Introduction
Liver cancer is one of the most common malignancies
worldwide. In China in 2012, liver cancer accounted for 12.9% of
all novel cancer cases, 17.4% of all cases of cancer-associated
mortality, and ranked second in cancer incidence and third in
mortality rate (1). According to
the latest data issued by the World Health Organization, liver
cancer is the second most frequent cause of cancer-associated
mortality worldwide (2). Several
risk factors for liver cancer have been identified, including
hepatitis B and C virus, aflatoxin, alcohol consumption, tobacco
smoking, obesity and diabetes. Despite primary and secondary
preventative measures, such as effective health education,
hepatitis B vaccination and early detection in numerous areas of
China, liver cancer mortality has only increased (3). The current treatment options for
human hepatocellular carcinoma include surgery, radiotherapy,
chemotherapy and liver transplantation, with limited evidence of a
successful cure. The hepatosis of patients with hepatocellular
carcinoma affects drug metabolism, which intensifies the side
effects of chemotherapy and may induce multidrug resistance.
Therefore, novel therapeutic options for human hepatocellular
carcinoma have focused on natural products as an increasingly
important source of potential anticancer agents that target liver
cancer (4).
Sulforaphane (SFN) is a natural isothiocyanate that
is present in cruciferous plants, with the highest content found in
broccoli. SFN has received a great deal of attention due to its
chemopreventive activity and potent anticancer effects (5). The chemopreventive activity of SFN
has been investigated in various chemically induced cancer models
(6). SFN modulates the metabolism
of carcinogens via inhibition of cytochrome P450-dependent
monooxygenases and/or induction of Phase II detoxification enzymes
in chemically induced cancer. Previous studies have also
demonstrated that SFN may inhibit proliferation of cancer cells
in vitro by inducing apoptosis and/or cell cycle arrest
(7–9). SFN suppresses growth in PA-1 human
ovarian cancer (10), LNCaP and
PC-3 human prostate cancer (11),
T24 human urinary bladder cancer (12), pre-B acute lymphoblastic leukemia
(ALL) (Nalm-6, REH and RS-4), and T-cell ALL cells (Jurkat, RPMI,
DND41 and KOPTK1) (13). In PC-3
prostate cancer cells, SFN has been revealed to arrest cancer cells
at the G2/M phase, which is associated with checkpoint
kinase 2-mediated phosphorylation of cell division cycle 25C, and
further induces caspase-9 and −8-mediated apoptosis (14). Furthermore, SFN reduces ovarian
cancer cell migration and increases apoptotic cell death via
increased B-cell lymphoma 2 (Bcl-2) antagonist killer/Bcl-2 ratio,
and cleavage of procaspase-9 and poly (adenosine
diphosphate-ribose) polymerase (15). Several studies have reported that
SFN exerts a relatively strong effect on HepG2 human liver cancer
cells, and evidently inhibits the proliferation of HepG2 cells
(16–19).
A previous study demonstrated that SFN induces
apoptosis of HepG2 cells via the mitochondrial pathway, through
unknown molecular mechanisms (20). Endoplasmic reticulum (ER) stress is
known to serve an important role in apoptosis mediated by several
anticancer agents. However, whether SFN induces apoptosis of HepG2
cells via the ER pathway remains unclear. The present study aimed
to explore the antiproliferative and apoptotic effects of SFN, and
to determine the underlying mechanisms in HepG2 human liver cancer
cells. SFN is transported by relevant proteins of the ER pathway;
therefore, the role of the ER in SFN-induced apoptosis of HepG2
cells was explored.
Materials and methods
Reagents, drugs and assay kits
SFN (purity, 98.3%) was purchased from Alexis
Biochemicals; Enzo Life Science (Farmingdale, NY, USA). The
following additional materials were obtained: Adriamycin (ADR;
110826; Zhejiang Hisun Pharmaceutical Co., Ltd., Taizhou, China);
RPMI-1640 culture medium and pancreatin (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA); Annexin V-fluorescein
isothiocyanate (FITC) Apoptosis Detection kit, rabbit anti-human
binding immunoglobulin protein (Bip)/glucose-regulated protein 78
(GRP78) antibody (cat. no. AF0171), prestained protein molecular
weight marker (cat. no. P0062), cell lysis buffer, and alkaline
phosphatase-labeled sheep anti-rabbit immunoglobulin (Ig) G
(catalog no. A0239) (Beyotime Institute of Biotechnology, Shanghai,
China); mouse anti-β-actin antibody (cat. no. TA-09),
peroxidase-conjugated affinipure goat anti-mouse IgG (H+L; cat. no.
ZB-2305; Beijing Zhongshan Golden Bridge Biotechnology, Co., Ltd.,
Beijing, China); Tween 20 and bovine serum albumin (BSA;
Sigma-Aldrich; Merck Millipore, Darmstadt, Germany); Coomassie
Brilliant Blue G250, Triton X-100 and paraformaldehyde (Beijing
Chemical Reagents Company, Beijing, China); rabbit anti-human X-box
binding protein-1 (XBP-1; cat. no. bs-1668R), C/EBP homologous
protein (CHOP)/growth arrest- and DNA damage-inducible gene 153
(GADD153; cat. no. bs-8875R), BH3 interacting domain death agonist
(Bid; cat. no. bs-1153R), caspase-12 (cat. no. bs-23014R), and
FITC-conjugated goat anti-rabbit antibodies (cat. no.
bs-0295G-FITC; BIOSS, Beijing, China).
