKK3 overexpression. In addition, the
immunoblot assay showed that MKK3 overexpression did not affect the
expression of CDK2 inhibitors, p27 Cip1 and p27 Kip (Fig. 3B). Notably, p16 INK4A and p15 INK4B
were upregulated in HepG2 and PLC-PRF-5 cells transfected with the
MKK3 expression plasmid (Fig. 3C).
These results indicate that MKK3 may affect cell cycle by
upregulating the CDK4/6 inhibitors, p16 INK4A and p15 INK4B in
HCC.
MKK3 tumor suppressor activity is
dependent on Bmi-1 downregulation and p38 activation
Bmi-1 is a member of the polycomb group (PcG) of
proteins and is important in the regulation of cell proliferation
and senescence through repression of the p16 INK4A and p15 INK4B
genes (21–24). A recent study also indicated its
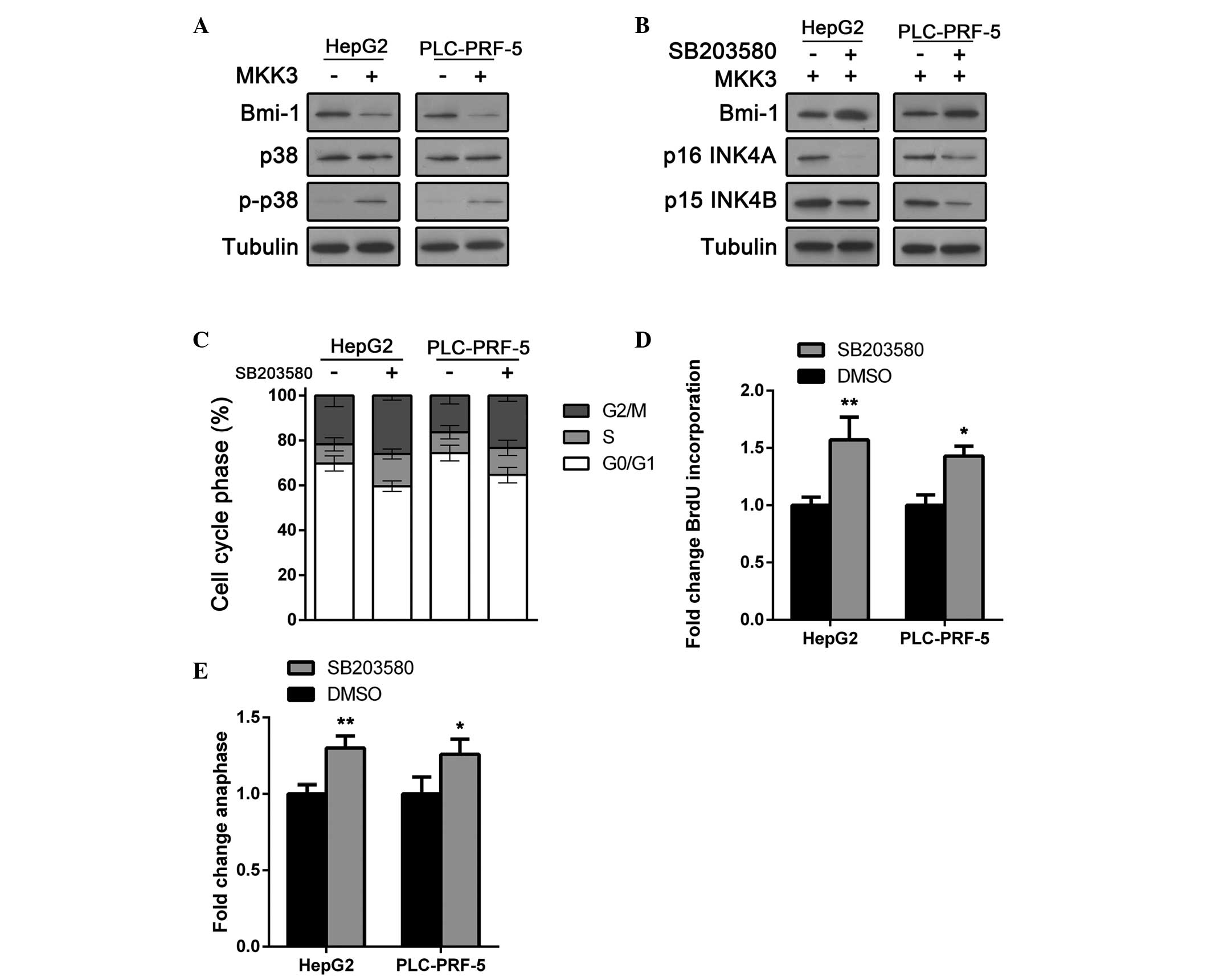
oncogenic role (25,26). Bmi-1 expression was examined in
MKK-overerpressing cells and control cells. As expected, Bmi-1 was
downregulated in HepG2 and PLC-PRF-5 cells following MKK3
overexpression (Fig. 4A).
