Introduction
Cardiovascular disease induces the highest
incidence, morbidity and mortality worldwide (1). The resulting cardiac remodeling is
associated with the underlying pathological changes in most heart
diseases (myocardial infarction, heart failure and atrial
fibrillation), and myocardial fibrosis is the most important
pathological feature of cardiac tissue remodeling (2). Cardiac fibroblasts (CFbs) account for
2/3 of heart cells, which plays an important role in the
homeostasis of the cardiac extracellular matrix metabolism and
remodeling of cardiac tissue (3).
Studies on the biological activity of CFbs (proliferation,
differentiation and migration) may provide the basis for exploring
the mechanism of cardiac remodeling and developing new therapeutic
strategies.
It has been widely confirmed that the transforming
growth factor-β1 (TGFβ1) signal transduction pathway plays an
important role in the process of cardiac fibrosis (3). TGFβ1 acts on downstream transcription
factors and then regulates the expression of target genes and
proteins through the binding on their receptors namely TGFβ
receptor one and two (TGFβRI/TGFβRII) (4). It can promote the transformation of
CFbs into its active myofibroblasts form, which enhances the
migration, proliferation and collagen synthesis, and thus, leads to
fibrosis (5).
However, microRNAs (miRNAs), a kind of small RNA of
18–24 bp length, can inhibit such a process. Indeed, mature miRNAs
combine with 3′ untranslated regions (3′-UTR) of the gene and
inhibit the target gene transcription and/or degrade the target
gene mRNAs, which affects the expression of the target proteins
(6). Accordingly, an increasing
number of studies have indicated that miRNAs play an important role
in many kinds of heart diseases (7).
The present study mainly focused on the role of
miR-370 in the myocardial remodeling after myocardial
infarction
Materials and methods
Rat myocardial infarction models
Experiments were in compliance with the council of
China on Animal Care and were approved by the Animal Ethics
Committee of the Medical College of He Xi University.
Sprague-Dawley (SD) male rats (180–250 g) were randomly divided
into two groups including, the myocardial infarction group (n=3)
and sham operation group (n=3). Rats were anesthetized with
pentobarbital sodium (0.1%) and were assisted breathing with small
animal ventilator. Their electrocardiograms were recorded with II
leads and the third, fourth rib gap was open to expose the left
atrial appendage. Anterior descending artery was ligated with 7–0
ligation line 2 mm at the lower edge of the right in the left
atrial appendage. Two weeks later, rats were anesthetized, and the
heart was quickly removed. After the residual blood was washed with
normal saline, the samples were stored at −80°C.
Hematoxylin and eosin (H&E) and
Masson staining
Rats of both groups were anesthetized, and their
heart was taken out. Saline was used to wash residual blood.
Following 24 h of fixation with 4% poly-formaldehyde, tissues were
embedded with paraffin, sliced and stained using H&E and Masson
procedures.
Cell culture and treatment
The epicardium of heart of SD neonatal rats (1–3
days) was tore with pincett. The residual blood was washed with 1X
PBS. The sample was cut with a pair of scissors (about 1
mm3), and double enzymes were added (0.25% pancreatin +
0.1% collagenase B) to digest at 37°C using shock for a total of 10
times, initially for 10 min, and then 6 min each time. The
supernatant of each collection was terminated digestion with DMEM
culture medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) containing 10% fetal bovine serum (FBS, Gibco). After
centrifugation of cell suspension at 800 × g for 10 min, the cells
were cultured in 50 ml culture flask after resuspension with 10%
FBS, and then stored at 37°C in a 5% CO2 incubator. CFb
was obtained after differential for 75 min and discarding the
supernatant. Cells were cultured with 10% FBS DMEM and 5%
CO2 at 37°C. Cell passage occurred in 1–2 days, and
cells between passages 2–3 were used for experiments. Serum-free
DMEM was used to starve cells for 24 h.
Cell transfection
After washing with serum-free DMEM, the cells were
cultured with 2 ml serum-free DMEM for 4–6 h (6-well plate).
miR-370 mimics (5′-CAGGUCACGUCUCUGCAGUUACAC-3′), miR-370 antisense
inhibitor (5′-GTCCAGTGCAGAGACGTCAATGTG-3′), NC
(5′-CACAUUGTGCUCUCUGCACUGCTC-3′) (Guangzhou RioBio Co., Ltd.,
Guangzhou, China) and transfection reagent
lipofectmine®2000 (Invitrogen Life Technologies,
Carlsbad, CA, USA) and 500 µl Opti-MEM (Gibco) were mixed, with a
standing time of 5 min. Two kinds of mixture mixed and stood for 20
min, and then transfected cells. 6 h later, culture medium was
replaced and corresponding stimulation was added.