Equipment
The following laboratory equipment was used
throughout the experiments of the present study. Super-clean bench
(JJT-900/1300; Suzhou SuJie Purifying Equipment Co., Ltd., Suzhou,
China); microplate reader (Model 680; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA); electrophoresis apparatus (DYY-24D, Beijing
Liuyi Instrument Factory, Beijing, China); high-speed centrifuge at
low temperature (Beckman Coulter, Inc., Brea, CA, USA); flow
cytometer (COULTER® EPICS® XL™; Beckman
Coulter, Inc.); CO2 incubator (MC0175; SANYO Electric
Co., Ltd., Moriguchi, Japan); and gel imaging system (GIS-2019,
Tanon Science & Technology Co., Ltd., Shanghai, China).
Cell line and cell culture
The HepG2 human hepatocellular carcinoma cell line
was obtained from the Research Center of Life Sciences and
Environmental Sciences, Harbin University of Commerce (Harbin,
China). HepG2 cells were maintained in culture flasks containing
RPMI-1640 supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.). The cultures were incubated at 37°C in an
atmosphere containing 5% CO2 with saturated humidity.
Cells were transferred to fresh medium once every 2 to 3 days.
MTT assay
Logarithmic phase HepG2 cells were seeded in a
96-well plate at a density of 5×104 cells/ml (100
µl/well). Following a 24 h incubation at 37°C, 100 µl of the drugs
were added to each well. The final concentrations of SFN used were
2.5, 5, 10, 20, 40 and 80 µmol/l; the final concentrations of ADR
were 0.01, 0.1, 1, 10 and 100 µmol/l. Following treatment with SFN
or ADR for 24, 48 and 72 h, drugs were discarded and the cells were
incubated with 100 µl MTT (0.5 mg/ml) for 4 h at 37°C. After 4 h,
the supernatant was aspirated, and 200 µl dimethyl sulfoxide was
added. Finally, the absorbance was measured at 570 nm using a
microplate reader, which was used to calculate the rate of
inhibition and the half maximal inhibitory concentration
(IC50).
Fluorescence microscopy of apoptosis
in HepG2 cells
After placing coverslips in a 6-well plate,
3×105 HepG2 cells (1 ml) were seeded in each well and
allowed to attach overnight. Cells were cultured with various
concentrations of SFN (10, 20 and 40 µmol/l). ADR was added as a
positive control at a final concentration of 0.5 µmol/l, and the
control group was supplemented with equal volumes of RPMI-1640
culture medium. Following 48 h of treatment with SFN or ADR at
37°C, the cells were digested with pancreatin and washed once with
phosphate-buffered saline (PBS). The cells were then fixed with a
fixing solution composed of methyl alcohol and glacial acetic acid
(3:1) for 10 min at 4°C. After further washing with PBS, 5 mg/l
Hoechst 33258 fluorescent probe was added to the cells and
incubated for 15 min. The cover slips were hand washed with PBS and
placed on glass slides containing drops of glycerin. Finally, the
cells were visualized and images were captured under an inverted
fluorescence microscope.
Detection of apoptotic rate of HepG2
cells by flow cytometry (FCM)
Logarithmic phase HepG2 cells were seeded in 6-well
plates at a density of 3×105 cells/ml (1 ml/well) and
were allowed to attach overnight. SFN was added to the wells (1 ml
per well) at a final concentration of 10, 20 or 40 µmol/l. ADR was
added as a positive control at a final concentration of 0.5 µmol/l.
An equal volume of medium was added to the wells of the control
group. The plates were incubated at 37°C in an atmosphere
containing 5% CO2 for 48 h. The cells were digested with
pancreatin, collected and washed with PBS (4°C), and adjusted to
1×105 cells/ml. The cells were resuspended in binding
buffer and then stained with Annexin V-FITC and propidium iodide
(PI) solution in an ice bath in the dark, according to the Annexin
V-FITC Apoptosis Detection kit instructions. A nylon mesh filter
(300 µm) was used to filter the cell samples to ensure cells were
detected at single cell suspension. Following filtration, the cell
samples of each group were analyzed by a flow cytometer at a
wavelength of 488 nm and the results were analyzed by Expo 32 ADC
Analysis software (Beckman Coulter, Inc.).
Western blot analysis of Bip/GRP78,
XBP-1 and Bid protein expression
The HepG2 cells were plated in culture flasks and
allowed to attach overnight. The cells were treated with various
concentrations of SFN (10, 20 and 40 µmol/l) or ADR (0.5 µmol/l).
The cells of the control group were treated with an equal volume of
medium. After 48 h of treatment, cells were collected, lysed, and
proteins were extracted. The Bradford method was used to quantify
protein content. Equal amounts of protein (2 µg/ml; 20 µl loading
volume) from the various groups were separated by 12% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis. The proteins
were transferred onto nitrocellulose membranes at 200 mA for 30
min. The membranes were then incubated in blocking buffer [5%
nonfat dry milk in Tris-buffered saline containing Tween-20 (TBST)]
for 2 h at room temperature, and the blots were incubated with
rabbit anti-human Bip/GRP78, XBP-1 and Bid antibodies (1:200) and
β-actin antibody (1:5,000) overnight at 4°C. The membranes were
rinsed twice with TBST, undergoing 10 min of oscillation with each
wash, and were rinsed once with TBS for 10 min. Subsequently, the
membranes were incubated with alkaline phosphatase-labeled
anti-mouse IgG antibody (1:2,000 dilution) for 2 h at room
temperature. The membranes were rinsed three times as
aforementioned and were stained with a diaminobenzidine chromogenic
system. Finally, images were captured using the gel imaging system,
and the protein content was quantified and analyzed using a GIS-
2019 gel imaging system software, version 3.14 (Tanon Science &
Technology Co., Ltd.).