Furthermore, as MKK3 is an MAPK kinase that activates p38, p38
activation was investigated. As shown in Fig. 4A, p38 expression was not altered.
However, phosphor-p38, the active form of p38, was significantly
upregulated in MKK-overexpressing HCC cells. These results indicate
that MKK3 may regulate Bmi-1 and cell cycle arrest by p38
activation.
To test whether the tumor suppressive role of MKK is
p38 dependent, cells were treated with SB203580, a p38 inhibitor.
As shown in Fig. 4B, Bmi-1
expression was rescued by SB203580 treatment in HCC cells.
Furthermore, p16 INK4A and p15 INK4B were also downregulated to
normal levels in MKK-overexpressing HCC cells (Fig. 4B). The impact of SB203580 on HCC
cell cycle arrest was then determined. HepG2 and PLC-PRF-5 cells
transfected with the MKK3 expression plasmid were treated with
SB203580 or vehicle and cell cycle staging was conducted using flow
cytometric analysis. Results showed that SB203580 suppressed
MKK3-induced cell cycle arrest in HepG2 and PLC-PRF-5 cells
(Fig. 4C). Moreover, the impact of
SB203580 on HCC cell proliferation was also determined. The BrdU
incorporation assay showed that SB203580 restored proliferation in
MKK3-overexpressing cells (Fig.
4D). These results were confirmed by an anaphase cell count
assay (Fig. 4E) and suggested that
the tumor suppressive role of MKK3 is dependent on Bmi-1
downregulation and p38 activation.
Discussion
In the development of cancer, tumor cells acquire
six capabilities, which are shared by the majority of/perhaps all
types of human tumor. These six capabilities are the well-known
hallmarks of cancer (27,28). Three of the six hallmarks,
self-sufficiency in growth signals, insensitivity to
growth-inhibitory signals and limitless replicative potential are
associated with cell cycle control. Thus, understanding of this
process is important for developments in cancer therapy. In the
current study, a novel mechanism by which HCC cells control
proliferation and cell cycle transition was determined. The results
suggest that by activating p38, MKK3 suppresses the expression of a
PcG protein, Bim-1, which is a negative regulator of the expression
of CDK4/6 inhibitors p16 INK4A and p15 INK4B. In this way, MKK3
upregulates p16 INK4A and p15 INK4B and therefore, induces HCC cell
cycle arrest. A recent study reported that MKK3 was downregulated
in HCC (16). Together with our
results, these data suggest that HCC may pass through the cell
cycle checkpoint by downregulating MKK3.
The role of p38 in cancer depends on the cell type
and cancer stage. Certain studies have reported that p38 increases
cell proliferation, whereas in others, the activation of the MAPK
p38 pathway is described as tumor suppressive (29–34).
