Introduction
Sepsis is a systemic inflammatory response syndrome
caused by severe infection, which is characterized by inflammation
occurring in tissues that are remote from the infection. The
inflammatory responses in sepsis are primarily initiated by the
bacterial lipopolysaccharide (LPS), known as an endotoxin. During
endotoxemia, LPS acts as a ligand for pattern recognition receptors
known as toll-like receptors (TLRs), specifically TLR4 (1,2). The
binding of LPS to TLR4 activates either myeloid differentiation
primary response 88 (MyD88) or Toll/IL-1 receptor domain-containing
adaptor inducing interferon-β (TRIF) downstream pathways (2,3).
MyD88 is an adaptor protein that is critical for various TLR
activities. Stimulation of MyD88 increases the systemic and tissue
levels of pro-inflammatory cytokines, including tumor necrosis
factor-α (TNF-α) and interleukin-6 (IL-6) through the
trans-location of nuclear factor (NF)-κB to the nucleus. The
increase in inflammatory cytokines and mediators following LPS
exposure contributes to generalized inflammation (4,5). A
severe immune response may lead to septic shock (6) along with a reduction in cardiac
output and multiple organ injury (7), including lung and liver failure
(8). The rate of mortality due to
sepsis among intensive care unit patients is 30–50% (6).
Previous studies have identified that the activation
of AMP-activated protein kinase (AMPK) led to suppressed expression
levels and activation of TLR4 in heart tissues, in conditions
associated with inflammation, such as myocardial infarction
(9,10). Numerous studies have demonstrated
that AMPK activation prevents the inflammatory reaction, and a
reduction in AMPK activity has been associated with increased
inflammation (11–13). However, the association of AMPK
activity and TLRs in inflammation, particularly in vital tissues,
including lung and heart, remains unknown. AMPK is a
serine-threonine protein kinase that has a critical role in
cellular metabolism and function (14). It acts as a sensor of energy in
cells and is activated when the nutrient supply or ATP is limited,
or upon an increase in the demand of cellular energy. Therefore,
metabolic inhibitors, hypoxia, myocardial ischemia, hypoglycemia,
exercise, heat shock, osmotic stress, peroxynitrite and oxidative
stress are notable AMPK activators (15). Following AMPK activation,
energy-consuming processes, such as protein and glycogen synthesis
are suppressed, and ATP generating pathways such as glucose uptake,
glycolysis and fatty acid oxidation are activated (16).
At the molecular level, AMPK is a heterotrimer
complex comprised of α, β and γ subunits (17). Mammalian AMPK is sensitive to the
AMP:ATP ratio and an increase in the ratio activates the enzyme.
AMP binds to the γ subunit of AMPK and induces a conformational
change in the structure, that allosterically activates the α
catalytic subunit, enhances phosphorylation of the Thr172 residue
in the α subunit by upstream AMPK kinases, and inhibits the action
of protein phosphatase 2C to dephosphorylate Thr172 (18,19).
A-769662 is a small non-nucleoside thienopyridine molecule with
high specificity for AMPK. It directly binds to the β subunit of
AMPK to activate it (20)
independently of the AMP:ATP ratio (21,22).
Furthermore, A-769662 activates the eukaryotic elongation factor
kinase subsequent to AMPK activation and inhibits the
energy-requiring protein synthesis, thus promoting ATP preservation
during ischemia (20). In
chondrocytes, A-769662 suppresses the matrix degradation response
to inflammatory cytokines and the biochemical injury in which
peroxisome proliferator-activated receptor-γ coactivator 1-α
(PGC-1α) and forkhead box O3a mediate chondroprotection by
A-769662-induced AMPK activation (23). A previous study suggested that
preserving the AMPK activity by A-769662 in injured chondrocytes
protects the cartilage matrix integrity and inhibits caspase-3
activation and catabolic response (24). A-769662 is a novel agent and
compared with other activators of AMPK, including metformin and
AICAR, few studies have investigated its anti-inflammatory effect.
Therefore, for the present study, the effect of A-769662 on
LPS-induced inflammation and tissue injury was investigated.
Materials and methods
Animals
Male Wistar rats (240±10 g, 8-weeks old) were
purchased from Pasteur Institute of Iran (Tehran, Iran). A total of
15 rats were used (5 animals in each group). Animals were
administered food and water ad libitum and were housed in
the animal house of Tabriz University of Medical Sciences (Tabriz,
Iran) at a controlled ambient temperature of 22±2°C with 50±10%
relative humidity and a 12-h light/12-h dark cycle. The animals
were anesthetized by natrium pentobarbital (50 mg/kg; KELA
Laboratoria NV, Hoogstraten, Belgium). The present study was
performed in accordance with the Guide for the Care and Use of
Laboratory Animals of Tabriz University of Medical Sciences,
Tabriz, Iran (National Institutes of Health Publication No. 85–23,
revised 1985).
Chemical reagents
Escherichia (serotype k235)
lipopolysaccharide (LPS) and myeloperoxidase (MPO) were purchased
from Sigma-Aldrich (St. Louis, MO, USA), and A-769662 from Tocris
Bioscience (Bristol, UK). Rabbit monoclonal antibodies against
phosphorylated (p)-AMPKα (Thr172; cat. no. 2535;
1:1,000), AMPKα (cat. no. 5832; 1:1,000) and MyD88 (cat. no. 4283;
1:1,000) were obtained from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Mouse monoclonal GAPDH primary antibody (cat.
no. mAbcam9484; 1:5,000), and peroxidase-conjugated goat
anti-rabbit IgG - H&L (HRP; cat. no. ab6721; 1:5,000) and
rabbit anti-mouse IgG - H&L (HRP; cat. no. ab6728; 1:5,000)
secondary antibodies were obtained from Abcam (Cambridge, MA, USA),
and Bender Med rat TNF-α ELISA from eBioscience, Inc. (San Diego,
CA, USA). The protease inhibitor cocktail was purchased from Roche
Diagnostics GmbH (Mannheim, Germany).
