Introduction
Glucose is the primary metabolic fuel of the brain,
and its availability is directly associated with neuronal activity
(1). Recurrent episodes of
hypoglycemia; a condition where blood glucose level decrease to
below normal levels as a result of the administration of an
incorrect insulin dosage, prolonged fasting or strenuous exercise,
may result in the loss of neurons, impaired intelligence quotient
and may ultimately lead to morbidity (2). Hernandez-Fonseca et al
(3) demonstrated that the cerebral
cortex and hippocampus, which are critically important for
cognition, are vulnerable to injury as a result of hypoglycemia.
Very little is known about the effects of hypoglycemia on the
function of the hypothalamus, which is a region of the brain that
is critically important for regulating feeding and energy balance,
and is rich in glucose-sensing neurons (4).
Previously, ovarian steroid hormones have been
demonstrated to exhibit important effects in the maintenance of
brain function (5). Estrogen (E2)
has been demonstrated to possess anti-apoptotic effects (6,7), and
neuroprotective effects against stroke (ischemic injury) (8), memory-associated behavioral tasks
(9) and hypertension (10). Postmenopausal women taking hormone
replacement therapy demonstrate a reduction in counter-regulatory
responses to hypoglycemia when compared with women not taking E2
(11). The actions of E2 are
mediated through: i) The increased synthesis and activity of growth
factors (12); ii) a reduction in
the activity of apoptotic factors (13); iii) protection from several
neurotoxic agents (14–16); iv) potential antioxidant effects,
leading to a reduction in neuronal nitric oxide synthase in brain
homogenates (6); and iv) a
reduction in glutamate receptors (17).
A number of previous studies have reported the
neurotrophic and neuroprotective effects of E2 (6,16,18–20).
For instance, E2 has been demonstrated to stimulate the growth of
hypothalamic explants, as well as those in the preoptic area of the
basal diencephalons (18). In
addition, E2 protects neuroblastoma cells from serum deprivation
(19), increases the mRNA
expression levels of the gene encoding the anti-apoptotic
NEP1-interacting protein in neuroblastoma (16), completely attenuates hemoglobin
neurotoxicity in cortical cultures (20), and protects primary cerebrocortical
neurons from hypoglycemia (6).
It has been suggested that E2 affects cell
viability, survival and proliferation, and regulates the expression
of target genes through two mechanisms: i) By binding to and
activating the estrogen receptor (ER)-α and ER-β receptors, which
belong to the steroid/thyroid hormone superfamily of transcription
factors (21,22), and are responsible for mediating
the classical or genomic pathways of E2 to regulate gene
transcription; and ii) by mediating intracellular signaling of
membrane receptors [e.g. G-protein, Ca2+, cyclic
adenosine monophosphate (cAMP) and protein kinase C] (23,24),
which are located in the cytoplasm or the plasma membrane and are
involved in the non-genomic or membrane-initiated steroid signaling
pathways of E2.
Survival stimuli generally activate transmembrane
receptors through G-protein coupled receptors located in the plasma
membrane (25,26). As a result,
phosphotidylinositol-3-OH kinase (PI3K) is activated, which
subsequently activates a cascade of signaling molecules leading to
alterations in the AKT/protein kinase B signaling pathway (27); a key regulator of neuronal cell
death. AKT is a proto-oncogene, which is activated by a variety of
substrates and has numerous effects, including regulating cell
survival, proliferation and protein synthesis. The first step in
the AKT pathway is the activation of the receptor tyrosine kinase
(RTK). RTK activates PI3K, which phosphorylates
phosphatidylinositol phosphate (PIP)-2 to PIP3. PIP3 then
phosphorylates and activates AKT (28) at threonine 308 and serine 473
residues. Transfection of granule cells with a dominant-negative
AKT allele abolished the ability of insulin-like growth factor-1 to
promote cell survival (29). By
contrast, transfecting granule cells with wild-type AKT alleles
promoted cell survival (29). A
previous study suggested that AKT inhibits apoptosis induced by
death stimuli, and its activity is required for growth
factor-induced survival (30).
PRAS40 is a substrate of AKT, and due to this functional
association, PRAS40 expression is considered to be a marker of AKT
activity. PRAS40 is phosphorylated by AKT and binds to 14-3-3
family proteins (30,31).
While a number of in vitro and in vivo
models exist to study the toxicity of hypoglycemia and investigate
the protective effects of E2 (7,32) to
the best of our knowledge, the present study is the first to report
hypoglycemic damage to hypothalamic cells and demonstrate that E2
protects hypothalamic cells from hypoglycemic conditions. Using
this system, E2 was observed to protect hypothalamic cells from
hypoglycemic shock. The authors hypothesize that the
neuroprotective effects of E2 under hypoglycemic conditions may be
mediated through the AKT signaling pathway.
