Introduction
There are >400 types of syndromic hearing loss,
and Waardenburg syndrome (WS) is the most common, accounting for
2–5% of congenital deafness (1).
WS is a type of auditory-pigmentary syndrome, the clinical
manifestations of which include congenital neurosensory deafness,
change in iris pigmentation, telecanthus, abnormal distribution of
hair and skin pigmentation (prematurely white hair, eyebrows and
eyelashes at <30-years old; facial freckles; skin depigmentation
leukoplakia), wide root of the nose, and synophrys. WS is
classified into four types, according to various clinical
manifestations.
The pathogenesis of WS is largely attributed to the
mutations in genes including PAX3, MITF, SNAI2, SOX10, ENDRB, and
EDN3. Dysregulation of these genes causes abnormal development of
neural crest cells, change in iris pigmentation, deafness, hair
graying, and abnormal skin pigmentation. Notably, the distinct
subtypes of WS differ in the pathogenic mutations of relevant genes
(2).
The present study investigated three WS pedigrees,
and clinical classification was made according to the patients'
symptoms. Written informed consent was obtained from the family
members who were willing to participate in the present study. The
known candidate disease-causing genes were selected for mutation
detection, in order to obtain a genetic diagnosis. The current
study was approved by the ethics committee of the Family Planning
Institute of Hunan Province (Changsha, China)
Materials and methods
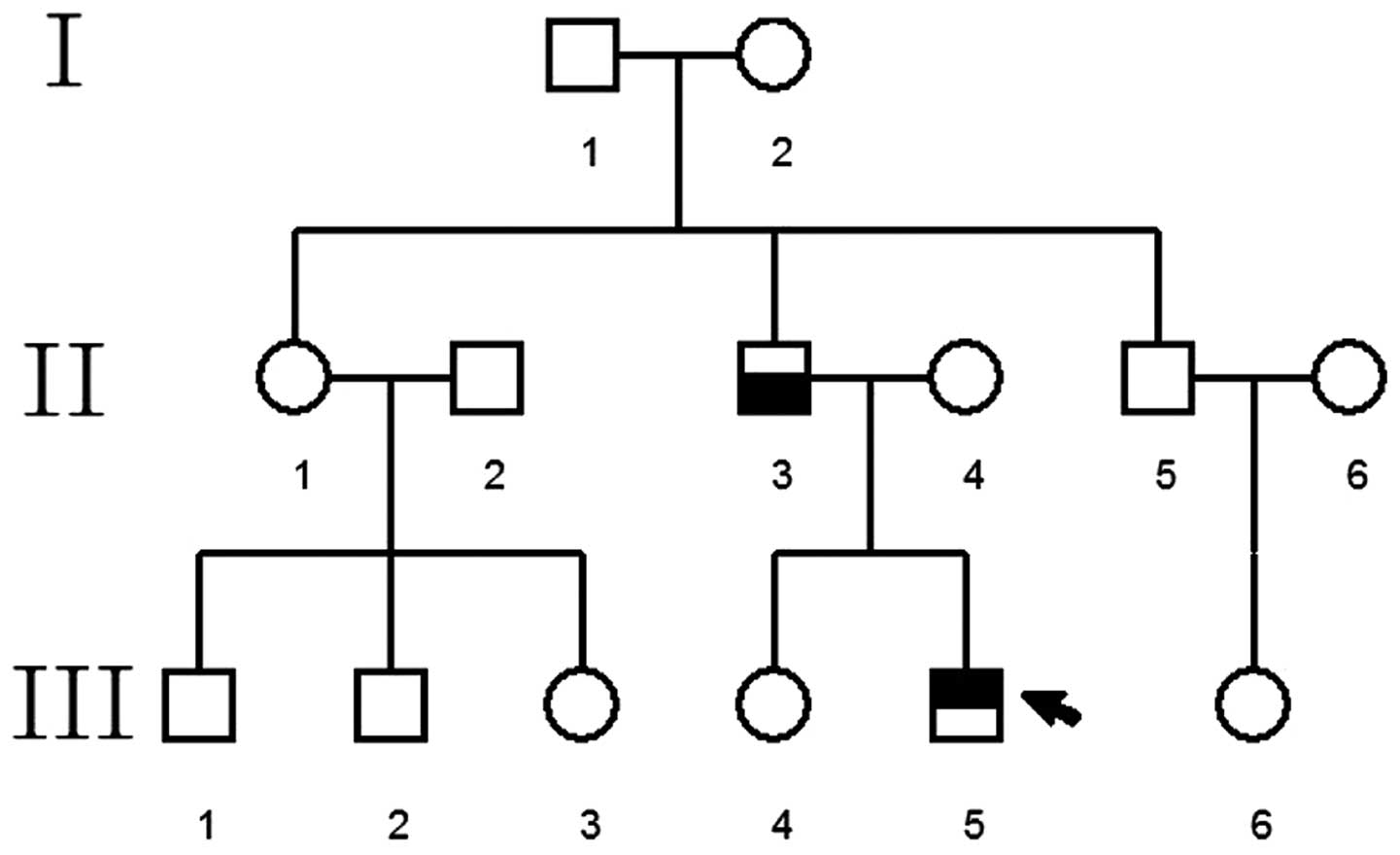
Pedigree 1
The proband was a patient at The Special Clinic of
Neonate Hearing Screening, The Second Xiangya Hospital (Changsha,
China), who planned to undergo fitting of a cochlear implant. The
family consisted of three generations (nine individuals) with five
cases of WS, including the proband, the proband's grandmother,
mother, aunt and uncle (Fig.