In the present study, the p38 pathway was observed to exhibit a
tumor suppressive role in HCC. MKK3-induced p38 activation impaired
cancer cell growth. In addition, p38 inhibition by SB203580
restored HCC cell G1-S transition and proliferation. p38 MAPK
targeting inhibitors and drugs are currently in development to
treat cancer (13). However, a
recent study reported the antitumor effect of another p38
inhibitor, SB202190 in colon adenocarcinoma (35). These conflicting results may
reflect the differences in the effects of p38, depending on cell
type. Furthermore, a recent study in breast cancer reported that
MKK3 regulates cell cycle transition by p21 Cip1 and p27 Kip1.
However, it was suggested that MKK3 regulates the cell cycle via
p16 INK4A and p15 INK4B. Together, these results highlight the
importance of understanding the cell type-specific differences of
the effects of p38. It is important to carefully consider the type
of tumor prior attempting to modulate this pathway for cancer
therapy.
In conclusion, the present results indicate that
MKK3 exhibits a critical suppressive role in hepatocarcinogenesis
through the control of Bim-1 expression and p38 activation. It also
reports a previously undescribed MKK3 dependent HCC cell cycle
control mechanism. These results shed light on the regulation of
HCC cell cycle and identify a novel target for HCC treatment.
Acknowledgments
This study was supported by the National Clinical
Key Subject Construction Project of NHFPC Fund, China.
References
|
1
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yeoman AD, Al-Chalabi T, Karani JB,
Quaglia A, Devlin J, Mieli-Vergani G, Bomford A, O'Grady JG,
Harrison PM and Heneghan MA: Evaluation of risk factors in the
development of hepatocellular carcinoma in autoimmune hepatitis:
Implications for follow-up and screening. Hepatology. 48:863–870.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mohamed AE, Kew MC and Groeneveld HT:
Alcohol consumption as a risk factor for hepatocellular carcinoma
in urban southern African blacks. Int J Cancer. 51:537–541. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsukuma H, Hiyama T, Tanaka S, Nakao M,
Yabuuchi T, Kitamura T, Nakanishi K, Fujimoto I, Inoue A, Yamazaki
H, et al: Risk factors for hepatocellular carcinoma among patients
with chronic liver disease. N Engl J Med. 328:1797–1801. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Badvie S: Hepatocellular carcinoma.
Postgrad Med J. 76:4–11. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Alazawi W, Cunningham M, Dearden J and
Foster GR: Systematic review: Outcome of compensated cirrhosis due
to chronic hepatitis C infection. Aliment Pharmacol Ther.
32:344–355. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: From genes to environment. Nat Rev Cancer.
6:674–687. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roux PP and Blenis J: ERK and p38
MAPK-activated protein kinases: A family of protein kinases with
diverse biological functions. Microbiol Mol Biol Rev. 68:320–344.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ono K and Han J: The p38 signal
transduction pathway: Activation and function. Cell Signal.
12:1–13. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zarubin T and Han J: Activation and
signaling of the p38 MAP kinase pathway. Cell Res. 15:11–18. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cerezo-Guisado MI, del Reino P, Remy G,
Kuma Y, Arthur JS, Gallego-Ortega D and Cuenda A: Evidence of
p38gamma and p38δ involvement in cell transformation processes.
Carcinogenesis. 32:1093–1099. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kyriakis JM and Avruch J: Mammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammation. Physiol Rev. 81:807–869.
2001.PubMed/NCBI
|
|
16
|
MacNeil AJ, Jiao SC, McEachern LA, Yang
YJ, Dennis A, Yu H, Xu Z, Marshall JS and Lin TJ: MAPK kinase 3 is
a tumor suppressor with reduced copy number in breast cancer.
Cancer Res. 74:162–172. 2014. View Article : Google Scholar
|
|
17
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tian Y, Wan H and Tan G: Cell
cycle-related kinase in carcinogenesis. Oncol Lett. 4:601–606.