Materials and methods
Cell culture and glucopenic
insult
The N38 murine hypothalamic cell line (Cedarlane,
Cellutions Biosystems, Burlington, Ontario, Canada) was cultured in
Dulbecco's modified Eagle's medium (DMEM, consisting of 4.5 g/l
glucose, L-glutamine, sodium pyruvate and phenol red; 1X;
Mediatech; Corning Life Sciences, Corning, NY, USA) containing 0.5%
fetal bovine serum (FBS, Mediatech; Corning Life Sciences) and 1%
penicillin-streptomycin (Mediatech; Corning Life Sciences) until
the cells reached 70% confluence. N38 cells were subsequently
washed with DMEM, trypsinized with trypsin EDTA (1X; Mediatech;
Corning Life Sciences) and divided into the following groups based
on different media formulations: i) DMEM group, consisting of DMEM
plus 0.5% FBS; ii) DMEM-G group, cultured in DMEM (1X; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) with sodium
pyruvate plus 5% FBS in the absence of glucose; iii) DMEM-PR group,
cultured in DMEM (1X; Mediatech; Corning Life Sciences) with sodium
pyruvate plus glucose and 5% FBS in the absence of phenol red; iv)
DMEM+E2 group, cultured in DMEM plus 1 mM E2 (beta-estradiol;
Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) and 5% FBS; v)
DMEM-G+E2 group, cultured in DMEM plus 1 mM E2 and 5% FBS in the
absence of glucose; and vi) DMEM-PR+E2 group, cultured in DMEM in
absence of phenol red, with 1 mM E2 and 5% FBS. These media
formulations were selected in order to assess cell growth and
proliferation under the following conditions: i) normal; ii)
hypoglycemic; iii) in the absence of phenol red, as phenol red is
known to exhibit mild estrogenic effects, but is commonly used as
an indicator of pH in cell growth medium (33); iv) in the presence of estrogen
only; v) in hypoglycemic conditions plus estrogen; vi) in the
absence of phenol red and the presence of estrogen.
Assessment of cell morphology and
number
Cells were cultured in 6-well plates
(~1×104-1×105 cells/well) and maintained in
the different media formulations for 24, 48, 72 and 96 h at 37°C
and a relative humidity of 95%. At each time point, the cells were
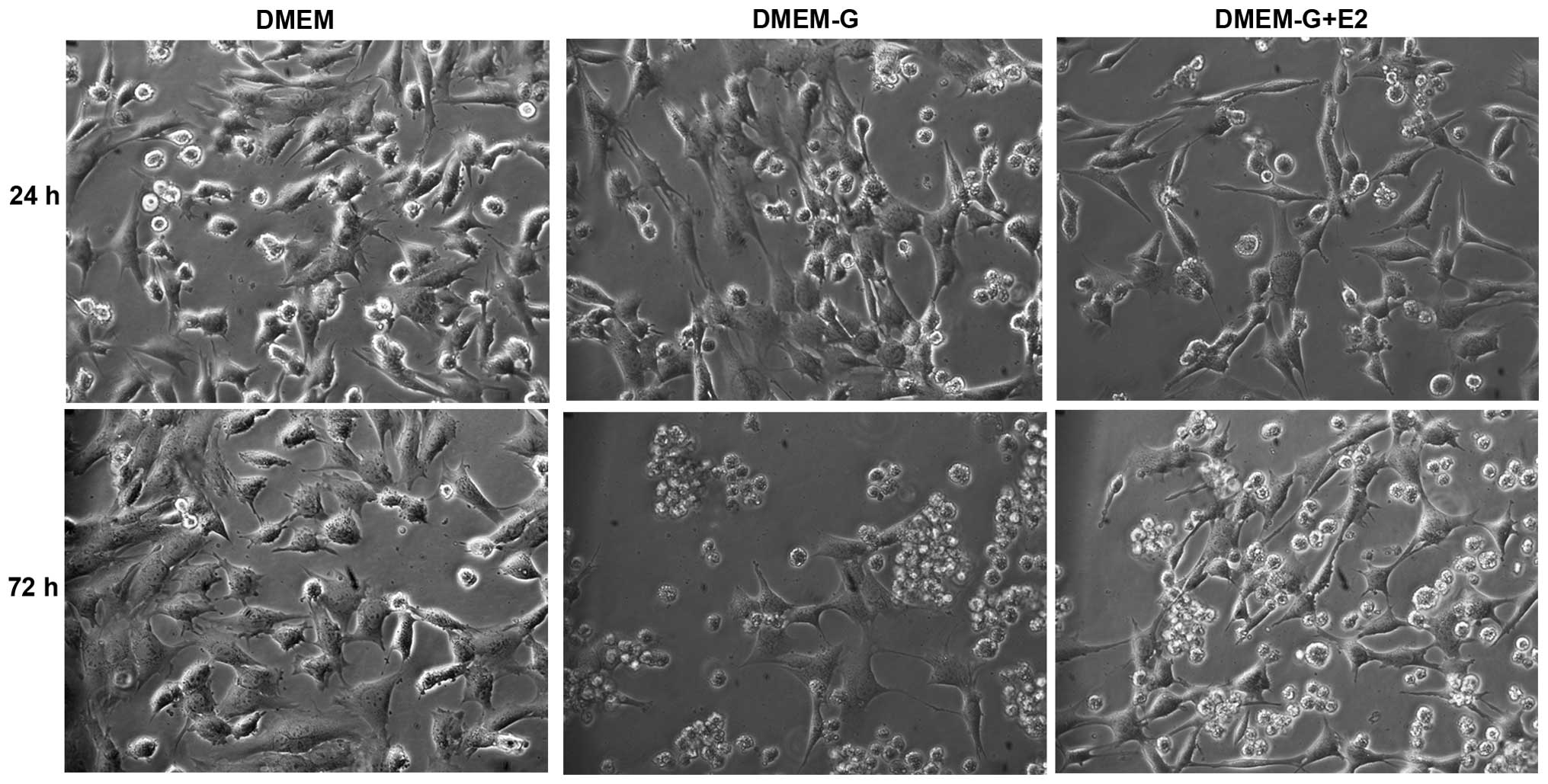
trypsinized and counted with a hemocytometer. Under the microscope
N38 cells were oval-shaped cell bodies connected to extensions.
Rounded cells with no extensions were considered to be dead.
Preliminary experiments were conducted using trypan blue in order
to determine the viability of cells. Photomicrographs of cells
cultured in DMEM, DMEM without glucose and DMEM plus 1 mM E2 in the
absence of glucose at 24 and 72 h were obtained in order to detect
morphological changes in the cells.