FCM of caspase-12 and CHOP/GADD153
expression
Logarithmic phase HepG2 cells were seeded in 6-well
plates at a density of 3×105 cells/ml (1 ml/well) and
were allowed to attach overnight. Following treatment with various
concentrations of SFN (10, 20 and 40 µmol/l) or ADR (0.5 µmol/l)
for 48 h, the cells were collected by centrifugation for 10 min at
4°C at a speed of 558 × g and washed twice with PBS. The
cells were then fixed with paraformaldehyde (500 µl) for 30 min at
room temperature, collected by centrifugation for 5 min at room
temperature, at a speed of 558 × g and then washed with PBS
for 45 min. The cells were permeabilized with 200 µl PBS containing
0.1% Triton X-100 for 10 min and blocked with 200 µl blocking
solution (PBS containing 5% BSA) for 60 min. After rinsing once
with PBS, the cells were incubated with rabbit anti-human
CHOP/GADD153 or rabbit anti-human caspase-12 antibodies (1:400) for
2 h at room temperature. Subsequently, the cells were washed once
with PBS and incubated with FITC-labeled goat anti-rabbit secondary
antibody for 60 min in the dark at room temperature. The samples
were finally resuspended in PBS and filtered via a 300 µm nylon
mesh filter. The protein content was analyzed using flow cytometry
and Expo 32 ADC Analysis software (Beckman Coulter Inc.).
Statistical analysis
Data are presented as the mean ± standard deviation
of three independent experiments. Data were analyzed using SPSS
Software for Windows Version 11.5 (SPSS, Inc., Chicago, IL, USA).
Data from the various groups were compared using one-way analysis
of variance and Fisher's least significant difference test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
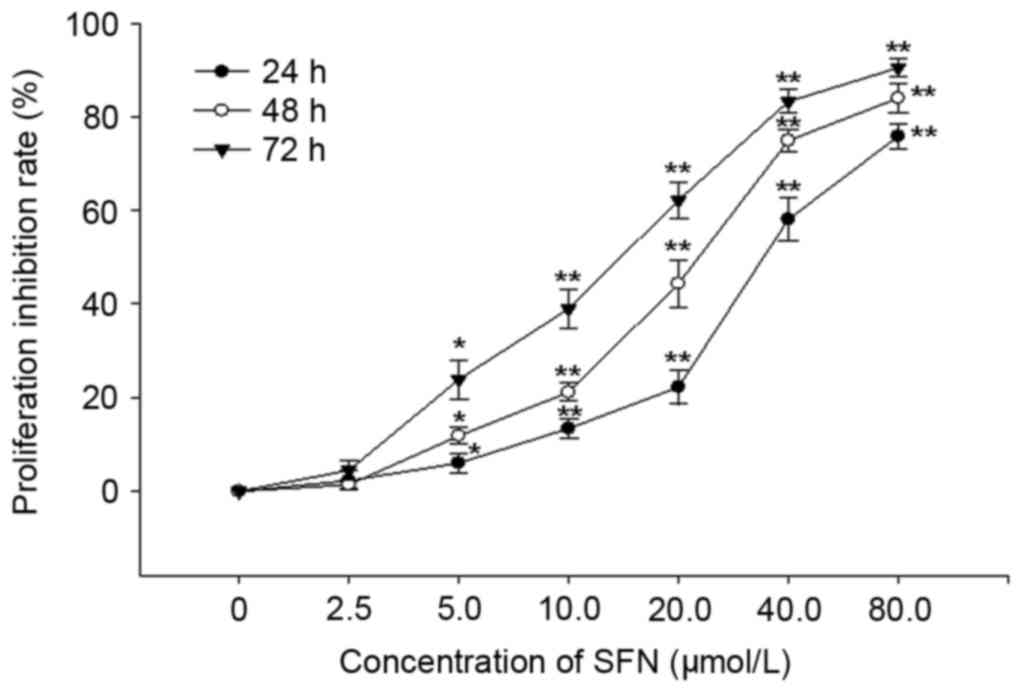
Antiproliferative effects of SFN
Following treatment with various concentrations of
SFN for 24, 48 and 72 h, the proliferation of HepG2 cells was
effectively inhibited in a dose- and time-dependent manner. The
IC50 values of SFN treatment were 32.03±0.96, 20.90±1.96
and 13.87±0.44 µmol/l following treatment for 24, 48 and 72 h,
respectively (Fig. 1). The
IC50 values of ADR were 0.78±0.12, 0.43±0.09 and
0.32±0.40 µmol/l following treatment for 24, 48 and 72 h,
respectively.