Experimental protocols
The rat model of LPS-induced inflammation was used
as previously described (25) with
minor modifications. The rats were divided into three groups (n=4)
as follows: i) The normal control group, a vehicle-only, 80
µl dimethyl sulfoxide (Merck Millipore, Darmstadt, Germany)
to final volume of 1 ml with normal saline; intraperitoneally
injection (i.p.); ii) the LPS-treated group, LPS (0.5 mg/kg; i.p.);
and iii) the LPS + A-769662-treated group, LPS (0.5 mg/kg; i.p.)
and A-769662 (10 mg/kg; i.p.). The rats were weighed prior to
treatment (time set at zero) and at the end of the experiment. At 9
h post LPS injection, the heart and lung tissues were removed. The
harvested tissues were immediately rinsed in cold saline,
snap-frozen in liquid nitrogen and stored at -70°C, or were
directly fixed in formalin (Chem-Lab NV, Zedelgem, Belgium) after
rinsing for further analysis.
Measurements of TNF-α serum levels by
ELISA
Serum levels of TNF-α were quantified using the
ELISA kit according to the manufacturer's instructions. Briefly,
blood was collected in a non-heparinized tube from the hepatic
portal vein and serum was separated by centrifugation within 15 min
of collection at 238.97 × g for 10 min at 15°C. Serum was
immediately aliquoted and stored at −70°C until further analysis.
The concentration of TNF-α serum levels are expressed as pg/ml of
serum.
Neutrophil count
Prior to euthanasia, venous blood samples were
collected to determine the number of neutrophils in the blood. A
blood sample was smeared on a glass slide and the percentage of
neutrophils was counted at a magnification of ×100 using a CX31
optical microscope (Olympus Corporation, Tokyo, Japan) following
Giemsa (Labtron Co., Tehran, Iran) staining. The percentage of
neutrophils was calculated as a percentage of total white blood
cells.
Measurement of MPO activity in heart and
lung tissues
MPO activity was measured to quantify the activity
of neutrophils in the tissues of interest as previously described
(9), with minor modifications.
Briefly, the tissues were sectioned in 50 mM potassium phosphate
buffer (pH 6; Merck Millipore), containing 0.5% hexadecyl-trimethyl
ammonium bromide (HTAB; Sigma-Aldrich) and homogenized for 3 min at
7,673.7 × g. The homogenates were sonicated using an ultrasonic
cleaner (Starsonic 18–35, Bologna, Italy) for 10 sec, frozen and
thawed 3 times, and then centrifuged at 2,150.7 × g at 4°C for 45
min. An aliquot of the supernatant (0.1 ml) or standard was added
to 2.9 ml phosphate-buffered saline containing 0.167 mg/ml of
O-dianisidine dihydrochloride and 0.0005%
H2O2 (Merck Millipore). After 5 min, the
reaction was stopped with 0.1 ml 1.2 M HCl (Merck Millipore) and
absorbance was measured with a spectrophotometer (Cecil 9000, Cecil
Instruments, Cambridge, UK) at 400 nm. The concentrations were
calculated using calibration curves and expressed as units of MPO
in 100 mg weight of wet tissue (mU/100 mg).
Lung histopathological examination
For the histopathological examination, samples of
lung tissue were removed at the end of the experiment and fixed in
10% neutral-buffered formalin. The tissues were embedded in
paraffin, sectioned at 5 µm and stained with hematoxylin and
eosin (Labtron Co.) for assessment of tissue injury and neutrophil
accumulation in the microvasculature of injured lungs.
Western blot analysis
Western blotting was performed as previously
described (9), with minor
modifications. Following the experimental procedure, myocardial and
lung tissues were removed and immediately deep-frozen in liquid
nitrogen. The tissue samples were homogenized in ice-cold solution
(pH 7.4) containing 50 mM Tris-HCl (Merck Millipore)., 150 mM NaCl
(Merck Millipore)., 5 mM sodium pyrophosphate (Sigma-Aldrich), 50
mM NaF (Sigma-Aldrich), 1 mM EDTA (Merck Millipore)., 1 mM
dithiothreitol (Sigma-Aldrich), 0.1% sodium dodecyl sulfate (SDS;
Merck Millipore) (w/v), 1% TXT-100 (v/v; Sigma-Aldrich) and
protease inhibitor cocktail. Lung tissue contains extracellular
matrix that is resistant to homogenization. Thus, prior to tissue
lysis, tissue was ground thoroughly with a pestle and mortar, in
liquid nitrogen. Following homogenization in lysis buffer (Merck
Millipore), to completely destruct the cell membrane, samples were
sonicated 8–10 times, for 3–5 sec. Homogenized heart and lung
samples were centrifuged at 10,621 × g at 4°C for 10 min and
2,150.7 × g at 4°C for 45 min, respectively. The supernatant was
aliquoted and stored at -70°C for further analysis. The Bradford
Protein Assay kit (Sigma-Aldrich) was used to evaluate the protein
concentrations in the supernatant. The samples were mixed with
loading buffer [1 g SDS, 7 cc 1 M Tris (pH 6.8), 3 cc glycerin and
Bromophenol blue (all from Merck Millipore)] and subsequently
boiled for 10 min, at 100°C. Protein samples (50 µg) were
loaded onto a SDS-polyacrylamide gel (Sigma-Aldrich) using a
Min-Protean Tetra Cell system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) to be separated by electrophoresis at 120 mA.
Separated proteins were transferred to an Immobilon-P membrane (EMD
Millipore, Billerica, MA, USA) and blocked in 5% non-fat milk in
Tris-buffered saline with Tween-20 (all from Merck Millipore) at
room temperature with gentle shaking, for 1 h. The membranes were
washed with the wash buffer [Tris base (6.05 g,) + NaCl (8.76 g) +
Tween-20 (1%) to 1 L by deionized water, pH 7.4] all from (Merck
Millipore). The membranes were then incubated with the primary
antibodies against p-AMPKα (Thr172), AMPKα, MyD88 (1:1,000) and
GAPDH (1:5,000) at 4°C, with gentle shaking, overnight. The
membranes were then washed and incubated with the
peroxidase-conjugated goat anti-rabbit and rabbit anti-mouse
secondary antibodies (1:5,000), at room temperature, with gentle
shaking, for 1 h. For phosphorylated proteins, blocking buffer and
antibodies diluents contained 50 mM NaF as anti-phosphatase.
Subsequent to washing, antibodies were visualized using the BM
Chemiluminescence Western Blotting kit (Roche Diagnostics GmbH).