Assessment of mitochondrial activity
using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphrenyltetrazolium
bromide (MTT) assay
The MTT assay provides a quantitative measure of
mitochondrial activity and is an indicator of cell viability. The
assay is based on the conversion of a tetrazolium salt to the
colored product formazan by dehydrogenase enzymes, which can be
detected spectrophotometrically (34). Under glucopenic stress, cells may
exhibit altered mitochondrial activity, which can be detected using
the MTT assay. For the purposes of the present study, cell
viability was assessed using the MTT Cell Proliferation assay kit
(cat. no. 30–1010K; American Type Culture Collection, Manassas, VA,
USA) according to the manufacturer's instructions. N38 cells were
cultured in 6-well plates (10,000 cells/well) and maintained in the
different media formulations for 24, 48, 72 and 96 h before MTT
reagent was added. The absorbance was read at 570 nm. Since, no
significant alterations in cell viability were observed at 48 and
96 h, cells cultured for 24 and 72 h were used for further
analyses.
Assessment of cell injury by lactate
dehydrogenase (LDH) activity
Cell injury was assessed using the Cytotoxicity
Detection kitPLUS (LDH; cat. no. 04 744 926 001, Roche
Diagnostics, Indianapolis, IN, USA), which measures LDH activity
released from cells with damaged plasma membranes, according to the
methods described previously by Regan and Panter (35). Briefly, the cells were cultured in
6-well plates (1×106 cells/ml/well) and in the different
media formulations for 24 and 72 h. For each experimental condition
one well was designated as a high control (maximum LDH activity
produced in cells=maximum LDH release) and one as a low control
(LDH activity produced by untreated normal cells=spontaneous LDH
release). Prior to the addition of lysis buffer to the high control
all cells were incubated for ~15 min at 37°C, 5% CO2 and 95%
humidity. A mixture of catalyst and dye was then added to each of
the wells and incubated for 20 min at 25°C in the dark, before the
stop solution was added. The optical density (OD) was determined at
490 nm and the level of cytotoxicity was calculated using the
following formula: Cytotoxicity (%)=[(sample OD-low control
OD)/(high control OD-low control OD)]x100.
Assessment of the non-genomic pathway
of neuroprotection by E2 using a PRAS40 enzyme-linked immunosorbent
assay (ELISA)
N38 cells were first exposed to glucopenic stress
for 24 h, using the aforementioned methods. As the neuroprotective
effect of E2 was most pronounced at 24 h, the mechanism of action
of E2 was studied only at that time point. Using the PRAS40 STAR
ELISA kit (cat. no. 17–477; EMD Millipore, Billerica, MA, USA), the
cells were treated according to the manufacturer's protocol and the
supernatants were extracted. Using the anti-PRAS40 antibody (cat.
no. 17-477B) and the horseradish peroxidase-conjugated anti-rabbit
immunoglobulin G secondary antibody (cat. no. 17-477E) included in
the ELISA kit, PRAS40 expression was detected, according to the
manufacturer's protocol.
Statistical analysis
The data obtained from the cell count, MTT, LDH and
PRAS40 ELISA assays were expressed as the mean ± standard
deviation. Statistical differences among groups were analyzed using
one-way analysis of variance with a post-hoc Tukey test (JMP
software, version 7; SAS Institute, Inc., Cary, NC, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
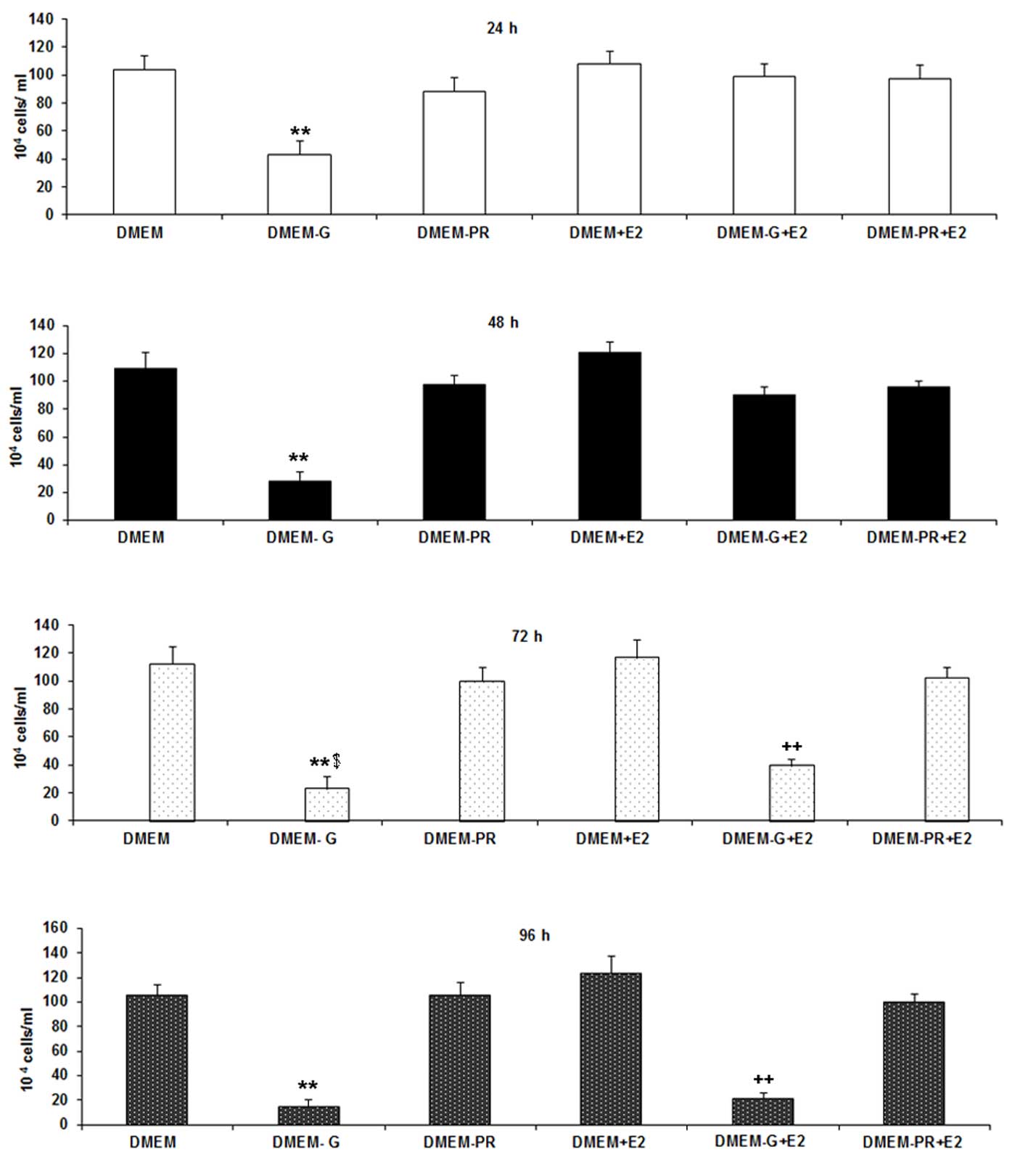