1).
Pedigree 2
The proband was a patient at The Special Clinic of
Neonate Hearing Screening, The Second Xiangya Hospital. The family
consisted of three generations (14 individuals) with two cases of
WS, including the proband and the proband's father (Fig. 2).
Pedigree 3
The proband was a patient at The Special Clinic of
Neonate Hearing Screening, The Second Xiangya Hospital, who planned
to undergo fitting of a cochlear implant. The family consisted of
three generations (four individuals) with two cases of WS,
including the proband and the proband's father (Fig. 3).
Pedigree investigation and sample
collection
According to the principle of informed consent,
following approval from all family members, detailed examinations
were performed on all patients by medical specialists. The
following tests were conducted: Observation of skin pigmentation,
hair color, joints, skeletomuscular system, digestion, nerves,
ophthalmology and otology, and an assessment of intelligence. In
addition, a detailed audiological examination was conducted on the
probands. The clinical audiology assessment included pure tone
test, acoustic immitance, auditory steady-state response, auditory
brainstem response (ABR), otoacoustic emission, test of study
ability and psychiatric behavior development, and ossa temporale
computerized tomography and magnetic resonance imaging. Peripheral
vein blood samples (5 ml) were collected from all subjects and were
placed in vacuum heparin anticoagulant tubes.
DNA extraction
Whole genomic DNA was isolated from the blood
samples using the phenol-chloroform extraction, and was quantified
by an ultraviolet spectrophotometer Du800 (Beckman Coulter, Inc.,
Brea, CA, USA). The DNA was subsequently stored at −20°C until
further use.
Analysis of genetic mutations
The probands and their healthy brother, sister or
relatives were selected as the subject of mutation analysis. The
sequences of the candidate genes, paired box 3 (PAX3),
microphthalmia-associated transcription factor (MITF),
sex-determining region Y-box 10 (SOX10), snail family zinc finger 2
(SNAI2), were used as the template. The online primer design
software Primer 3 (http://primer3.ut.ee/) was used to design the
polymerase chain reaction (PCR) primers to amplify all exons, and
the PCR primers (Invitrogen; Thermo Fisher Scientific, Inc.,
Beijing, China) to amplify boundary sequence of exon/intron of the
candidate gene (Tables I–IV). The amplification reaction was set
in a total volume of 20 µl containing the PrimeSTAR Max DNA
polymerase and PCR buffer (TaKaRa Biotechnology Co. Ltd., Dalian,
China) and specific primers. The reaction was then performed for 30
cycles under 95°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec
on an ABI 9700 thermal cycler (Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Following amplification, the
PCR products were subjected to agarose gel electrophoresis and
purified using MiniBest Agarose Gel DNA Extraction kit Ver4.0
(TaKaRa Biotechnology Co. Ltd.). The upstream and downstream
amplifying primers were used for sequence detection by Sanger
sequencing on a 3730 DNA analyzer (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The gene sequences were compared and
analyzed using GenBank (http://www.ncbi.nlm.nih.gov/genbank/), in order to
identify any mutations. Sanger sequencing was conducted for
verification on all family members
 | Table IPolymerase chain reaction primer
sequence of the exons of paired box 3 gene. |
Table I
Polymerase chain reaction primer
sequence of the exons of paired box 3 gene.
| Exon | Sequence of upstream
primer (5′–3′) | Sequence of
downstream primer (5′–3′) | Fragment size
(bp) |
|---|
| 1 |
ACTCGGTGTCACCACAGGA |
CCTGGAAGCACCAAAGGAG | 564 |
| 2 |
TACGTGCTGCTGTTCTTTGC |
TTACGCACCTTCACAAACCTC | 443 |
| 3 |
TCTGGTCTGCCCCTTTCTAA |
ATTGGGGTGATTACGTCTGG | 389 |
| 4 |
GTCCAGAGATGCAGGAGGAG |
CTGCCGTCAGATCACCAA | 369 |
| 5 |
TGTCTTGCAGTCGGAGAGAG |
GGTGGACTTCTGTGTGTCGT | 493 |
| 6 |
AATTCGCCCAAACAACACA |
CAGAGAAATCGCCTGGAAGT | 370 |
| 7 |
TGGCTGATGAACTTTTGCAC |
TGGTTTAAATTTGGCAATTCAT | 392 |
| 8.1 |
AGCTGTAGGCTGCAATCTGG |
GTGGCAATCAGGTTTCACGT | 550 |
| 8.2 |
TTTTGCAAAGCCAGCTGACT |
TAGGCTGCGAAGACCAGAAA | 468 |
| 8.3 |
GAATTGTCCCAGCATGACCT |
TAGAACAGTCTGCTTGCCCAA | 497 |
| Table IVPolymerase chain reaction primer
sequence of the exons of snail family zinc finger 2 gene. |
Table IV
Polymerase chain reaction primer
sequence of the exons of snail family zinc finger 2 gene.