Densitometric analysis of the immunoblots was performed using Image
J software (version 1.41; National Institutes of Health, Bethesda,
MD, USA). The densitometric values of p-AMPKα were normal-ized to
AMPKα and in the case of MyD88 to GAPDH.
Statistical analysis
Data are presented as the mean ± standard error. One
way analysis of variance (ANOVA) was used for comparison among the
groups. If the ANOVA analysis indicated significant differences,
the Fisher's least significant difference post-hoc test was
performed to compare the mean values between the treatment groups
and control. P<0.05 was considered to indicate a statistically
significant difference.
Results
Changes in body weight
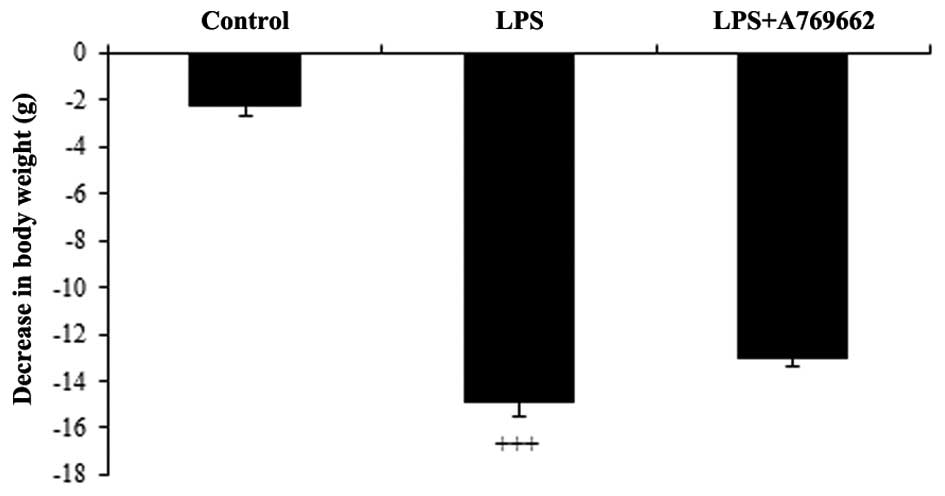
Following administration of LPS, the rats
demonstrated a general loss of appetite and reduction in water
consumption. This resulted in a significant reduction in body
weight of 14.9±1.6 g, at 9 h subsequent to LPS injection, compared
with the control group (P<0.001; Fig. 1). As demonstrated in Fig. 1, compared with the LPS-only treated
group, animals treated with A-769662 exhibited reduced weight
loss.
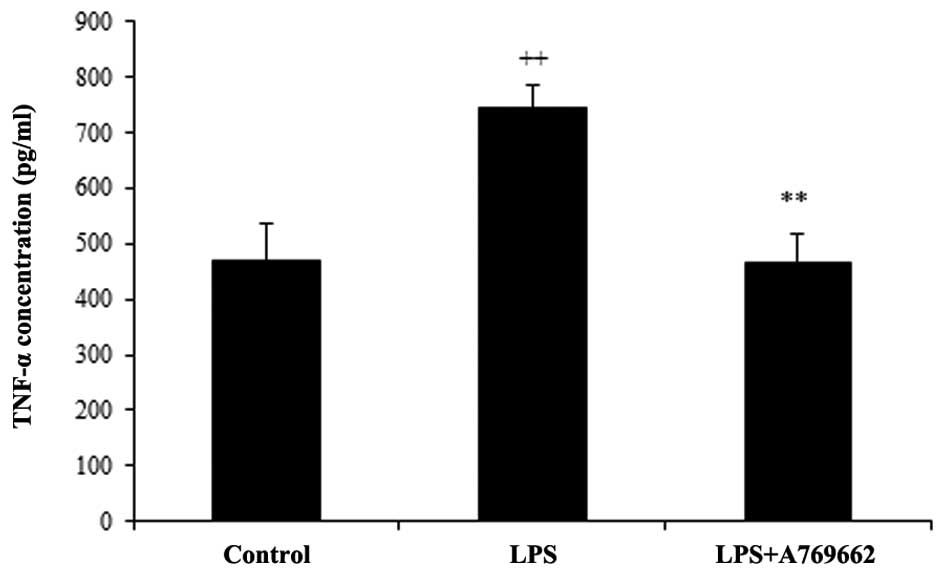
Effect of A-769662 on the serum levels of
TNF-α following LPS injection
As demonstrated in Fig.
2, the serum levels of TNF-α were significantly increased from
468±69.4 pg/ml in the normal control group to 743±42.9 pg/ml in the
LPS-treated group (P<0.01). The concentration of TNF-α in the
serum of the LPS + A-769662 group was reduced to a level similar to
that of the normal control group (467.2±51 pg/ml; P<0.01
compared with the LPS-only group).
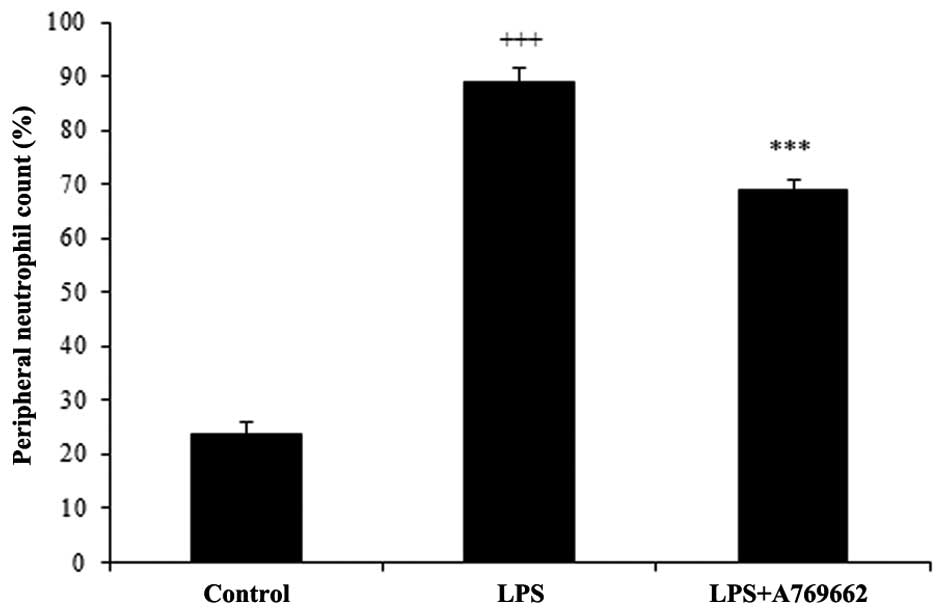
Efect of A-769662 on the blood neutrophil
count
Injection of LPS resulted in a prominent elevation
in the percentage of neutrophils from 23.7±2.2% in the normal
control group to 88.7±2.7% (P<0.001; Fig. 3). Administration of A-769662
significantly reduced the percentage of peripheral neutrophil to
68.9±2 compared with the LPS-treated group (P<0.01; Fig. 3).