Microscopic and quantitative analysis
of the neuroprotective effects of E2 on cell survival following
hypoglycemic injury
N38 cells in different groups were cultured in their
respective media formulations for 24, 48, 72 and 96 h prior to
visualization under a light microscope. As shown in Figs. 1 and 2, a qualitative and quantitative decrease
in cell number under hypoglycemic conditions was observed. A
significant increase in cell number was observed following exposure
to E2 under hypoglycemic conditions at 24 h when compared with
untreated cells under hypoglycemic conditions (42.8×104
vs. 98.5×104 cells; P<0.0001). Within 72 h the effect
dissipated and there was no significant difference in in cell
number between cells exposed to hypoglycemic conditions in presence
or absence of E2. The optimum concentration of E2 on reversing the
effects of hypoglycemia in N38 cells was determined in preliminary
experiments, by testing three different concentrations (0.1, 1 and
10 mM) of E2 (data not shown). At 0.1 mM E2 the number of cells was
significantly lower compared with 1 mM E2 (P=0.0096), and a 10-fold
increase in the concentration of E2 did not have any significant
effect on cell number (data not shown). Therefore, the optimum
concentration of E2 used in the present study was 1 mM. Cells in
the DMEM-PR group exhibited no significant alterations in cell
number when compared with cells in the DMEM group at all time
points, which indicated that phenol red had no effect on cell
number. No additive effects were observed following exposure of
cells to DMEM+E2 when compared with cells exposed to DMEM-PR+E2 at
all-time points (Fig. 1). The
small decrease in cell number observed between cells exposed to
DMEM-PR+E2 and DMEM+E2 did not reach statistical significance,
which suggests that phenol red demonstrated no significant effect
on the experimental results.
Assessing the effects of E2 on
mitochondrial function in N38 cells under hypoglycemic
conditions
Mitochondrial function was used to assess the
viability of cells in each treatment group, as shown in Fig. 3. At 24 h, a significant reduction
in mitochondrial function was observed in cells that were exposed
to hypoglycemic conditions when compared with cells cultured in
control media (DMEM vs. DMEM-G, P<0.0001). By contrast, a ~80%
increase in mitochondrial function was observed in cells in the
presence of E2 under hypoglycemic conditions when compared with
cells exposed to hypoglycemic conditions alone (DMEM-G vs.
DMEM-G+E2, P<0.0001). The neuroprotective effects of E2 were
gradually lost within 72 h of treatment, and cells exhibited a
reduction in mitochondrial function even in the presence of E2
(Fig. 3, P<0.0001). However,
the restoration of mitochondrial function by E2 did not prevent
cell death but may have prolonged cellular survival, since
mitochondrial function and cell viability were observed to decrease
gradually over time in the presence of E2. A small population of
cells was able to survive in the absence of glucose for 72 h
(Fig. 3). A gradual decrease in
cell viability in the majority of experimental conditions was
observed, likely due to the gradual depletion of growth factors in
the media. Similar to E2-treated cells under hypoglycemic
conditions, the number of viable cells in the DMEM-PR+E2 group
decreased over the 72 h period, which indicated that phenol red had
no additive effects on cell viability. Similar to 72 h, no
neuroprotective effects of estrogen on cell viability were observed
at 96 h.