| Exon | Sequence of upstream
primer (5′–3′) | Sequence of
downstream primer (5′–3′) | Fragment size
(bp) |
|---|
| 1 |
GCTGTGATTGGATCTTTCTTGC |
TGTAAGCTCCCTTTCAGGACAC | 449 |
| 2 |
TGTGTGTATACTTGCGTGTGG |
CTTCATGCAAATCCAACAGCC | 700 |
| 3 |
ATTTCTGTATGATTGGCAGCAG |
GCTTCGGAGTGAAGAGAAATGC | 471 |
Results
Pedigree analysis
Pedigree 1 consisted of three generations (nine
persons) and five patients with WS. The following characteristics
were observed: (i) The disease was observed in each subsequent
generation following the proband's grandfather; (ii) one of the
patient's parents was affected; (iii) the offspring of individuals
without the disease did not suffer from the disease; (iv) and male
and female children exhibited the same probability of suffering
from the disease. Therefore, WS in pedigree 1 exhibited an
autosomal dominant inheritance pattern. The mode of inheritance of
WS in pedigrees 2 and 3 remains to be confirmed as there weren't
enough patients in these families to gain reliable results.
Clinical diagnosis
The proband in pedigree 1 had blue irises,
prelingual deafness and hearing loss without progressive
aggravation. Acoustic conductance detected 'A' type wave, and ossa
temporale CT and MRI demonstrated an increase in density in the
middle ear cavity. Chronic otomastoiditis and traumatic brain
injury were not observed on the MRI, with no other major
abnormality. Both ears did not pass the distortion product
otoacoustic emission (DPOAE) test, suggesting pathology of the