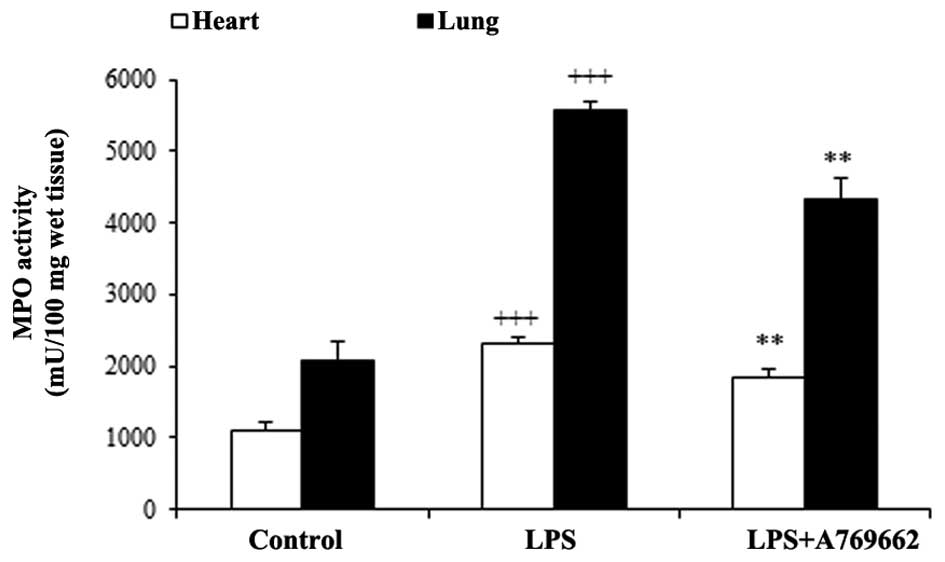
Effect of A-769662 on the heart and lung
MPO activity following LPS injection
A characteristic feature of acute endotoxemia is the
accumulation of neutrophils in the target tissues, thus MPO
activity was utilized as an index of neutrophil infiltration. As
demonstrated in Fig. 4, MPO
activity significantly increased in heart and lung tissues in the
LPS groups compared with the control groups (P<0.001).
Additional treatment with A-769662 significantly reduced the MPO
activity in the heart and lung tissues compared with the LPS-only
group (P<0.01; Fig. 4).
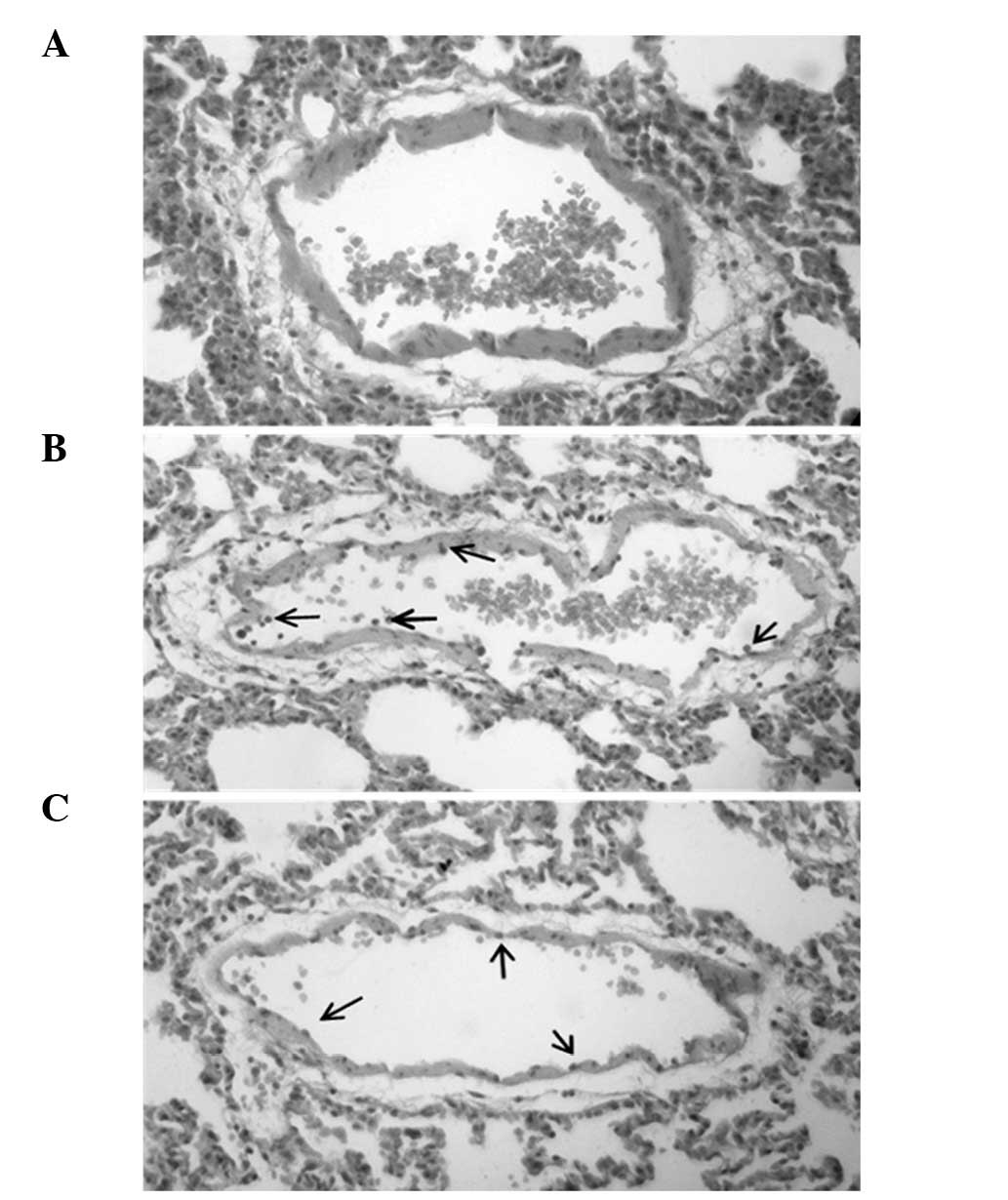
Histopathological examination of lung
tissue
Microscopic examination of the endothelium of the
lung tissue of LPS + A-769662-treated rats demonstrated reduced
neutrophil accumulation compared with the LPS-only treated group
(Fig. 5).