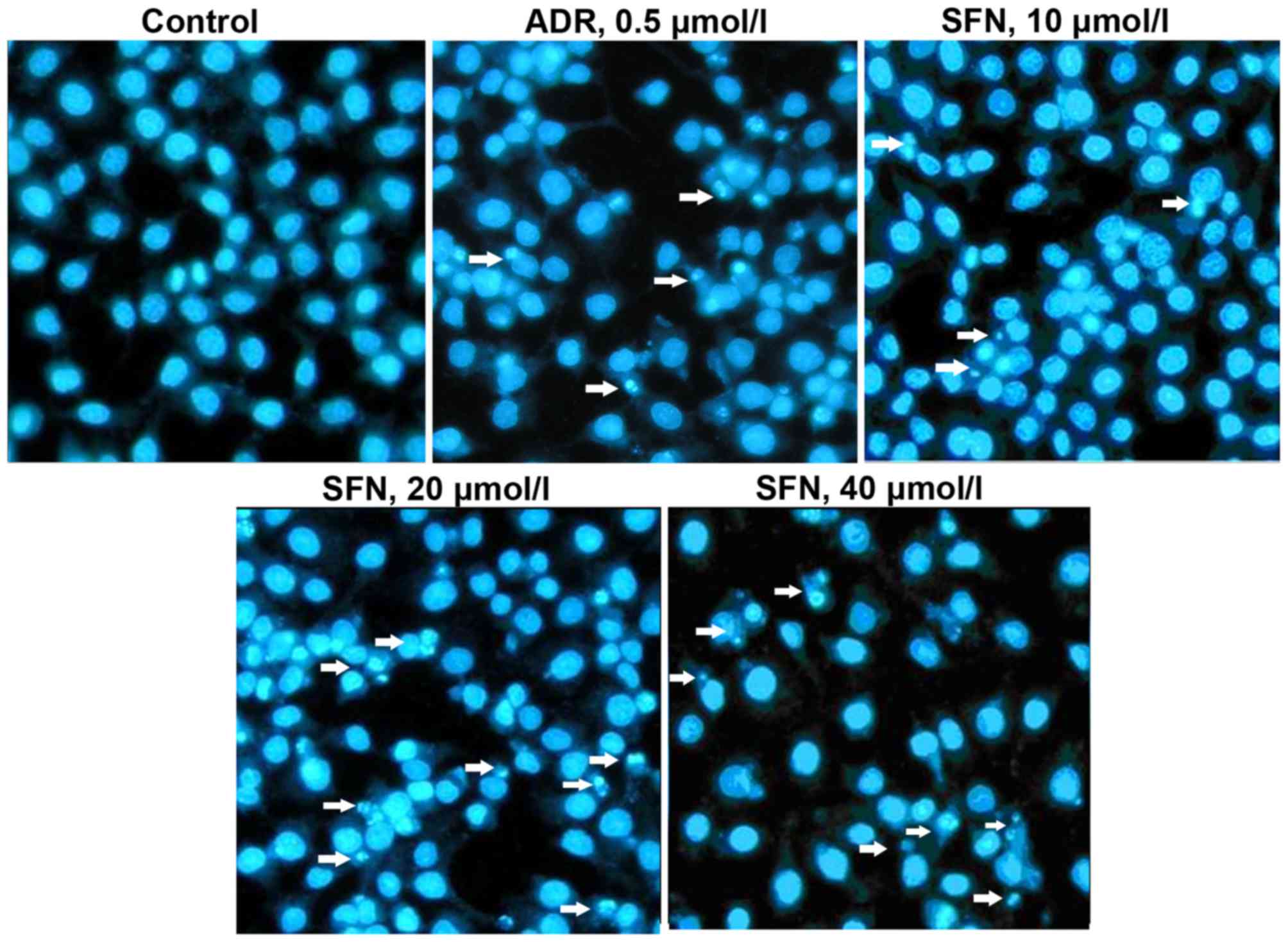
Effects of SFN on HepG2 cellular
morphology
Following exposure to various concentrations of SFN
or 0.5 µmol/l of ADR for 48 h, HepG2 cells displayed typical
apoptotic morphology, including chromatin condensation and the
formation of apoptotic bodies (Fig.
2). These results indicated that HepG2 cells were undergoing
apoptosis.
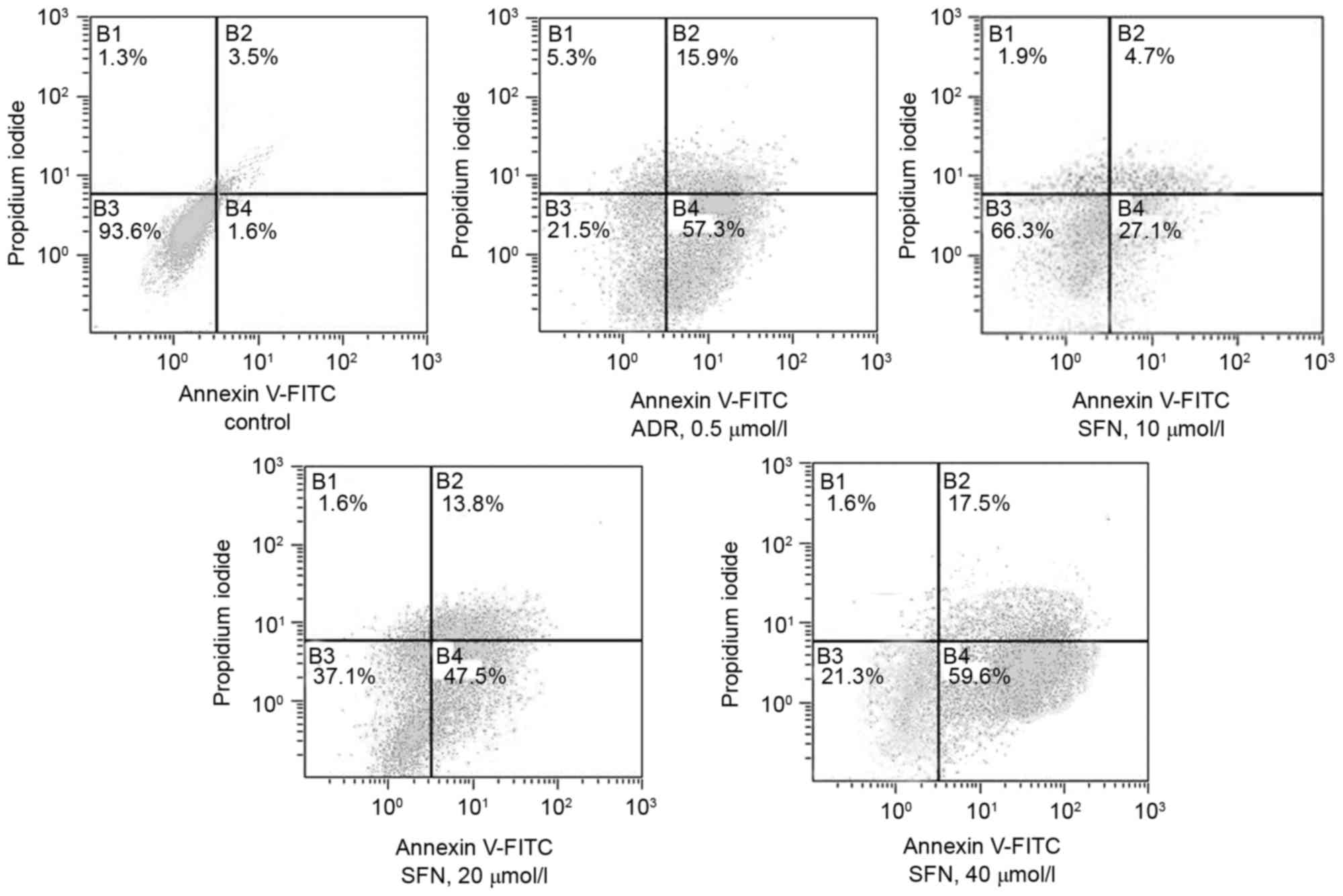
Effects of SFN on the apoptotic rate
of HepG2 cells
FCM demonstrated that treatment with 10, 20, 40
µmol/l SFN or 0.5 µmol/l of ADR significantly increased the
apoptotic rate of HepG2 cells (Annexin V+