Measurement of collagen protein
After treatment with the Sircol collagen assay kit
(Biocolor), cell samples were stored at 4°C for 24 h. After
centrifugation, 100 µl supernatant was added with 1 ml Sircol dye,
mixing, 4°C, 30 min. After centrifugation, the supernatant was
removed and 700 µl Sircol alkali reaction fluid was resuspended.
Data were obtained using a spectrophotometer (540 nm; Hitachi,
Tokyo, Japan).
Real-time fluorescence quantitative
polymerase chain reaction (qRT-PCR)
Total RNA in cardiac tissue and CFbs was extracted
with TRIzol (Invitrogen Life Technologies). Levels of GAPDH,
ColIa1, ColIIIa1, TGFβ1, TGFβRII and miR-370 were detected with
qRT-PCR and SYBR-Green (Takara Bio, Dalian, China) (Table I).
 | Table I.Primer sequence of miR-370 and mRNAs
of TGFβ1, TGFβRII, ColIa1, ColIIIa1 and GAPDH.a |
Table I.
Primer sequence of miR-370 and mRNAs
of TGFβ1, TGFβRII, ColIa1, ColIIIa1 and GAPDH.a
| Genes | Primers | Sequence |
|---|
| mir-370 | RT primers |
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGTGTAA-3′ |
|
| Sense |
5′-AGACCAGGTCACGTCTCTG-3′ |
|
| Antisense |
5′-GACAGACAAACCAGGTTCCA-3′ |
| U6 | RT primers |
5′-CGCTTCACGAATTTGCGTGTCAT-3′ |
|
| Sense |
5′-GCTTCGGCAGCACATATACTAAAAT-3′ |
|
| Antisense |
5′-CGCTTCACGAATTTGCGTGTCAT-3′ |
| TGFβ1 | Sense |
5′-GCGCCTGCAGAGATTCAAGTCAAC-3′ |
|
| Antisense |
5′-GTATCAGTGGGGGTCAGCAGCC-3′ |
| TGFβRII | Sense |
5′-TCACTAGGCACGTCATCAGC-3′ |
|
| Antisense |
5′-AGGACAACCCGAAGTCACAC-3′ |
| ColIa1 | Sense |
5′-TTCACCTACAGCACGCTTGT-3′ |
|
| Antisense |
5′-TTGGGATGGAGGGAGTTTAC-3′ |
| ColIIIa1 | Sense |
5′-TTGAATATCAAACCGCAAGGC-3′ |
|
| Antisense |
5′-GGTCACTTTCACTGGTTGACGA-3′ |
| GAPDH | Sense |
5′-AGACAGCCGCATCTTCTTGT-3′ |
|
| Antisense |
5′-TGATGGCAACAATGTCCACT-3′ |
Western blotting
Total protein in cardiac tissue and CFbs was
extracted with protein lysis buffer (Beyotime Biotech, Jiangsu,
China). Proteins were isolated using 60 µg protein system and 10%
SDS polyacrylamide gel, and transferred to the polyvinylidene
fluoride membrane. After three hours of blocking with 5% skim milk,
TGFβ1, TGFβRII, a-SMA (a-smooth muscle actin, Gene Tex) and
internal reference APDH (Anti Gene) primary antibody diluent was
used to seal the membrane for 16 h. 1X TBST washing membrane for 15
min each time for a total of three times. The secondary antibody
(Anti Gene) dilution was incubated for 2 h and then membranes were
washed in 1X TBST for 15 min each time for a total of three times.
Thereafter, ECL (Thermo Fisher Scientific, Waltham, MA, USA)
luminescence was applied. The Bio-Rad Biology system was used to
capture images and the results were analyzed.
Target gene prediction of miRNA
We used two databases, TargetScan6.2 (http://www.targetscan.org/index.html)
and PicTar (http://pictar.mdc-berlin.de), to predict the
combination of miR-370 and TGFβII mRNA.