cochlea. ABR, multi-frequency stable evoked potential, and acoustic
field measurement results suggested severe neurosensory deafness in
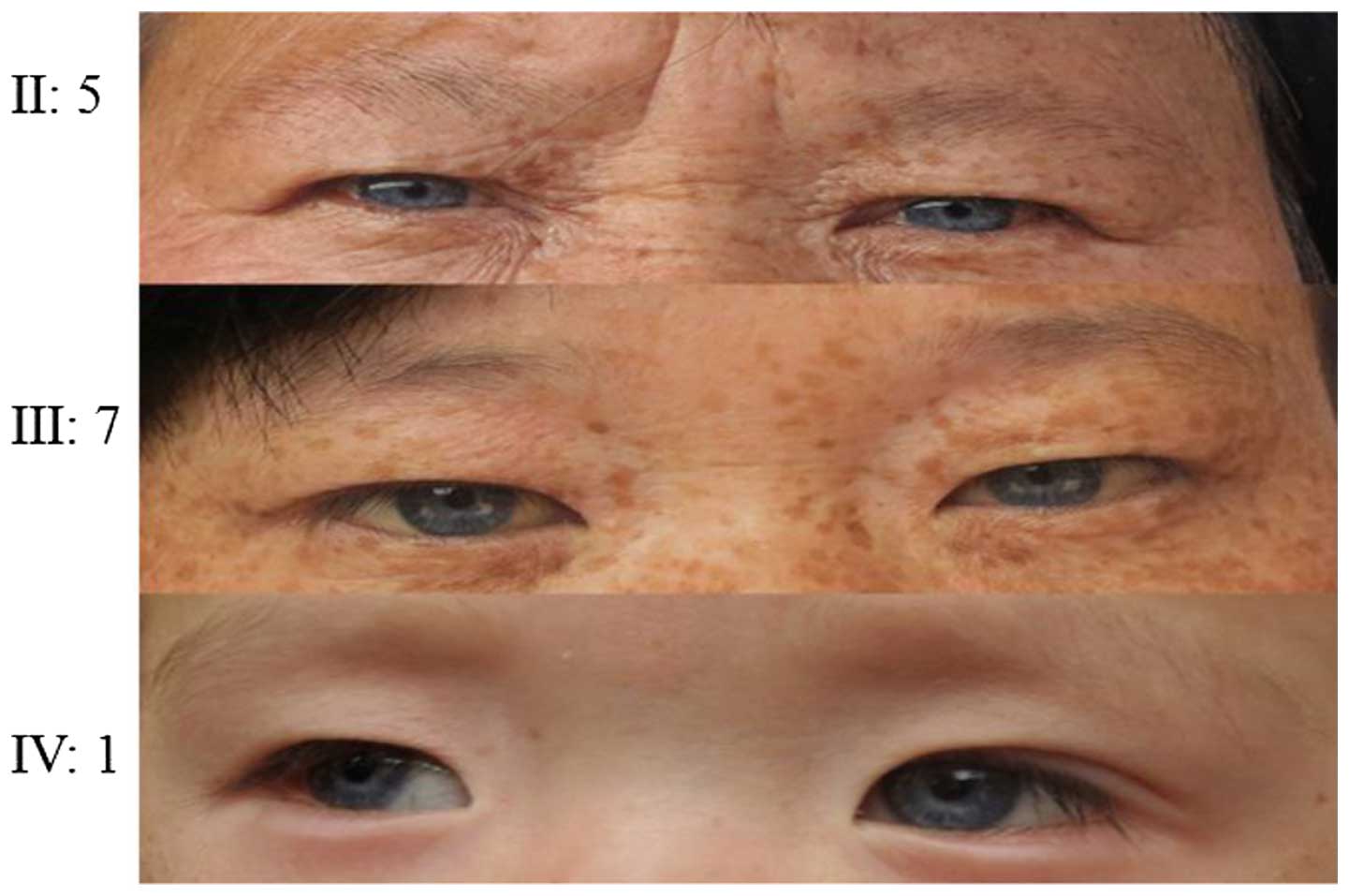
the proband. Probands of pedigrees 2 and 3 had blue irises,
prelingual deafness and hearing loss without progressive
aggravation (Figs. 5Figure 6–7). Acoustic conductance detected 'A' type
wave and ossa temporale CT and high-resolution MRI observed no
abnormality, suggesting a normal middle ear. Both ears did not pass
the DPOAE test, suggesting pathology of the cochlea. ABR,
multi-frequency stable evoked potential, and acoustic field
measurement results suggested severe neurosensory deafness in both
ears of the probands. According to the WS diagnostic standards (W
index values should be <1.95) produced by Faarer et al
(3) and Liu et al (4) in 1995, which is recommended by the
Waardenburg association, the W index values of the probands in the
present study were all <1.95, and probands also demonstrated
congenital neurosensory deafness, different colored irises, and no
angulus oculi medialis translocation, thus suggesting all three
families conform to the WS type II diagnosis.
Analysis of mutations in candidate
disease-causing genes
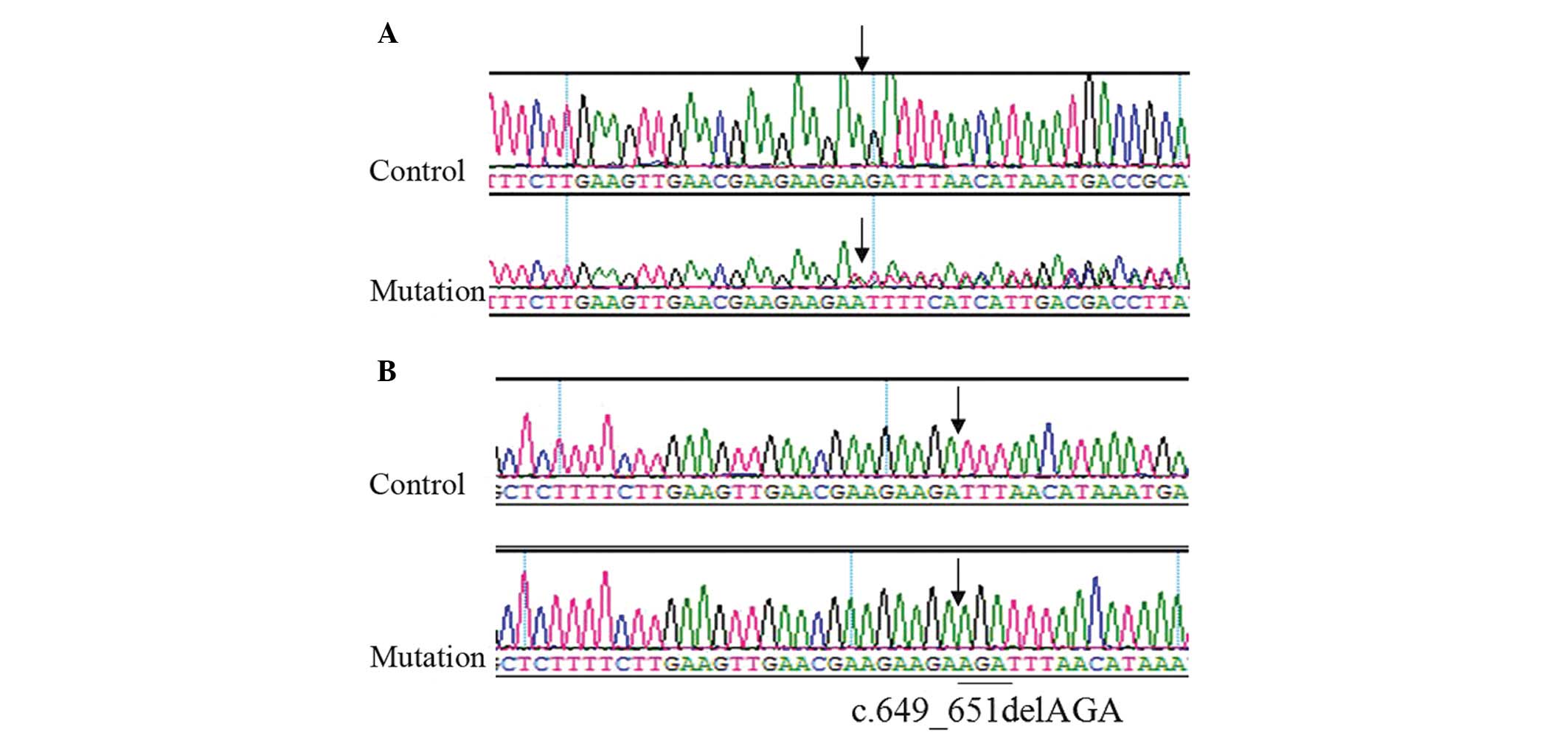
Genetic analysis of the proband of pedigree 1
observed mixed peaks in exon 7 of the MITF candidate gene; however,
the unrelated relatives exhibited a single peak, suggesting a loss
of heterozygosity (Fig. 4A).