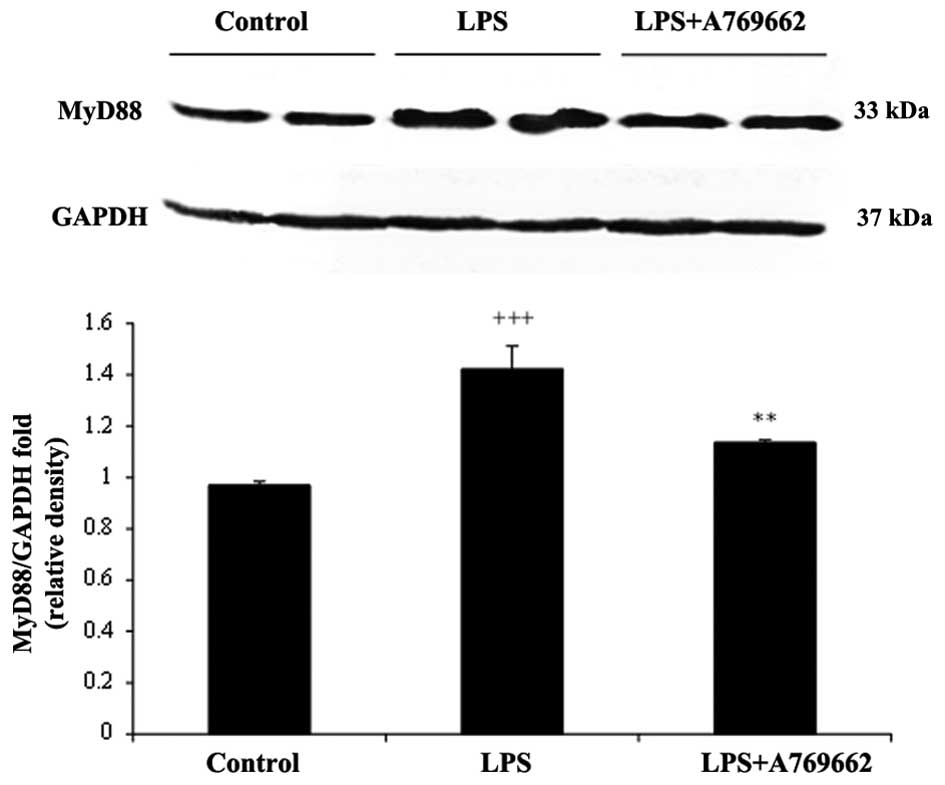
Effect of A-769662 on MyD88 protein
expression levels in the heart and lung tissues of the rats
injected with LPS
The protein expression levels of MyD88 were assessed
to determine the effect of the treatments. As demonstrated in
Fig. 6, 9 h subsequent to LPS injection, the
protein levels of myocardial MyD88 were significantly increased
compared with the control group (P<0.001). Additional treatment
with A-769662 led to a significant reduction in the MyD88 protein
expression levels compared with the LPS-only group (P<0.01;
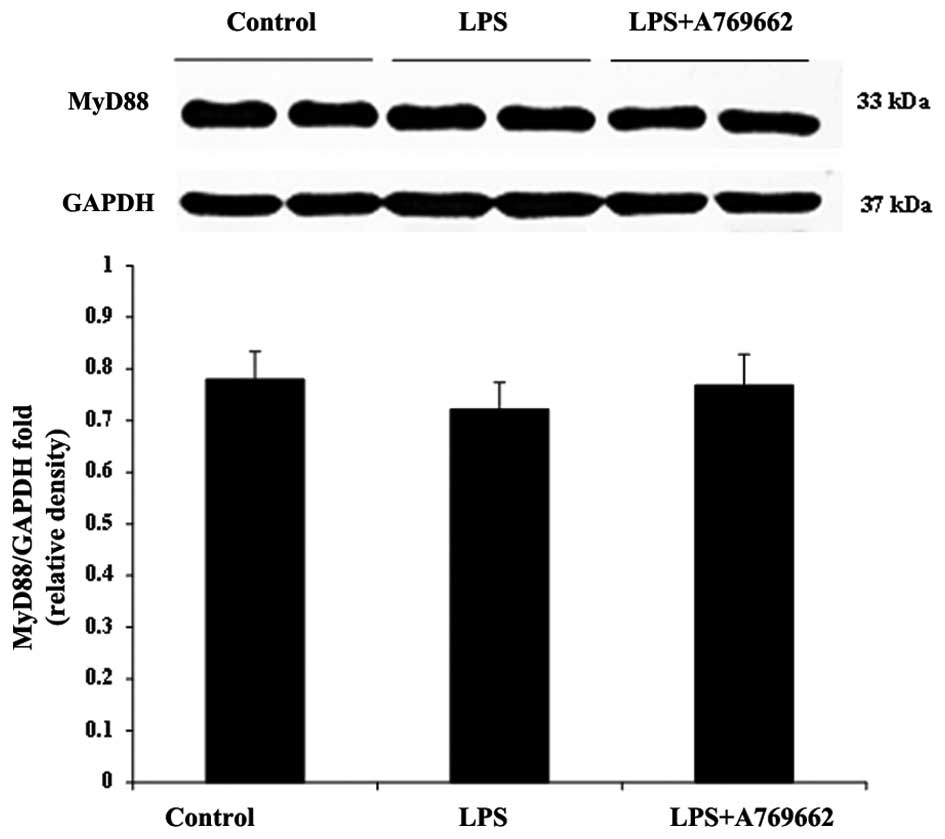
Fig. 6). Compared with the heart
tissue, LPS was observed to have no effect on the content of MyD88
in the lung and there was no significant difference in the lung
MyD88 levels between the LPS-only and LPS + A-769662 groups
(P>0.05; Fig. 7).