PI−) compared with that of the control group (Table I and Fig. 3).
 | Table I.Effects of SFN on the apoptotic rate
of HepG2 cells. |
Table I.
Effects of SFN on the apoptotic rate
of HepG2 cells.
| Group | Concentration
(µmol/l) | Apoptotic rate
(%) |
|---|
| Control | – | 5.1 |
| ADR | 0.5 | 73.2 |
| SFN | 10 | 31.8 |
| SFN | 20 | 61.3 |
| SFN | 40 | 77.1 |
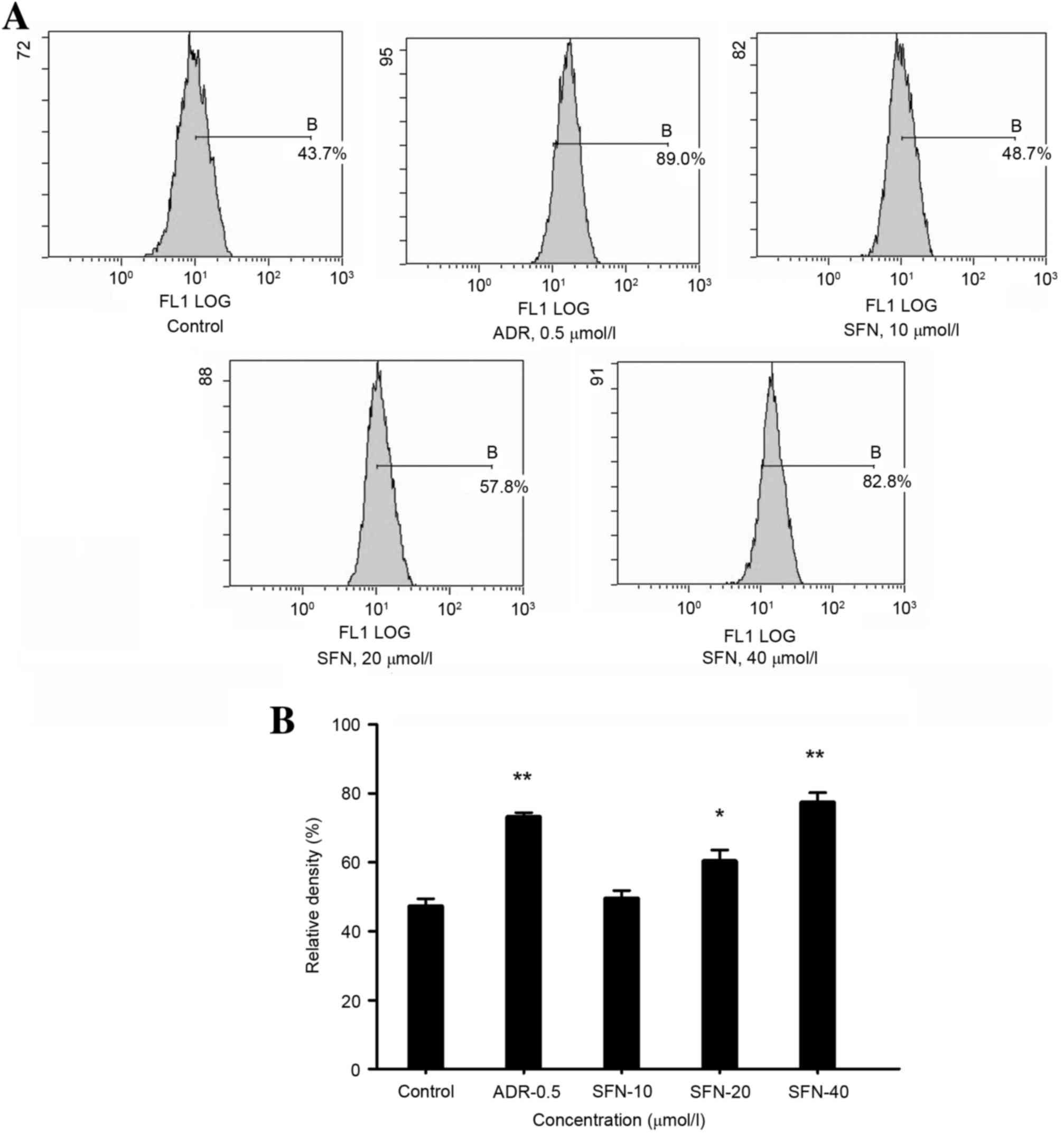
Effects of SFN on the protein
expression levels of Bip/GRP78, XBP-1 and Bid
Following treatment with 20 or 40 µmol/l SFN or 0.5
µmol/l ADR for 48 h, the protein expression levels of Bip/GRP78,
Bid and XBP-1 were significantly increased (P<0.01; Fig. 4).