Sequence detection revealed the c.649_651delAGA mutation (Fig. 4B). Members II:5, III:7 and III:9 of
pedigree 1 all exhibited the same genetic mutation. Following exon
detection of the candidate mutant genes, PAX3, SOX10 and SNAI2 in
family 1, no disease-causing mutations were observed in these
genes. Following exon detection of the candidate genes, PAX3, MITF,
SOX10 and SNAI2 in family 2 and family 3, no disease-causing
genetic mutations were observed.
Discussion
The five predominant diagnostic criteria for WS are:
(i) Congenital sensorineural hearing loss; (ii) iris pigment
abnormalities, including complete heterochromia iridis where the
eyes are different colors, partial or fragmentary heterochromia
iridis where blue is present in parts of the iris, and bright blue
irises; (iii) hypochromic change of hair, presented by white hair
on the forehead; (iv) no angulus oculi medialis malposition; (v)
and a first-degree relative with the condition. WS type II is
diagnosed by the presence of two of the five predominant diagnostic
criteria with no angulus oculi medialis malposition. Following
observation of the probands from the three pedigrees investigated
in the present study, they all exhibited the three most common and
marked phenotypic characteristics of WS type II: Sensorineural
prelingual deafness, bright blue irises, white hair at the
forehead, and W indexes <1.95, thus suggesting that all probands
had WS type II.
Due to the genetic heterogeneity and reduced
penetrance of WS, differences are often observed between patients
from both different families and the same family (5). Among the nine patients in the three
families tested in the present study, deafness and different iris
colors were the most common clinical phenotypes of WS, with a
penetrance of 77% (7/9 cases) and 88% (8/9 cases), respectively.