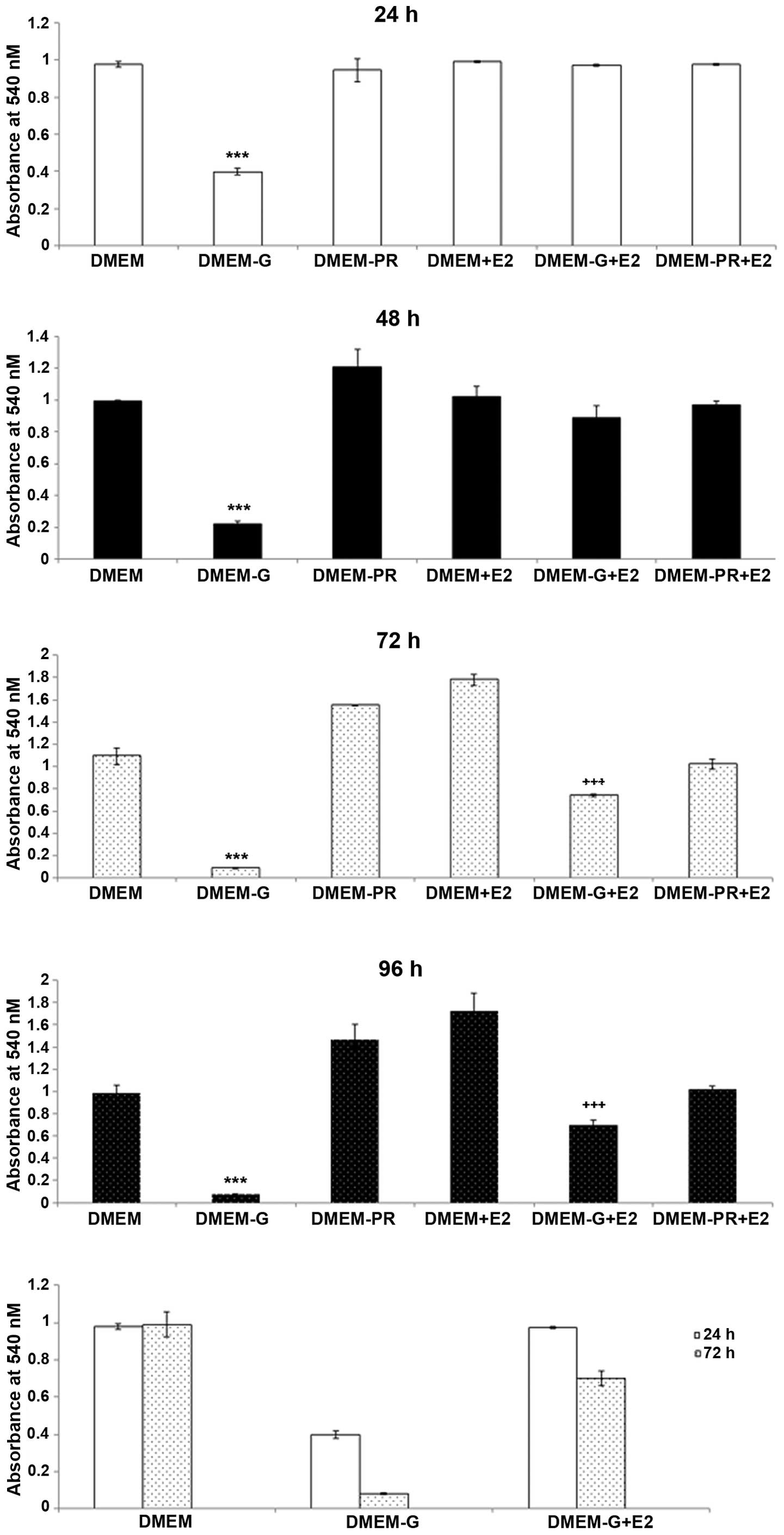 | Figure 3.Mitochondrial function in N38 cells.
The mitochondrial function of N38 cells under hypoglycemic
conditions following exposure to E2 at 24, 48, 72 and 96 h was
determined using the
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphrenyltetrazolium bromide
assay. Minimal mitochondrial function was observed in the DMEM-G
group, which resulted in extensive cell death when compared with
the other experimental groups at all time points (DMEM-G vs. DMEM,
DMEM-PR, DMEM+E2, DMEM-G+E2, DMEM-PR+E2). Exposure to E2
demonstrated the highest mitochondrial function at 24 h
(P<0.0001). Exposure of cells under hypoglycemic conditions to
E2 demonstrated a significant increase in mitochondrial function
when compared with untreated cells under hypoglycemic conditions at
24 h. The effect was similar at 48 h. However, within 72 h, a
significant reduction in mitochondrial function was observed in
DMEM-G+E2 cells when compared with DMEM-G cells. A similar effect
was observed at 96 h. ***P<0.0001 vs. all other groups at all
time points; +++P<0.0001 vs. all other groups at all
time points. DMEM; Dulbecco's modified Eagle's medium plus phenol
red; DMEM-G, DMEM without glucose; DMEM-PR, DMEM without phenol
red; DMEM+E2, DMEM plus 1 mM estrogen; DMEM-G+E2, DMEM plus 1 mM E2
without glucose; DMEM-PR+E2, DMEM plus 1 mM E2 without phenol
red. |
Assessing the effects of E2 on
hypoglycemia-induced cytotoxicity in N38 cells
Consistent with the MTT assay at 24 h, an increase
in the release of LDH (% cytotoxicity) from dead cells was observed
in N38 cells cultured under hypoglycemic conditions when compared
with those cultured under normal conditions (Fig. 4). A significant decrease
(P<0.0001) in LDH activity was observed in cells under
hypoglycemic conditions following exposure to E2, at 24 h when
compared with untreated cells under hypoglycemic conditions.
However, the protective effects of E2 on hypoglycemic shock were
reduced within 72 h. A marked increase in LDH activity was observed
between cells exposed to E2 at 24 h compared with cells exposed to
E2 for 72 h, whereas the level of cytotoxicity was consistent in
cells cultured under normal or hypoglycemic conditions at these
time points (Fig. 4). These
results suggested that E2 may prolong the viability of cells under
hypoglycemic conditions.
Effect of E2 on PRAS40 expression in
N38 cells under hypoglycemic conditions
The AKT protein is a critical regulator of cell
survival, the levels of which may be affected by E2 in the absence
of glucose availability, and can be assessed by the level of PRAS40
expression. At 24 h, no significant alterations in PRAS40
expression were observed in cells under hypoglycemic conditions
following exposure to E2 when compared with untreated cells under
hypoglycemic conditions. In addition, a slight decrease in PRAS40
expression was observed in cells under hypoglycemic conditions
compared with those cultured under normal conditions. These results
suggest that additional signaling pathways may mediate the effects
of E2 under hypoglycemic conditions.