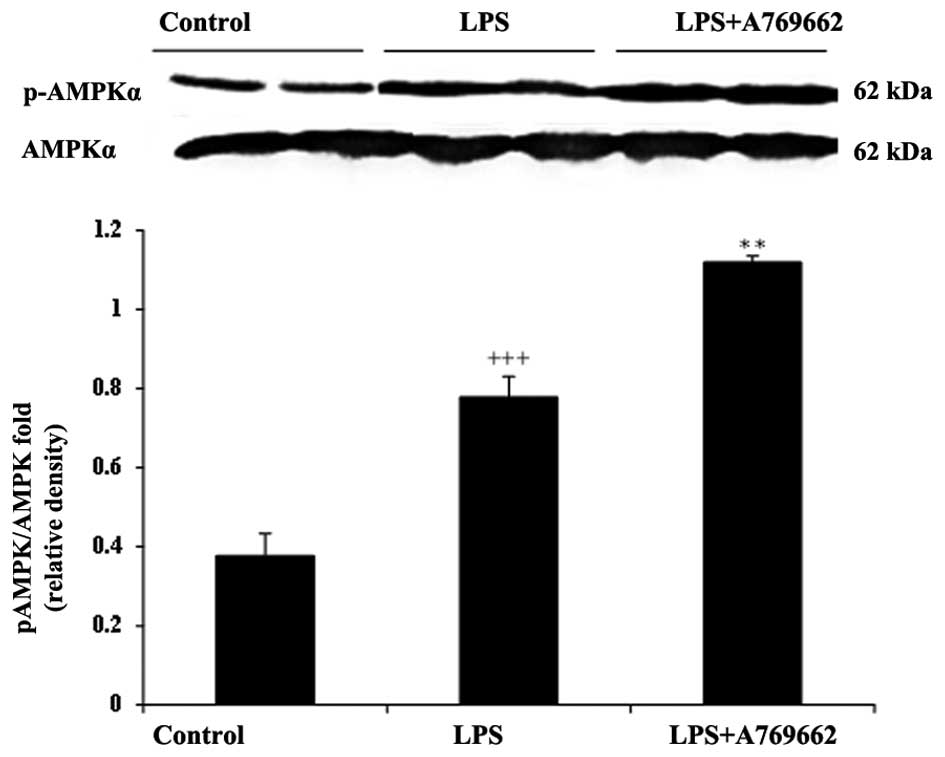
Effect of A-769662 on p-AMPKα protein
expression levels in the heart and lung tissues of LPS-injected
rats
AMPK is an energy regulator present in various cells
and its activation during metabolic stress, particularly
inflammation, serves a role in cell survival. A-769662 is an
established AMPK agonist, thus it was utilized for experimental
purposes. As acute endotoxemia is associated with inflammation, the
protein expression levels of p-AMPKα were determined in the
myocardial and lung tissues of LPS-injected rats with or without
A-769662 treatment.
As demonstrated in Fig.
8, LPS treatment induced a notable AMPK activation in the heart
tissue of rats. The relative expression of p-AMPKα to AMPKα in the
LPS-treated group was significantly increased compared with that of
the control group (P<0.001; Fig.
8). Co-administration with A-769662 in the heart tissue of rats
significantly enhanced the AMPK activation by LPS (P<0.01;
Fig. 8). However, as demonstrated
in Fig. 9, no significant effect
was observed in lung tissues following treatment with LPS or LPS +
A-769662.
Discussion
The present study demonstrated that A-769662
inhibited the LPS-induced increase in the peripheral neutrophil
count and MPO activity in the heart and lung tissues of the rats
injected with the endotoxin. In addition, the administration of
A-769662 significantly reduced the LPS-induced elevation of TNF-α
concentration levels in the serum of the rats. Following LPS
injection, levels of pro-inflammatory cytokines with a prominent
role in endotoxin-induced organ injury (8), such as TNF-α, rapidly increased in
the blood (25,26). For the results of the present
study, MPO activity was used as an index of neutrophil infiltration
and an increase in the activity levels was demonstrated in heart
and lung tissues. Neutrophil accumulation in lung and heart tissues
is a noticeable feature of acute endotoxemia (8). Excessive levels of LPS result in
acute endotoxemia associated with the systemic inflammation and
accumulation of macrophages in targeted tissues. Endotoxemia leads
to septic shock, multiple organ damage and death (6–8), and
pro-inflammatory cytokines, reactive oxygen and nitrogen species,
proteases and bioactive lipids are considered as tissue damaging
factors in endotoxemia (8).
AMPK serves a role in cellular energy homeostasis,
and as a metabolic regulation enzyme, its activation during
metabolic stress is important for cell survival (15). Previous studies have suggested that
AMPK activation has a protective effect in inflammatory conditions
(9,11–13).
Furthermore, AICAR and metformin are indirect and nonspecific AMPK
activators with a certain AMPK-independent effect (20), compared with A-769662 that is able
to selectively and directly activate AMPK (21,22)
by binding to its β subunit. This subunit is a site distinct from
those of AMP, however, in a similar process to that of AMP,
A-769662 allosterically activates AMPK and renders the
phosphorylated Thr172 residue resistant to protein phosphatases
(20).
AMPK activation has been demonstrated to be involved
in the anti-inflammatory effect of the agonists in different models
(11–13). In the current study, administration
of A-769662 prior to LPS injection was demonstrated to suppress the
neutrophil infiltration into the heart and lung tissues, and reduce
the peripheral neutrophil count. A-769662 was demonstrated to
activate AMPK through a mechanism involving the phosphorylation of
a subunit of the enzyme (20), and
previous studies indicated that the phosphorylation of AMPK may
suppress the TLR4 expression and activity in conditions associated
with inflammation, such as myocardial infarction (9,10).