 | Figure 4.Effects of SFN on Bip/GRP78, XBP-1 and
Bid expression in HepG2 cells. (A) Following treatment with 10, 20
or 40 µmol/l SFN for 48 h, the expression levels of Bip/GRP78,
XBP-1 and Bid were analyzed by western blotting. Medium was used as
a vehicle control and ADR (0.5 µmol/l) was used as a positive
control. The relative density of (B) Bip/GRP78, (C) XBP-1 and (D)
Bid protein was calculated and statistically analyzed. *P<0.05
vs. the control; **P<0.01 vs. the control. ADR, Adriamycin; SFN,
sulforaphane; Bip, binding immunoglobulin protein; GRP78,
glucose-regulated protein 78; XBP-1, X-box binding protein-1; Bid,
BH3 interacting domain death agonist. |
Effects of SFN on the protein
expression of CHOP/GADD153 and caspase-12
The expression levels of CHOP/GADD153 and caspase-12
were significantly higher in HepG2 cells following exposure to
increasing concentrations of SFN or 0.5 µmol/l of ADR for 48 h
compared with control cells (P<0.01 or P<0.05). Protein
quantities relative to the control were calculated and plotted as
histograms. The results are presented in Figs. 5 and 6.
Discussion
SFN exerts chemoprotective effects due to its
antitumor activity, and exhibits no clinically adverse effects;
therefore, it has been the focus of intensive research worldwide
(21,22). Previous studies have demonstrated
that SFN may induce apoptosis of several tumor cell lines through
different pathways, and it has been shown to significantly reduce
the mitochondrial membrane potential of human gastric cancer cells
(23–25). Furthermore, SFN has been reported
to decrease the ratio of Bcl-2/Bcl-2-associated X protein, reduce
the expression of Bcl-2 in HepG2 cells, and activate caspase-3,
thus triggering apoptosis of tumor cells (26). In addition, SFN may induce tumor
cell apoptosis via the mitochondrial pathways. SFN was shown to
induce a significant reduction in the expression of
phosphorylated-extracellular signal-regulated kinase (ERK) in HepG2
cells, inhibit ERK/mitogen-activated protein kinase signaling, and
promote apoptosis of HepG2 cells in a dose-dependent manner
(27).
The present study aimed to investigate whether the
ER pathway is involved in SFN-induced apoptosis of HepG2 cells. The
Bip/GRP78 protein triggers ER stress; under normal physiological
conditions, Bip/GRP78 combines with inositol-requiring enzyme 1
(IRE1), protein kinase RNA-like endoplasmic reticulum kinase (PERK)
and activating transcription factor 6 (ATF6) to maintain its
stability in the ER. When cells are stimulated by an external
signal, Bip/GRP78 is released from IRE1, PERK and ATF6. By
increasing the levels of Bip/GRP78 protein expression, ER stress is
ameliorated through a self-regulatory mechanism (28). ER stress in cells is predominantly
resolved via the ATF6 and XBP-1-mediated pathways. At relatively
low levels of stress, the steady state of ER may only be restored
via the activation of ATF6 proteolysis; however, at markedly high
level of stress, XBP-1 system serves a major role. Therefore,
increased expression of XBP-1 protein is a marker of overwhelming
ER stress in cells (29). Such
stress cannot be relieved through self-regulatory mechanisms and
the role of apoptosis becomes predominant. The ER is able to
independently induce cellular apoptosis. The results of present
study demonstrated that 20–40 µmol/l SFN treatment markedly
upregulated the protein expression levels of Bip/GRP78 and XBP-1.
These findings suggested that SFN treatment induced ER stress of
HepG2 cells and the overexpression of XBP-1 suggested ER stress
reached a peak level and apoptosis was subsequently triggered by
the ER signaling pathway.
Out of the caspase family proteins, only caspase-12
is present in the ER, which is the key element for mediating the
stress response to apoptosis. Caspase-12 triggers the transcription
and expression of CHOP/GADD153 (30). CHOP/GADD153 is minimally expressed
in normal cells; however, under conditions of ER stress, its
expression is increased, resulting in apoptosis (31). Bid is predominantly expressed in
the ER, but also in the nucleus to a limited extent. Upon apoptotic
signaling, Bid translocates to the mitochondria and increases
mitochondrial membrane permeability, thus leading to the release of
cytochrome c, activation of caspase-9, and induction of
apoptosis (32). The results of
present studies revealed that 20–40 µmol/l SFN treatment markedly
upregulated the protein expression levels of caspase-12,
CHOP/GADD153 and Bid. Following SFN treatment for 48 h, the
apoptosis rate of HepG2 cells significantly increased. These
findings suggested that SFN may induce ER stress-mediated apoptosis
in HepG2 cells. A previous study demonstrated that SFN decreases
the expression of Bcl-2 in HepG2 cells, but increases Bax levels,
resulting in the release of cytochrome c and the enhanced
activity of caspase-3, resulting in the induction of apoptosis via
the mitochondrial pathway (20).