Discussion
The present study is the first, to the best of our
knowledge, to report the neuroprotective effects of E2 on
hypoglycemic injury in a hypothalamic cell line. The results
indicated three novel observations: i) E2 exhibits neuroprotective
effects, which dissipate over time; ii) a subset of neuronal cell
exist that can resist hypoglycemic insult even after 72 h; and iii)
E2 may not exert the neuroprotective effects through AKT/PRAS40
signaling pathway.
In the present study, the neuronal viability and
neurotoxicity analyses demonstrated that E2 exhibited
neuroprotective effects in N38 cells under hypoglycemic conditions
within 24 h of exposure. In the absence of glucose, multiple death
stimuli activate different neuronal death pathways (6,36,37).
In the present study, the gradual reduction in neuronal cell number
in the absence of glucose, correlated with the observed decrease in
mitochondrial function. Mitochondrial dysfunction serves a critical
role in necrotic cell death and apoptosis. The high demand for
adenosine triphosphate (ATP) in neurons is dependent on ATP
production by the mitochondria (38,39).
Damage to mitochondria leads to the disruption of ATP production
and a concomitant increase in reactive oxygen species, which can
lead to cell death (38,39). In the current study, the observed
increase in mitochondrial function in the presence of E2 under
hypoglycemic conditions may have been due to the stabilization of
mitochondria under Ca2+ loading, which maintains the
mitochondrial membrane potential and preserves mitochondrial
function (40). Whether the
observed increase in cell death under glucopenic stress was due to
mitochondrial dysfunction or activation of apoptotic pathways (or
both) remains unknown. However, E2 may facilitate mitochondrial
membrane stabilization and may delay the activation of cell death
signaling pathways. In the present study, the attenuation in the
hypoglycemia-induced reduction in cell number was only observed
within 24 h of E2 exposure and gradually decreased within 72 h,
which indicated that E2 exhibits short-term neuroprotective
effects. A previous study demonstrated that E2 may facilitate cell
survival rather than cellular proliferation (11), and following activation of the
apoptotic signaling cascade, E2 demonstrated no effect on cell
viability. In the present study, a small population of cells was
unexpectedly observed to survive for >72 h in the absence of
glucose. Previous studies have demonstrated that astrocytes present
in primary hippocampal cultures possess glycogen stores that
provide a buffer to alterations in glucose reserves (41,42).
However, the hypothalamic model used in the present study consisted
of immortalized hypothalamic neurons and no supporting cells.
Therefore, additional pathways that facilitate the survival of
these cells during hypoglycemic shock may have been activated.
The classical signaling pathway by which E2
modulates the expression of downstream target genes, involves the
binding of E2 to the nuclear ER, which subsequently activates a
signaling cascade and the transcription of a number of genes
(16). However, several reports
have indicated that E2 exhibits antiapoptotic effects in number of
cell systems (5,43–45),
via non-genomic signaling pathways. A previous study demonstrated
that alterations in blood glucose levels led to fluctuations in the
phosphorylation states of members of the AKT pathway in the
cerebral cortex and hippocampus (46). Therefore, alterations in AKT
protein levels following E2 exposure under hypoglycemic conditions
in the present study were investigated by assessing PRAS40
expression. Cell survival in the presence of E2 under hypoglycemic
conditions, may have been due to activation of the AKT signaling
pathway involving PRAS40, or the inhibition of apoptotic pathways
or additional pathways by caveolin. Several studies have
demonstrated that E2 activates multiple kinases in various types of
brain cells within min, including ERK, AKT and the cAMP response
element binding protein (47–49).
In addition, it has been reported that AKT is activated in the
presence of E2 in rat adrenal medulla pheochromocytoma PC12 cells,
which protects neurons against β-amyloid-induced apoptosis by
upregulating telomerase activity (50). Preconditioning of neuronal cells by
exposure to compounds, such as growth factors, E2 and free
radicles, is an effective neuroprotective strategy against stroke
(51), hypothermia (52), hypoxia (53) and involves the AKT signaling
pathway. In the cerebral cortex and the hippocampus, AKT-glycogen
synthase kinase 3β (GSK3β) coupling is influenced by fluctuations
in blood glucose concentrations (46). Therefore, the authors speculate
that E2 may be able to affect cell survival by upregulating
pAKT/pPRAS40, but may have a limited effect on total AKT/PRAS40
activity, since the role of AKT in signal transduction is the
amplification of signals induced by RTK (54). However, an increase in pAKT levels
may serve a more significant role in determining the actual
physiological effects of E2, which requires further
investigation.
Taking into account the biochemical and molecular
results obtained in in the present and in previous studies, it is
possible that one of the mechanisms by which E2 demonstrates
neuroprotective effects against hypoglycemic injury may be through
activation of the pAKT signaling pathway. This would lead to the
phosphorylation and inactivation of the BCL2 associated agonist of
cell death protein, which prevents BCL2 associated X-mediated
release of cytochrome c from the mitochondria, thus protecting it
from mitochondrial dysfunction and cell death. In order to
elucidate the precise mechanisms of action by E2, future studies
should aim to confirm whether E2 activates this signaling pathway,
potentially by inhibiting distinct AKT-GSK3β pathway members using
the LY294002 PI3K inhibitor.
In conclusion, the present data indicate that E2
exhibits significant neuroprotective effects under glucopenic
stress conditions in hypothalamic cells, and demonstrates that the
neuroprotective efficacy of E2 is time-dependent. The precise
mechanisms by which E2 mediates these neuroprotective effects
remains to be explored in future studies. The present study raises
the possibility of novel pharmacological interventions against
hypoglycemia and the use of E2 against neuronal deficits. In
addition, the results support the role of this ovarian steroid
hormone in modulating the response to recurring glucodeprivation in
the central nervous system, and its mechanism of action through
membrane receptors.