Facial freckles and pigmentation on the neck, chest and abdomen,
and upper limbs were observed in II:5, II:7 and III:9 in family 1.
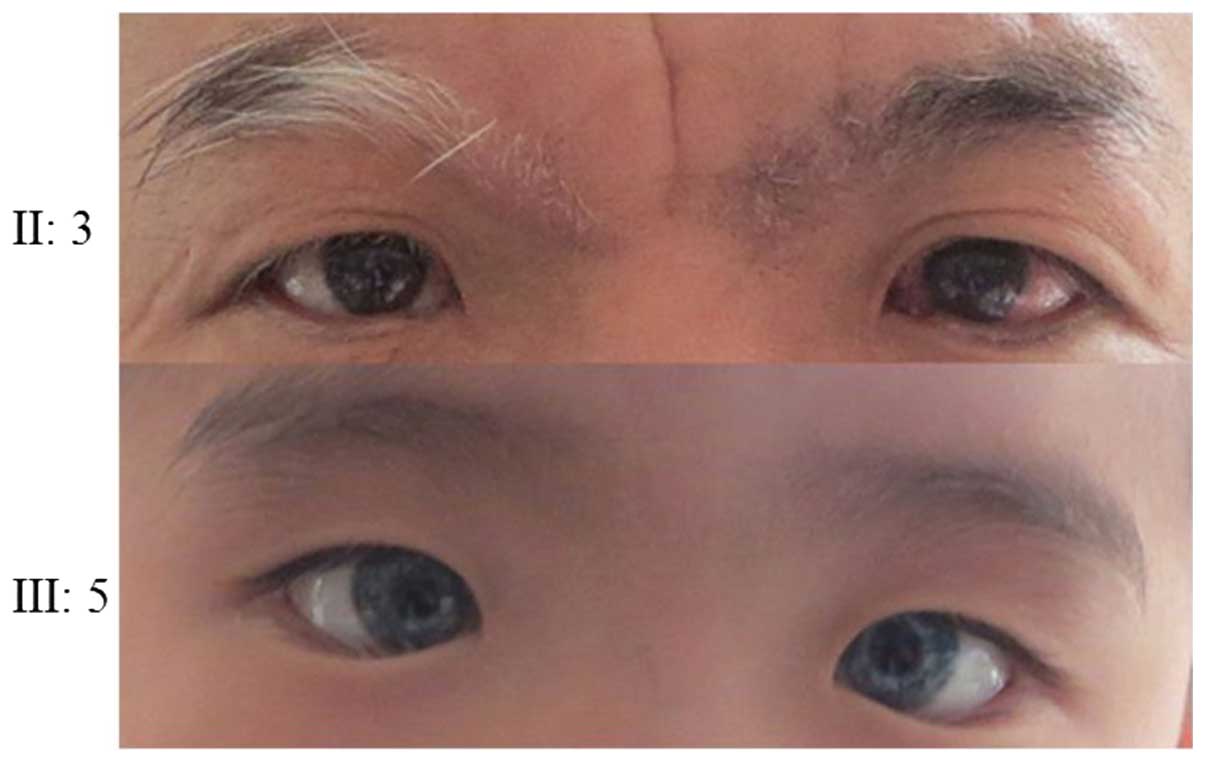
The father of the proband in family 2 had one blue iris, premature
white hair, mild deformity of the left elbow joint, limited
extension and mild fusion abnormality at the extreme ends of his
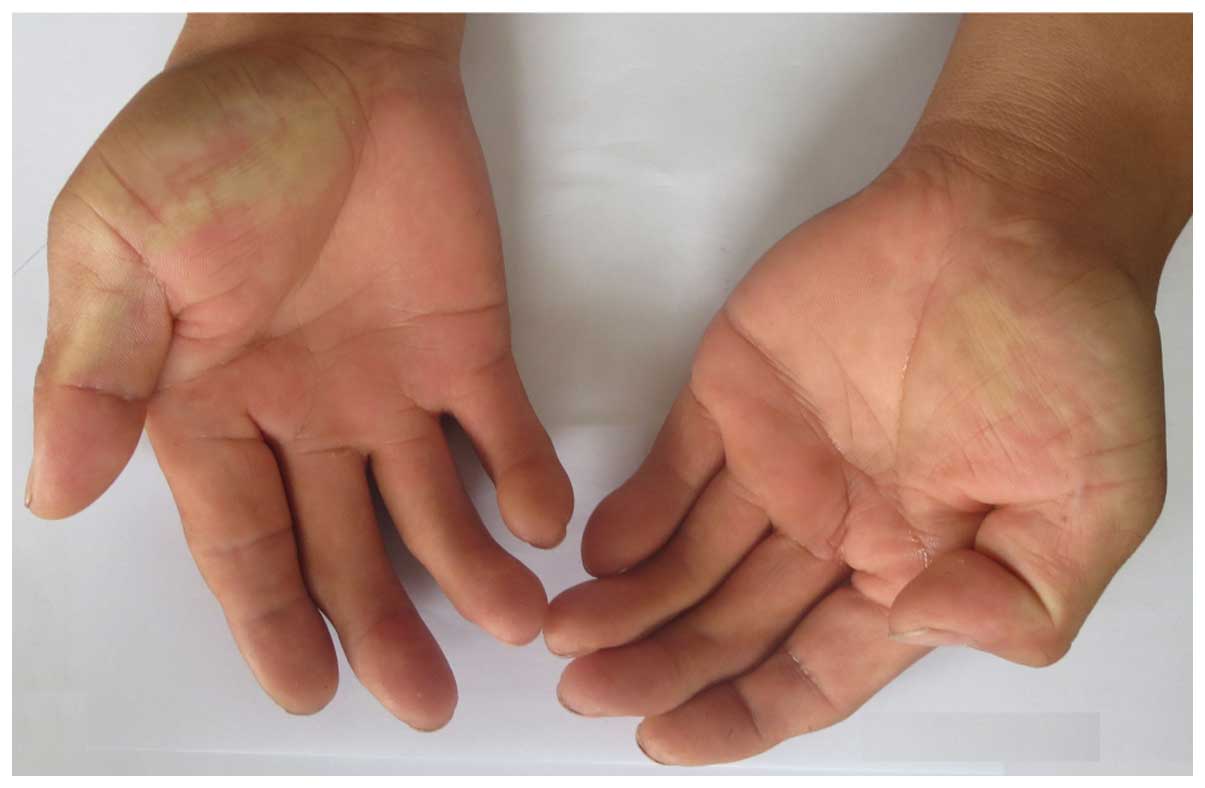
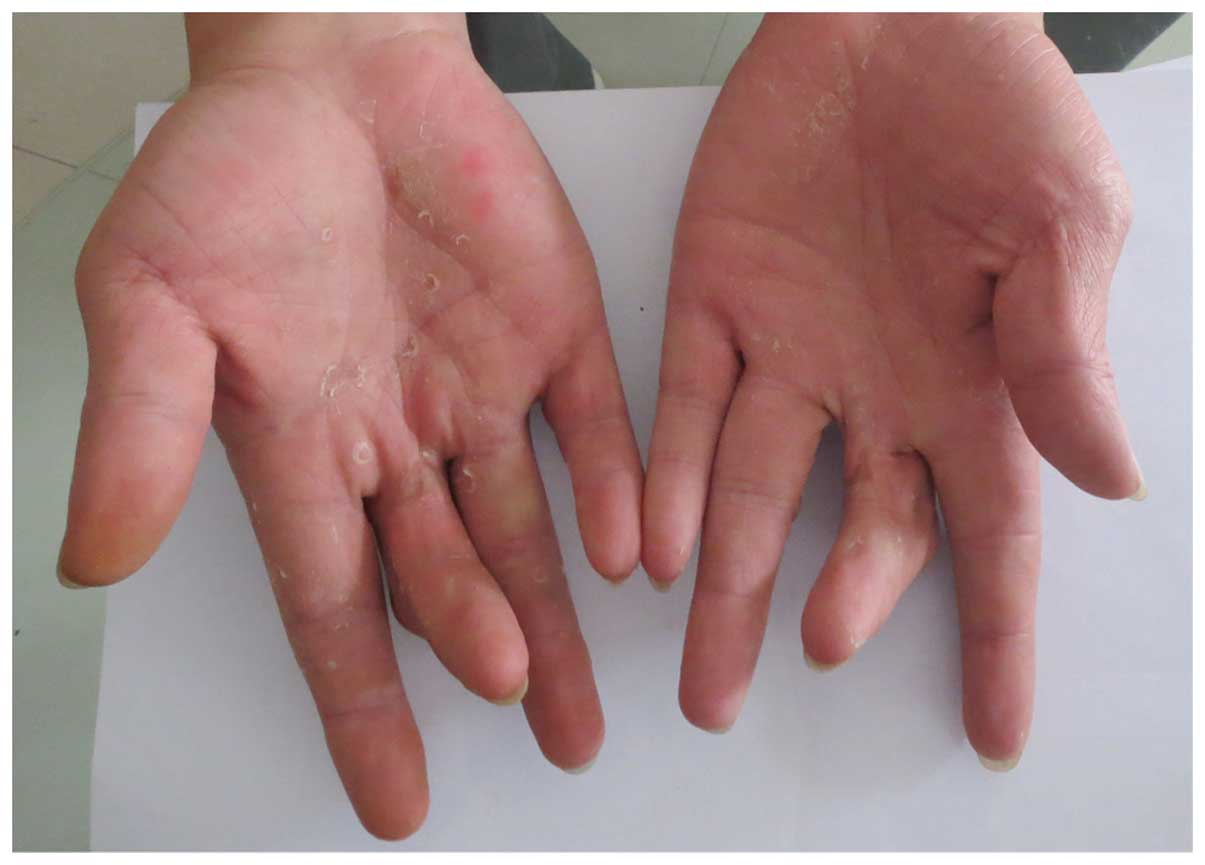
middle fingers, fourth fingers and little fingers (Fig. 8), but no clinical phenotype of
deafness. The father of the proband in family 3 exhibited normal
iris color and hearing, with no abnormal skin pigmentation but
limited extension of middle fingers (Fig. 9). His hair, right eyebrow and right
eyelashes were white by the age of 30. The other members of the two
families exhibited no abnormalities of limb joints, or the skeletal
or digestive systems, thus excluding WS type III, despite the
overlapping characteristics of each subtype. The known relevant
genes in WS are positioned on chromosomes 2, 3, 8, 13, 20 and 22,
and are PAX3, MITF, SNAI2, endothelin 3, endothelin receptor type B
and SOX10, respectively (6). PAX3
is the predominant disease-causing gene of WS types I and III
(7). The pathogenesis of WS type
II is associated with genetic mutations of MITF, SNAI2 and SOX10
(8–10); therefore, PAX3, MITF, SOX10 and
SNAI2 were considered the candidate disease-causing genes in the
three families of the present study, and were the focus of mutation
detection.
MITF is expressed in melanocytes and encodes a
transcription factor containing the helix-loop-helix-leucine zipper
structure. It is also a key factor in regulating the growth of
melanocytes. Defects in melanocytes result in abnormal pigment
distribution, whereas defects in melanocytes of the stria
vascularis lead to deafness (11).