CHOP/GADD153 is a classic marker of ER stress and primarily induces
apoptosis by inhibiting the expression of Bcl-2, which is
downregulated in SFN-treated HepG2 cells. The overexpression of Bid
induced by SFN may promote mitochondrial-mediated apoptosis and may
be the primary mechanism underlying the induction of
mitochondrial-mediated apoptosis by SFN.
In conclusion, in addition to directly inducing
apoptosis of HepG2 cells via mitochondrial pathways, the present
study demonstrated that SFN also triggers ER stress in HepG2 cells.
By modulating ER-related protein expression, SFN activates the
expression of Bid, which further activates mitochondrial apoptosis
and induces cellular apoptosis. These results indicated that the
mechanism underlying SFN-induced apoptosis is mediated by the
interaction between the ER and mitochondrial pathways, and Bid
serves an important role. Therefore, the ER pathway may be involved
in SFN-induced HepG2 cell apoptosis. The induction of cell
apoptosis is an important cancer therapeutic strategy. The present
study demonstrated SFN was able to induce cell apoptosis via the ER
stress-mediated pathway. SFN is considered a promising drug in the
treatment of different types of cancer due to its chemopreventative
and therapeutic effects in various cancer cells. The elucidation of
the underlying molecular mechanisms of SFN provides novel evidence
for the research and application of SFN-related anticancer
therapeutic agents.
Acknowledgements
The present study was supported by the National
Natural Science Funds of China (grant no. 81102858); the
Postdoctoral Science Foundation (grant no. 2013M321061); the Key
Project of Chinese Ministry of Education (grant no. 210059); the
Heilongjiang Postdoctoral Fund (grant no. LBH-Z11102); the
University Nursing Program for Young Scholars with Creative Talents
in Heilongjiang Province (grant no. UNPYSCT-2015070).
References
|
1
|
Wei KR, Yu X, Zheng RS, Peng XB, Zhang SW,
Ji MF, Liang ZH, Ou ZX and Chen WQ: Incidence and mortality of
liver cancer in China, 2010. Chin J Cancer. 33:388–394.
2014.PubMed/NCBI
|
|
2
|
Bernard WS and Chrstopher PW: World cancer
report 2014. IARC; Lyon: 2014
|
|
3
|
Fan YG, Wang JB, Jiang Y, Li P, Xiao HJ,
Chen WQ, Qiao YL and Boffetta P: Attributable causes of lung cancer
mortality and incidence in China. Asian Pac J Cancer Prev.
14:7251–7256. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ji YB, Qu ZY and Zou X: Juglone-induced
apoptosis in human gastric cancer SGC-7901 cells via the
mitochondrial pathway. Exp Toxicol Pathol. 63:69–78. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Traka MH, Melchini A and Mithen RF:
Sulforaphane and prostate cancer interception. Drug Discov Today.
19:1488–1492. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abdull Razis AF and Noor NM: Sulforaphane
is superior to glucoraphanin in modulating carcinogen-metabolising
enzymes in Hep G2 cells. Asian Pac J Cancer Prev. 14:4235–4238.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sawai Y, Murata H, Horii M, Koto K, Matsui
T, Horie N, Tsuji Y, Ashihara E, Maekawa T, Kubo T and Fushiki S:
Effectiveness of sulforaphane as a radiosensitizer for murine
osteosarcoma cells. Oncol Rep. 29:941–945. 2013.PubMed/NCBI
|
|
8
|
Li SH, Fu J, Watkins DN, Srivastava RK and
Shankar S: Sulforaphane regulates self-renewal of pancreatic cancer
stem cells through the modulation of Sonic hedgehog-GLI pathway.
Mol Cell Biochem. 373:217–227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sharma C, Sadrieh L, Priyani A, Ahmed M,
Hassan AH and Hussain A: Anti-carcinogenic effects of sulforaphane
in association with its apoptosis-inducing and anti-inflammatory
properties in human cervical cancer cells. Cancer Epidemiol.
35:272–278. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chang CC, Hung CM, Yang YR, Lee MJ and Hsu
YC: Sulforaphane induced cell cycle arrest in the G2/M phase via
the blockade of cyclin B1/CDC2 in human ovarian cancer cells. J
Ovarian Res. 6:412013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choi S, Lew KL, Xiao H, Herman-Antosiewicz
A, Xiao D, Brown CK and Singh SV: D,L-Sulforaphane-induced cell
death in human prostate cancer cells is regulated by inhibitor of
apoptosis family proteins and Apaf-1. Carcinogenesis. 28:151–162.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jo GH, Kim GY, Kim WJ, Park KY and Choi
YH: Sulforaphane induces apoptosis in T24 human urinary bladder
cancer cells through a reactive oxygen species-mediated
mitochondrial pathway: The involvement of endoplasmic reticulum
stress and the Nrf2 signaling pathway. Int J Oncol. 45:1497–1506.