References
|
1
|
McCall AL: Cerebral glucose metabolism in
diabetes mellitus. Eur J Pharmacol. 490:147–158. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mooradian AD, Chung HC and Shah GN: GLUT-1
expression in the cerebra of patients with Alzheimer's disease.
Neurobiol Aging. 18:469–474. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hernández-Fonseca K, Massieu L, de la
Cadena García S, Guzmán C and Camacho-Arroyo I: Neuroprotective
role of estradiol against neuronal death induced by glucose
deprivation in cultured rat hippocampal neurons.
Neuroendocrinology. 96:41–50. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Routh VH, Hao L, Santiago AM, Sheng Z and
Zhou C: Hypothalamic glucose sensing: Making ends meet. Front Syst
Neurosci. 8:2362014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Behl C, Skutella T, Lezoualc'h F, Post A,
Widmann M, Newton CJ and Holsboer F: Neuroprotection against
oxidative stress by estrogens: Structure-activity relationship. Mol
Pharmacol. 51:535–541. 1997.PubMed/NCBI
|
|
6
|
Belcredito S, Vegeto E, Brusadelli A,
Ghisletti S, Mussi P, Ciana P and Maggi A: Estrogen
neuroprotection: The involvement of the Bcl-2 binding protein
BNIP2. Brain Res Brain Res Rev. 37:335–342. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheng H, Isoda F and Mobbs CV: Estradiol
impairs hypothalamic molecular responses to hypoglycemia. Brain
Res. 1280:77–83. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wise PM, Dubal DB, Wilson ME, Rau SW and
Böttner M: Minireview: Neuroprotective effects of estrogen-new
insights into mechanisms of action. Endocrinology. 142:969–973.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Singh M, Meyer EM, Millard WJ and Simpkins
JW: Ovarian steroid deprivation results in a reversible learning
impairment and compromised cholinergic function in female
Sprague-Dawley rats. Brain Res. 644:305–312. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
De Nicola AF, Pietranera L, Bellini MJ,
Goya R, Brocca ME and Garcia-Segura LM: Protective effect of
estrogens on the brain of rats with essential and endocrine
hypertension. Horm Mol Biol Clin Investig. 4:549–557.
2010.PubMed/NCBI
|
|
11
|
Sandoval DA, Ertl AC, Richardson MA, Tate
DB and Davis SN: Estrogen blunts neuroendocrine and metabolic
responses to hypoglycemia. Diabetes. 52:1749–1755. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cardona-Gómez GP, Mendez P, DonCarlos LL,
Azcoitia I and Garcia-Segura LM: Interactions of estrogens and
insulin-like growth factor-I in the brain: Implications for
neuroprotection. Brain Res Brain Res Rev. 37:320–334. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Singh M, Dykens JA and Simpkins JW: Novel
mechanisms for estrogen-induced neuroprotection. Exp Biol Med
(Maywood). 231:514–521. 2006.PubMed/NCBI
|
|
14
|
Goodman RL: Neural systems mediating the
negative feedback actions of estradiol and progesterone in the ewe.
Acta Neurobiol Exp (Wars). 56:727–741. 1996.PubMed/NCBI
|
|
15
|
Green PS, Gridley KE and Simpkins JW:
Estradiol protects against beta-amyloid (25–35)-induced toxicity in
SK-N-SH human neuroblastoma cells. Neurosci Lett. 218:165–168.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meda C, Vegeto E, Pollio G, Ciana P,
Patrone C, Pellicciari C and Maggi A: Oestrogen prevention of
neural cell death correlates with decreased expression of mRNA for
the pro-apoptotic protein nip-2. J Neuroendocrinol. 12:1051–1059.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gazzaley AH, Siegel SJ, Kordower JH,
Mufson EJ and Morrison JH: Circuit-specific alterations of
N-methyl-D-aspartate receptor subunit 1 in the dentate gyrus of
aged monkeys. Proc Natl Acad Sci USA. 93:3121–3125. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Toran-Allerand CD: Mechanisms of estrogen
action during neural development: Mediation by interactions with
the neurotrophins and their receptors? J Steroid Biochem Mol Biol.
56:169–178. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bishop J and Simpkins JW: Estradiol
treatment increases viability of glioma and neuroblastoma cells in
vitro. Mol Cell Neurosci. 5:303–308. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Regan RF and Guo Y: Estrogens attenuate
neuronal injury due to hemoglobin, chemical hypoxia and excitatory
amino acids in murine cortical cultures. Brain Res. 764:133–140.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Enmark E and Gustafsson JA: Oestrogen
receptors-an overview. J Intern Med. 246:133–138. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao L and Brinton RD: Estrogen receptor
alpha and beta differentially regulate intracellular Ca(2+)
dynamics leading to ERK phosphorylation and estrogen
neuroprotection in hippocampal neurons. Brain Res. 1172:48–59.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gu Q and Moss RL: 17 beta-Estradiol
potentiates kainate-induced currents via activation of the cAMP
cascade. J Neurosci. 16:3620–3629. 1996.PubMed/NCBI
|
|
24
|
Watters JJ and Dorsa DM: Transcriptional
effects of estrogen on neuronal neurotensin gene expression involve
cAMP/protein kinase A-dependent signaling mechanisms. J Neurosci.