Genetic mutation analysis demonstrated that all patients in
pedigree 1 had c.649_651AGA fragment loss in exon 7 of the MITF
candidate gene. This mutation results in the loss of codon 217
encoding arginine, and does not appear in normal controls. Arg217
mutation influences the positioning of MITF protein in the cell
nucleus and inhibits wild-type MITF function (12), which is consistent with the cases
in family 1 (13,14).
MITF gene mutation is also observed in Tietz
syndrome (albinism-deafness syndrome) (15). All patients in pedigree 1 exhibited
congenital severe sensorineural hearing loss, bright blue irises,
marked brown freckle deposition on the face, neck and/or limbs, but
no whole-body pigmentation alterations; therefore, Tietz syndrome
was excluded. Detection of known candidate disease-causing genes
(MITF, SOX10, SNAI2 and PAX3) was also conducted on the patients in
pedigrees 2 and 3; however, no known genetic mutations associated
with WS were observed. At present, MITF and SOX10 mutations are
detected in 30% of patients with WS type II (8,10).
Yang et al (16) reported
that the genetic mutation rate of MITF in China may be higher than
in non-Asian people. The majority of cases of WS type II cannot be
diagnosed at a molecular level, thus suggesting that there are
other disease-causing genetic mutations requiring further study
that are important in the pathogenesis of WS.
Previous studies demonstrated that following
treatment with the electronic cochlear implant, the rehabilitation
efficacy in patients with WS is comparable to other deaf patients
(17,18). Following the relevant examination
of the probands of pedigrees 1 and 3, an electronic cochlear
implant was fitted. Following the surgery, the patients reacted to
sound and underwent language rehabilitation; however, the
rehabilitation effects require further follow-up.