2012.PubMed/NCBI
|
|
19
|
Abraham RT: Cell cycle checkpoint
signaling through the ATM and ATR kinases. Genes Dev. 15:2177–2196.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Murray AW: Recycling the cell cycle:
Cyclins revisited. Cell. 116:221–234. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jacobs JJ, Kieboom K, Marino S, DePinho RA
and van Lohuizen M: The oncogene and Polycomb-group gene bmi-1
regulates cell proliferation and senescence through the ink4a
locus. Nature. 397:164–168. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jacobs JJ, Scheijen B, Voncken JW, Kieboom
K, Berns A and van Lohuizen M: Bmi-1 collaborates with c-Myc in
tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF.
Genes Dev. 13:2678–2690. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cao R, Tsukada Y and Zhang Y: Role of
Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol
Cell. 20:845–854. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Molofsky AV, He S, Bydon M, Morrison SJ
and Pardal R: Bmi-1 promotes neural stem cell self-renewal and
neural development but not mouse growth and survival by repressing
the p16Ink4a and p19Arf senescence pathways. Genes Dev.
19:1432–1437. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yao XB, Wang XX, Liu H, Zhang SQ and Zhu
HL: Silencing Bmi-1 expression by RNA interference suppresses the
growth of laryngeal carcinoma cells. Int J Mol Med. 31:1262–1272.
2013.PubMed/NCBI
|
|
26
|
Wu SQ, Xu ZZ, Niu WY, Huang HB and Zhan R:
ShRNA-mediated Bmi-1 silencing sensitizes multiple myeloma cells to
bortezomib. Int J Mol Med. 34:616–623. 2014.PubMed/NCBI
|
|
27
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hanahan D and Weinberg RA: Hallmarks of
Cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rosenthal DT, Iyer H, Escudero S, Bao L,
Wu Z, Ventura AC, Kleer CG, Arruda EM, Garikipati K and Merajver
SD: p38 gamma promotes breast cancer cell motility and metastasis
through regulation of RhoC GTPase, cytoskeletal architecture and a
novel leading edge behavior. Cancer Res. 71:6338–6349. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He J, Liu Z, Zheng Y, Qian J, Li H, Lu Y,
Xu J, Hong B, Zhang M, Lin P, et al: p38 MAPK in myeloma cells
regulates osteoclast and osteoblast activity and induces bone
destruction. Cancer Res. 72:6393–6402. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu K, Yu D, Cho YY, Bode AM, Ma W, Yao K,
Li S, Li J, Bowden GT and Dong Z and Dong Z: Sunlight UV-induced
skin cancer relies upon activation of the p38 alpha signaling
pathway. Cancer Res. 73:2181–2188. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sakurai T, Kudo M, Umemura A, He G,
Elsharkawy AM, Seki E and Karin M: p38 alpha inhibits liver
fibrogenesis and consequent hepatocarcinogenesis by curtailing
accumulation of reactive oxygen species. Cancer Res. 73:215–224.
2013. View Article : Google Scholar :
|
|
33
|
Zhou Y, Liang Y, Wei J, Chen J and Tang Q:
Lentiviral-mediated p38 MAPK RNAi attenuates aldosterone-induced
myocyte apoptosis. Mol Med Rep. 8:493–498. 2013.PubMed/NCBI
|
|
34
|
Sun BK, Kim JH, Nguyen HN, Oh S, Kim SY,
Choi S, Choi HJ, Lee YJ and Song JJ: MEKK1/MEKK4 are responsible
for TRAIL-induced JNK/p38 phosphorylation. Oncol Rep. 25:537–544.
2011.
|
|
35
|
Paillas S, Boissière F, Bibeau F, Denouel
A, Mollevi C, Causse A, Denis V, Vezzio-Vié N, Marzi L, Cortijo C,
et al: Targeting the p38 MAPK pathway inhibits irinotecan
resistance in colon adenocarcinoma. Cancer Res. 71:1041–1049. 2011.
View Article : Google Scholar
|