In accordance with the activation of TLRs, in the present study the
injection of rats with LPS led to a marked increase in the MyD88
protein expression levels in the heart tissue and a considerable
elevation in the TNF-α serum levels. The LPS binding protein bound
to endotoxins is recognized by the CD14/TLR4-MD-2 complex in the
innate immune cells and delivers a signal through the plasma
membrane (27). Stimulation of
TLR4 facilitates the activation of MyD88, leading to nuclear
translocation of NF-κB and the production of pro-inflammatory
cytokines, including TNF-α and IL-6 (28). MyD88 is an adaptor molecule of the
TLR4 pathway and a prominent part of the LPS receptor complex
involved in the production of pro-inflammatory cytokines that lead
to tissue injury. Furthermore, reactive oxygen and nitrogen species
are destructive via products of endotoxemia (8), and their production through TLR4 and
MyD88-dependent signaling may lead to oxidative stress via AMPK
activation (15).
In support of these observations, the present study
demonstrated that the increase of MyD88 protein expression and
TNF-α serum levels in the heart tissue were significantly
attenuated by A-769662 administration, suggesting suppressed TLR
activity.
In addition to heart tissue, lung tissue is
sensitive to LPS-induced endotoxemia (8). In the present study, LPS
administration induced the elevation of MPO activity in the lung
tissue. This effect was confirmed by lung histopathological
analysis in which neutrophils sequestered onto the vessel firmly
adhered to the endothelial wall. However, the levels of MyD88 and
p-AMPK protein expression were not increased in the lung tissue
following LPS administration. In accordance with these results and
Lefort et al (29), LPS
administration (i.p.) may trigger a signal at the systemic or heart
level, but fails to induce a full signal to increase the levels of
MyD88 or p-AMPK in the lungs. Additionally, the results of the
current study demonstrated that the administration of A-769662 to
the LPS-injected rats resulted in a significant reduction of MPO
activity and neutrophil infiltration in the lung tissue, however,
no effect was observed in the levels of MyD88 or p-AMPK.
To the best of our knowledge, this is the first
study investigating the effect of A-769662 on the AMPK activity in
the lung tissue. AMPK is a heterotrimer complex comprised of α-, β-
and γ-subunits, each of which has two or more isoforms encoded by
multiple genes and are differentially expressed in various types of
tissue (17). The α2 and β2
isoforms are highly expressed in the myocardium, and the α1 and β1
isoforms are prominent in the lung (30). Additionally, the α1, β1 and γ1
isoforms are ubiquitously expressed. A-769662 selectively activates
the AMPK heterotrimeric complex containing α2/β1 subunits (31) that may be noticeable in the
myocardium and not in the lung tissue.
Previous studies demonstrated that activation of
AMPK by metformin diminishes the cardiac inflammatory responses
following myocardial infarction by suppressing the TLR4/MyD88
activity (9,10). Salminen et al (32) demonstrated that the activation of
AMPK inhibits NF-κB activity, suppresses the expression of the
pro-inflammatory cytokines and attenuates inflammatory injury
through phosphorylation of downstream targets, including silent
information regulator 1, PGC-lα, p53 and FoxOs. Furthermore, AMPK
activation inhibits acute and chronic colitis (11), autoimmune encephalomyelitis
(12), inflammation in cystic
fibrosis (33), pro-inflammatory
effects following lung injury (13) and LPS-induced expression of
pro-inflammatory molecules and mediators (32). Stimulating autophagy (34) or inhibiting NF-κB activation
(35) may be the mechanism
underlying the regulation of inflammation by AMPK activation
(32). The present study provided
evidence that A-769662 reduces the systemic feature of LPS-induced
endotoxemia.
In conclusion, the current study indicated that
A-769662 protects against LPS-induced inflammatory responses in
rats. The effect is associated with suppression of TLR activity in
the heart tissue, potentially due to the increase in AMPK activity.
Inhibition of neutrophil activity in the lung tissue was due to the
inhibition of systemic inflammation by treatment with A-769662. The
effect of A-769662 in the lung tissue was demonstrated to be
independent of the AMPK activation and TLR suppression. Therefore,
AMPK activation by A-769662 and the reduction of systemic features
of endotoxemia may be a promising target in the endotoxemia
treatment.
Acknowledgments
The present study was supported by the Research Vice
Chancellors of Tabriz University of Medical Sciences (Tabriz,
Iran). The study was written based on the data of Maryam
Rameshrad's Ph.D. thesis at Tabriz University of Medical Sciences
(no. 88).
References
|
1
|
Akira S and Takeda K: Toll-like receptor
signalling. Nat Rev Immunol. 4:499–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Takeda K: Evolution and integration of
innate immune recognition systems: The Toll-like receptors. J
Endotoxin Res. 11:51–55. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Beutler B: Inferences, questions and
possibilities in Toll-like receptor signalling. Nature.
430:257–263. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cristofaro P and Opal SM: Role of
Toll-like receptors in infection and immunity: Clinical
implications. Drugs. 66:15–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reitsma PH, Branger J, Van Den Blink B,
Weijer S, Van Der Poll T and Meijers JC: Procoagulant protein
levels are differentially increased during human endotoxemia. J
Thromb Haemost. 1:1019–1023. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ramana KV, Willis MS, White MD, Horton JW,
DiMaio JM, Srivastava D, Bhatnagar A and Srivastava SK:
Endotoxin-induced cardiomyopathy and systemic inflammation in mice
is prevented by aldose reductase inhibition. Circulation.
114:1838–1846. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jardin F, Brun-Ney D, Auvert B, Beauchet A
and Bourdarias JP: Sepsis-related cardiogenic shock. Crit Care Med.
18:1055–1060. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Connor AJ, Chen LC, Joseph LB, Laskin JD
and Laskin DL: Distinct responses of lung and liver macrophages to
acute endotoxemia: Role of toll-like receptor 4. Exp Mol Pathol.
94:216–227. 2013. View Article : Google Scholar :
|
|
9
|
Soraya H, Farajnia S, Khani S, Rameshrad
M, Khorrami A, Banani A, Maleki-Dizaji N and Garjani A: Short-term
treatment with metformin suppresses toll like receptors (TLRs)
activity in isoproterenol-induced myocardial infarction in rat: Are
AMPK and TLRs connected? Int Immunopharmacol. 14:785–791. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Soraya H, Clanachan AS, Rameshrad M,
Maleki-Dizaji N, Ghazi-Khansari M and Garjani A: Chronic treatment
with metformin suppresses toll-like receptor 4 signaling and
attenuates left ventricular dysfunction following myocardial
infarction. Eur J Pharmacol. 737:77–84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bai A, Ma AG, Yong M, Weiss CR, Ma Y, Guan
Q, Bernstein CN and Peng Z: AMPK agonist downregulates innate and
adaptive immune responses in TNBS-induced murine acute and
relapsing colitis. Biochem Pharmacol. 80:1708–1717. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nath N, Giri S, Prasad R, Salem ML, Singh
AK and Singh I: 5-aminoimidazole-4-carboxamide ribonucleoside: A
novel immunomodulator with therapeutic efficacy in experimental
autoimmune encephalomyelitis. J Immunol. 175:566–574. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao X, Zmijewski JW, Lorne E, Liu G, Park
YJ, Tsuruta Y and Abraham E: Activation of AMPK attenuates
neutrophil proinflammatory activity and decreases the severity of
acute lung injury. Am J Physiol Lung Cell Mol Physiol.