18:6672–6680. 1998.PubMed/NCBI
|
|
25
|
Anderson GM and Barrell GK: Effects of
thyroidectomy and thyroxine replacement on seasonal reproduction in
the red deer hind. J Reprod Fertil. 113:239–250. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Razandi M, Pedram A, Greene GL and Levin
ER: Cell membrane and nuclear estrogen receptors (ERs) originate
from a single transcript: Studies of ERalpha and Ebeta expressed in
chinese hamster ovary cells. Mol Endocrinol. 13:307–319. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rameh LE and Cantley LC: The role of
phosphoinositide 3-kinase lipid products in cell function. J Biol
Chem. 274:8347–8350. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: A play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Buckmaster PS and Dudek FE: Neuron loss,
granule cell axon reorganization, and functional changes in the
dentate gyrus of epileptic kainate-treated rats. J Comp Neurol.
385:385–404. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kovacina KS, Park GY, Bae SS, Guzzetta AW,
Schaefer E, Birnbaum MJ and Roth RA: Identification of a
proline-rich Akt substrate as a 14-3-3 binding partner. J Biol
Chem. 278:10189–10194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wiza C, Nascimento EB and Ouwens DM: Role
of PRAS40 in Akt and mTOR signaling in health and disease. Am J
Physiol Endocrinol Metab. 302:E1453–E1460. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang J, Zhang B, Yin Z, Chen F, Liu T, Xu
H, Liu Y and Zhou X: Effects of metformin on the estrogen-induced
proliferation and the expression of ER in human endometrial cancer
cells. Zhonghua Fu Chan Ke Za Zhi. 49:932–937. 2014.(In Chinese).
PubMed/NCBI
|
|
33
|
Welshons WV, Wolf MF, Murphy CS and Jordan
VC: Estrogenic activity of phenol red. Mol Cell Endocrinol.
57:169–178. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Regan RF and Panter SS: Traumatic neuronal
injury in cortical cell culture is attenuated by 21-aminosteroids.
Brain Res. 682:144–150. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma ZQ, Santagati S, Patrone C, Pollio G,
Vegeto E and Maggi A: Insulin-like growth factors activate estrogen
receptor to control the growth and differentiation of the human
neuroblastoma cell line SK-ER3. Mol Endocrinol. 8:910–918. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gazzaley AH, Benson DL, Huntley GW and
Morrison JH: Differential subcellular regulation of NMDAR1 protein
and mRNA in dendrites of dentate gyrus granule cells after
perforant path transection. J Neurosci. 17:2006–2017. 1996.
|
|
38
|
Dykens JA: Isolated cerebral and
cerebellar mitochondria produce free radicals when exposed to
elevated CA2+ and Na+: Implications for neurodegeneration. J
Neurochem. 63:584–591. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kroemer G and Reed JC: Mitochondrial
control of cell death. Nat Med. 6:513–519. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Simpkins JW and Dykens JA: Mitochondrial
mechanisms of estrogen neuroprotection. Brain Res Rev. 57:421–430.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gruetter R: Glycogen: The forgotten
cerebral energy store. J Neurosci Res. 74:179–183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tamrakar P and Briski KP: Estradiol
regulation of hypothalamic astrocyte adenosine
5′-monophosphate-activated protein kinase activity: Role of
hindbrain catecholamine signaling. Brain Res Bull. 110:47–53. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vegeto E, Pollio G, Pellicciari C and
Maggi A: Estrogen and progesterone induction of survival of
monoblastoid cells undergoing TNF-alpha-induced apoptosis. FASEB J.
13:793–803. 1999.PubMed/NCBI
|
|
44
|
Billig H, Furuta I and Hsueh AJ: Estrogens
inhibit and androgens enhance ovarian granulosa cell apoptosis.
Endocrinology. 133:2204–2212. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Behl C, Widmann M, Trapp T and Holsboer F:
17-beta estradiol protects neurons from oxidative stress-induced
cell death in vitro. Biochem Biophys Res Commun. 216:473–482. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Clodfelder-Miller B, De Sarno P, Zmijewska
AA, Song L and Jope RS: Physiological and pathological changes in
glucose regulate brain Akt and glycogen synthase kinase-3. J Biol
Chem. 280:39723–39731. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Filardo EJ, Quinn JA, Bland KI and
Frackelton AR Jr: Estrogen-induced activation of Erk-1 and Erk-2
requires the G protein-coupled receptor homolog, GPR30, and occurs
via trans-activation of the epidermal growth factor receptor
through release of HB-EGF. Mol Endocrinol. 14:1649–1660. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lazennec G, Thomas JA and Katzenellenbogen
BS: Involvement of cyclic AMP response element binding protein
(CREB) and estrogen receptor phosphorylation in the synergistic
activation of the estrogen receptor by estradiol and protein kinase
activators. J Steroid Biochem Mol Biol. 77:193–203. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Du B, Ohmichi M, Takahashi K, Kawagoe J,
Ohshima C, Igarashi H, Mori-Abe A, Saitoh M, Ohta T, Ohishi A, et
al: Both estrogen and raloxifene protect against
beta-amyloid-induced neurotoxicity in estrogen receptor
alpha-transfected PC12 cells by activation of telomerase activity
via Akt cascade. J Endocrinol. 183:605–615. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kato H, Araki T, Murase K and Kogure K:
Induction of tolerance to ischemia: Alterations in second-messenger
systems in the gerbil hippocampus. Brain Res Bull. 29:559–565.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao H, Sapolsky RM and Steinberg GK:
Phosphoinositide-3-kinase/akt survival signal pathways are
implicated in neuronal survival after stroke. Mol Neurobiol.
34:249–270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gervitz LM, Nalbant D, Williams SC and
Fowler JC: Adenosine-mediated activation of Akt/protein kinase B in
the rat hippocampus in vitro and in vivo. Neurosci Lett.
328:175–179. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|