2014.PubMed/NCBI
|
|
13
|
Suppipat K, Park CS, Shen Y, Zhu X and
Lacorazza HD: Sulforaphane induces cell cycle arrest and apoptosis
in acute lymphoblastic leukemia cells. PLoS One. 7:e512512012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Singh SV, Herman-Antosiewicz A, Singh AV,
Lew KL, Srivastava SK, Kamarh R, Brown KD, Zhang L and Baskaran R:
Sulforaphane-induced G2/M phase cell cycle arrest involves
checkpoint kinase 2-mediated phosphorylation of cell division cycle
25C. J Biol Chem. 279:25813–25822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bryant CS, Kumar S, Chamala S, Shah J, Pal
J, Seward S, Qazi AM, Morris R, Semaan A, Shammas MA, et al:
Sulforaphane induces cell cycle arrest by protecting RB-E2F-1
complex in epithelial ovarian cancer cells. Mol Cancer. 9:472010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zou X, Qu ZY and Ji YB: Study on JNK
pathway in human hapetocelluar carcinoma HepG-2 cells apoptosis
induced by sulforaphane. Journal of Harbin University of Commerce
Natural Sciences Edition. 27:532–535. 2011.
|
|
17
|
Zou X, Qu ZY, Gao P, Sun SN and Ji Yb:
Effects of sulforaphane on G_2/M phase arrest in HepG-2 cells and
the expression of Cdk1 and CyclinB1. Acta Chinese Medicine and
Pharmacology. 38:8–10. 2010.
|
|
18
|
Yeh CT and Yen GC: Effect of sulforaphane
on metallothionein expression and induction of apoptosis in human
hepatoma HepG2 cells. Carcinogenesis. 26:2138–2148. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jeon YK, Yoo DR, Jang YH, Jang SY and Nam
MJ: Sulforaphane induces apoptosis in human hepatic cancer cells
through inhibition of
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase4, mediated by
hypoxia inducible factor-1-dependent pathway. Biochim Biophys Acta.
1814:1340–1348. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zou X, Qu ZY, Gao P and Ji YB: Effects of
sulforaphane on gene transcription and protein expression of Bcl-2
and Bax in HepG-2 Cell. Acta Universitatis Traditionis Medicalis
Sinensis Pharmacologiaeque Shanghai. 24:76–80. 2010.
|
|
21
|
Herr I, Lozanovski V, Houben P, Schemmer P
and Büchler MW: Sulforaphane and related mustard oils in focus of
cancer prevention and therapy. Wien Med Wochenschr. 163:80–88.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Juge N, Mithen RF and Traka M: Molecular
basis for chemoprevention by sulforaphane: A comprehensive review.
Cell Mol Life Sci. 64:1105–1127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xiao D, Powolny AA, Antosiewicz J, Hahm
ER, Bommareddy A, Zeng Y, Desai D, Amin S, Herman-Antosiewicz A and
Singh SV: Cellular responses to cancer chemopreventive agent
D,L-sulforaphane in human prostate cancer cells are initiated by
mitochondrial reactive oxygen species. Pharm Res. 26:1729–1738.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang L, Tian Z, Yang Q, Li H, Guan H, Shi
B, Hou P and Ji M: Sulforaphane inhibits thyroid cancer cell growth
and invasiveness through the reactive oxygen species-dependent
pathway. Oncotarget. 6:25917–25931. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mondal A, Biswas R, Rhee YH, Kim J and Ahn
JC: Sulforaphene promotes Bax/Bcl2, MAPK-dependent human gastric
cancer AGS cells apoptosis and inhibits migration via EGFR,
p-ERK1/2 down-regulation. Gen Physiol Biophys. 35:25–34.
2016.PubMed/NCBI
|
|
26
|
Park SY, Kim GY, Bae SJ, Yoo YH and Choi
YH: Induction of apoptosis by isothiocyanate sulforaphane in human
cervical carcinoma HeLa and hepatocarcinoma HepG2 cells through
activation of caspase-3. Oncol Rep. 18:181–187. 2007.PubMed/NCBI
|
|
27
|
Zou X, Qu ZY, Bai J and Ji YB: Effect of
sulforaphane on protein expression of ERK and pathway in human
hapetocelluar carcinoma HepG-2 cells. Journal of Harbin University
of Commerce (Natural Sciences Edition). 27257–258. (266)2011.
|
|
28
|
Franceschelli S, Moltedo O, Amodio G,
Tajana G and Remondelli P: In the Huh7 hepatoma cells diclofenac
and indomethacin activate differently the unfolded protein response
and induce ER stress apoptosis. Open Biochem J. 5:45–51. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou Y, Lee J, Reno CM, Sun C, Chung J,
Lee J, Fisher SJ, White MF, Biddinger SB and Ozcan U: Regulation of
glucose homeostasis through a XBP-1-FoxO1 interaction. Nat Med.
17:356–365. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen X, Fu XS, Li CP and Zhao HX: ER
stress and ER stress-induced apoptosis are activated in gastric
SMCs in diabetic rats. World J Gastroenterol. 20:8260–8267. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harshberger CA, Harper AJ, Carro GW, Spath
WE, Hui WC, Lawton JM and Brockstein BE: Outcomes of computerized
physician order entry in an electronic health record after
implementation in an outpatient oncology setting. J Oncol Pract.
7:233–237. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Uchibayashi R, Tsuruma K, Inokuchi Y,
Shimazawa M and Hara H: Involvement of Bid and caspase-2 in
endoplasmic reticulum stress- and oxidative stress-induced retinal
ganglion cell death. J Neurosci Res. 89:1783–1794. 2011. View Article : Google Scholar : PubMed/NCBI
|