295:L497–L504. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shirwany NA and Zou MH: AMPK in
cardiovascular health and disease. Acta Pharmacol Sin.
31:1075–1084. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Young LH, Li J, Baron SJ and Russell RR:
AMP-activated protein kinase: A key stress signaling pathway in the
heart. Trends Cardiovasc Med. 15:110–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dyck JR and Lopaschuk GD: AMPK alterations
in cardiac physiology and pathology: Enemy or ally? J Physiol.
574:95–112. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hardie DG, Carling D and Gamblin SJ:
AMP-activated protein kinase: Also regulated by ADP? Trends Biochem
Sci. 36:470–477. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hardie DG: Minireview: The AMP-activated
protein kinase cascade: The key sensor of cellular energy status.
Endocrinology. 144:5179–5183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kemp BE: Bateman domains and adenosine
derivatives form a binding contract. J Clin Invest. 113:182–184.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim AS, Miller EJ, Wright TM, Li J, Qi D,
Atsina K, Zaha V, Sakamoto K and Young LH: A small molecule AMPK
activator protects the heart against ischemia-reperfusion injury. J
Mol Cell Cardiol. 51:24–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cool B, Zinker B, Chiou W, Kifle L, Cao N,
Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, et al:
Identification and characterization of a small molecule AMPK
activator that treats key components of type 2 diabetes and the
metabolic syndrome. Cell Metab. 3:403–416. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Göransson O, McBride A, Hawley SA, Ross
FA, Shpiro N, Foretz M, Viollet B, Hardie DG and Sakamoto K:
Mechanism of action of A-769662, a valuable tool for activation of
AMP-activated protein kinase. J Biol Chem. 282:32549–32560. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao X, Petursson F, Viollet B, Lotz M,
Terkeltaub R and Liu-Bryan R: Peroxisome proliferator-activated
receptor γ coactivator 1α and FoxO3A mediate chondroprotection by
AMP-activated protein kinase. Arthritis Rheumatol. 66:3073–3082.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Petursson F, Husa M, June R, Lotz M,
Terkeltaub R and Liu-Bryan R: Linked decreases in liver kinase B1
and AMP-activated protein kinase activity modulate matrix catabolic
responses to biomechanical injury in chondrocytes. Arthritis Res
Ther. 15:R772013. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meng X, Ao L, Meldrum DR, Cain BS, Shames
BD, Selzman CH, Banerjee A and Harken AH: TNF-alpha and myocardial
depression in endotoxemic rats: Temporal discordance of an
obligatory relationship. Am J Physiol. 275:R502–R508.
1998.PubMed/NCBI
|
|
26
|
Copeland S, Warren HS, Lowry SF, Calvano
SE and Remick D; Inflammation and the Host Response to Injury
Investigators: Acute inflammatory response to endotoxin in mice and
humans. Clin Diagn Lab Immunol. 12:60–67. 2005.PubMed/NCBI
|
|
27
|
Turyn D, Dominici FP, Sotelo AI and Bartke
A: Specific interactions of growth hormone (GH) with GH-receptors
and GH-binding proteins in vivo in genetically GH-deficient Ames
dwarf mice. Growth Horm IGF Res. 8:389–396. 1998. View Article : Google Scholar
|
|
28
|
Lu YC, Yeh WC and Ohashi PS: LPS/TLR4
signal transduction pathway. Cytokine. 42:145–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lefort J, Singer M, Leduc D, Renesto P,
Nahori MA, Huerre M, Créminon C, Chignard M and Vargaftig BB:
Systemic administration of endotoxin induces bronchopulmonary
hyperreactivity dissociated from TNF-alpha formation and neutrophil
sequestration into the murine lungs. J Immunol. 161:474–480.
1998.PubMed/NCBI
|
|
30
|
Kim M and Tian R: Targeting AMPK for
cardiac protection: Opportunities and challenges. J Mol Cell
Cardiol. 51:548–553. 2011. View Article : Google Scholar :
|
|
31
|
Timmermans AD, Balteau M, Gélinas R,
Renguet E, Ginion A, de Meester C, Sakamoto K, Balligand JL,
Bontemps F, Vanoverschelde JL, et al: A-769662 potentiates the
effect of other AMP-activated protein kinase activators on cardiac
glucose uptake. Am J Physiol Heart Circ Physiol. 306:H1619–H1630.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Salminen A, Hyttinen JM and Kaarniranta K:
AMP-activated protein kinase inhibits NF-κB signaling and
inflammation: Impact on healthspan and lifespan. J Mol Med Berl.
89:667–676. 2011. View Article : Google Scholar
|
|
33
|
Myerburg MM, King JD Jr, Oyster NM, Fitch
AC, Magill A, Baty CJ, Watkins SC, Kolls JK, Pilewski JM and
Hallows KR: AMPK agonists ameliorate sodium and fluid transport and
inflammation in cystic fibrosis airway epithelial cells. Am J
Respir Cell Mol Biol. 42:676–684. 2010. View Article : Google Scholar :
|
|
34
|
Egan DF, Shackelford DB, Mihaylova MM,
Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor
R, et al: Phosphorylation of ULK1 (hATG1) by AMP-activated protein
kinase connects energy sensing to mitophagy. Science. 331:456–461.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hattori Y, Nakano Y, Hattori S, Tomizawa
A, Inukai K and Kasai K: High molecular weight adiponectin
activates AMPK and suppresses cytokine-induced NF-kappaB activation
in vascular endothelial cells. FEBS Lett. 582:1719–1724. 2008.
View Article : Google Scholar : PubMed/NCBI
|