In conclusion, no mutations were identified in the
relevant genes, including PAX3, MITF, SOX10 and SNAI2 in two of the
three families with type II WS, which implies that novel pathogenic
mutations associated with type II WS may exist and require further
investigation.
Acknowledgments
The present study was supported by the Science and
Technology project of Hunan Province (grant no. 2014SK3020) and
Major National Scientific Research Projects (grant no.
2012CB967904).
References
|
1
|
Kochhar A, Hildebrand MS and Smith RJ:
Clinical aspects of hereditary hearing loss. Genet Med. 9:393–408.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Read AP and Newton VE: Waardenburg
syndrome. J Med Genet. 34:656–665. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Farrer LA, Grundfast KM, Amos J, Arnos KS,
Asher JH Jr, Beighton P, Diehl SR, Fex J, Foy C, Friedman TB, et
al: Waardenburg syndrome (WS) type I is caused by defects at
multiple loci, one of which is near ALPP on chromosome 2: First
report of the WS consortium. Am J Hum Genet. 50:902–913.
1992.PubMed/NCBI
|
|
4
|
Liu XZ, Newton VE and Read AP: Waardenburg
syndrome type II: Phenotypic findings and diagnostic criteria. Am J
Med Genet. 55:95–100. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Farrer LA, Arnos KS, Asher JH Jr, Baldwin
CT, Diehl SR, Friedman TB, Greenberg J, Grundfast KM, Hoth C,
Lalwani AK, et al: Locus heterogeneity for Waardenburg syndrome is
predictive of clinical subtypes. Am J Hum Genet. 55:728–737.
1994.PubMed/NCBI
|
|
6
|
Pingault V, Ente D, Dastot-Le Moal F,
Goossens M, Marlin S and Bondurand N: Review and update of
mutations causing Waardenburg syndrome. Hum Mutat. 31:391–406.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hoth CF, Milunsky A, Lipsky N, Sheffer R,
Clarren SK and Baldwin CT: Mutations in the paired domain of the
human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well
as Waardenburg syndrome type I (WS-I). Am J Hum Genet. 52:455–462.
1993.PubMed/NCBI
|
|
8
|
Bondurand N, Dastot-Le MF, Stanchina L,
Collot N, Baral V, Marlin S, Attie-Bitach T, Giurgea I, Skopinski
L, Reardon W, et al: Deletions at the SOX10 gene locus cause
Waardenburg syndrome types 2 and 4. Am J Hum Genet. 81:1169–1185.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sánchez-Martín M, Rodríguez-García A,
Pérez-Losada J, Sagrera A, Read AP and Sánchez-García I: SLUG
(SNAI2) deletions in patients with Waardenburg disease. Hum Mol
Genet. 11:3231–3236. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tassabehji M, Newton VE and Read AP:
Waardenburg syndrome type 2 caused by mutations in the human
microphthalmia (MITF) gene. Nat Genet. 8:251–255. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Curran K, Raible DW and Lister JA: Foxd3
controls melanophore specification in the zebrafish neural crest by
regulation of Mitf. Dev Biol. 332:408–417. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takebayashi K, Chida K, Tsukamoto I, Morii
E, Munakata H, Arnheiter H, Kuroki T, Kitamura Y and Nomura S: The
recessive phenotype displayed by a dominant negative
microphthalmia-associated transcription factor mutant is a result
of impaired nucleation potential. Mol Cell Biol. 16:1203–1211.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen H, Jiang L, Xie Z, Mei L, He C, Hu Z,
Xia K and Feng Y: Novel mutations of PAX3, MITF, and SOX10 genes in
Chinese patients with type I or type II Waardenburg syndrome.
Biochem Biophys Res Commun. 397:70–74. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tassabehji M, Newton VE, Liu XZ, Brady A,
Donnai D, Krajewska-Walasek M, Murday V, Norman A, Obersztyn E,
Reardon W, et al: The mutational spectrum in Waardenburg syndrome.
Hum Mol Genet. 4:2131–2137. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Smith SD, Kelley PM, Kenyon JB and Hoover
D: Tietz syndrome (hypopigmentation/deafness) caused by mutation of
MITF. J Med Genet. 37:446–448. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang S, Dai P, Liu X, Kang D, Zhang X,
Yang W, Zhou C, Yang S and Yuan H: Genetic and phenotypic
heterogeneity in Chinese patients with Waardenburg syndrome type
II. PLoS One. 8:e771492013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Amirsalari S, Ajallouyean M, Saburi A,
Haddadi Fard A, Abed M and Ghazavi Y: Cochlear implantation
outcomes in children with Waardenburg syndrome. Eur Arch
Otorhinolaryngol. 269:2179–2183. 2012. View Article : Google Scholar
|
|
18
|
Kontorinis G, Lenarz T, Giourgas A,
Durisin M and Lesinski-Schiedat A: Outcomes and special
considerations of cochlear implantation in waardenburg syndrome.
Otol Neurotol. 32:951–955. 2011. View Article : Google Scholar : PubMed/NCBI
|