Dual luciferase reporter gene
assay
3′-UTR in TGFβRII was sub-cloned based on PCR, and
the miRNA binding site in reporter gene vector was constructed. The
construct was inserted into multiple cloning sites. After miRNA
reporter vector (Ambion Life Technologies, Carlsbad, CA, USA)
expressed by pMIR-REPORT™ luciferase (HindIII and
SpeI sites) in the serum-free medium for 24 h, human
embryonic kidney cell (HEK-293) (1≤105/well) 1ug PGL3
target DNA (firefly luciferase vector) and 0.1 µg pRL-TK (Ranilla
luciferase vector driven by TK) were transfected using liposome
(Lipofectamine® 2000, Invitrogen Life Technologies).
Forty-eight hours after transfection of dual luciferase, double
luciferase activity was detected, strictly according to the method
provided by Promega (8).
Statistical analysis
Statistical analysis was performed using SPSS 13.0
(SPSS, Inc., Chicago, IL, USA) and GraphPad Prism (version 6.0;
GraphPad Software, Inc, La Jolla, CA, USA). All data were expressed
as mean ± standard error. Analysis of variance and Dunnett's test
were conducted in comparisons among multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
miR-370 expression decreases after
myocardial infarction, whereas the expression of TGFβ1 and TGFβRII
increases
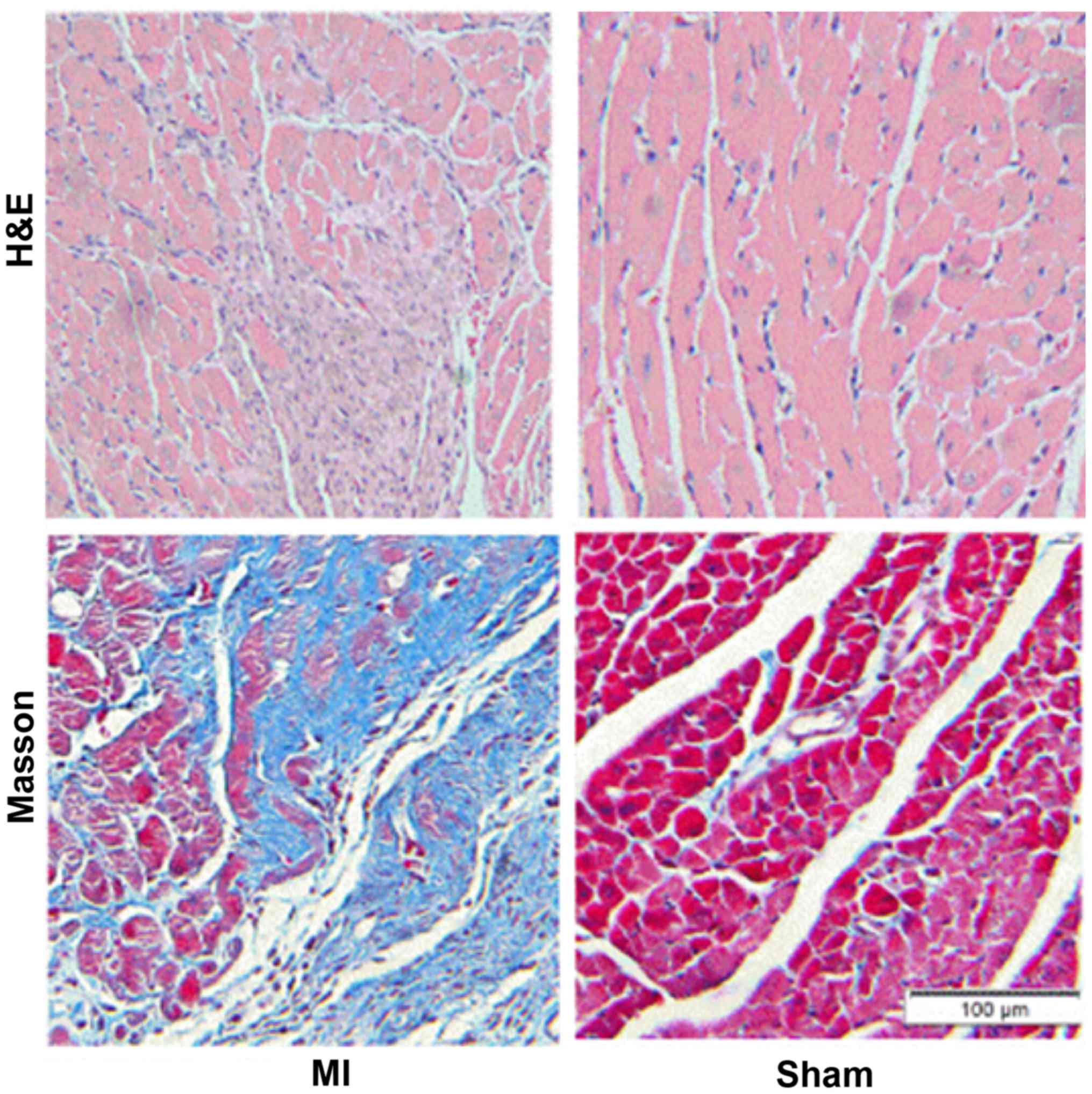
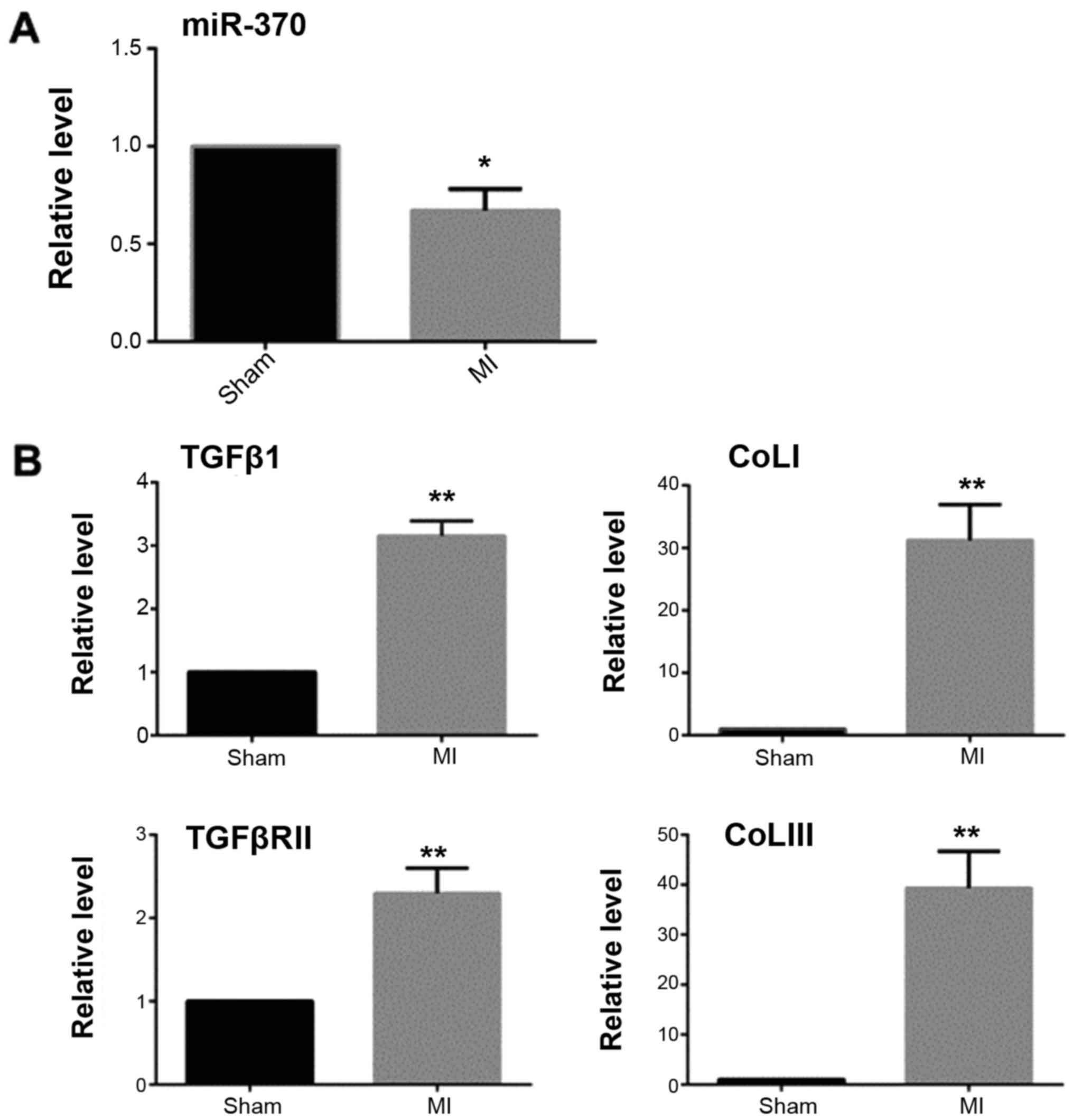
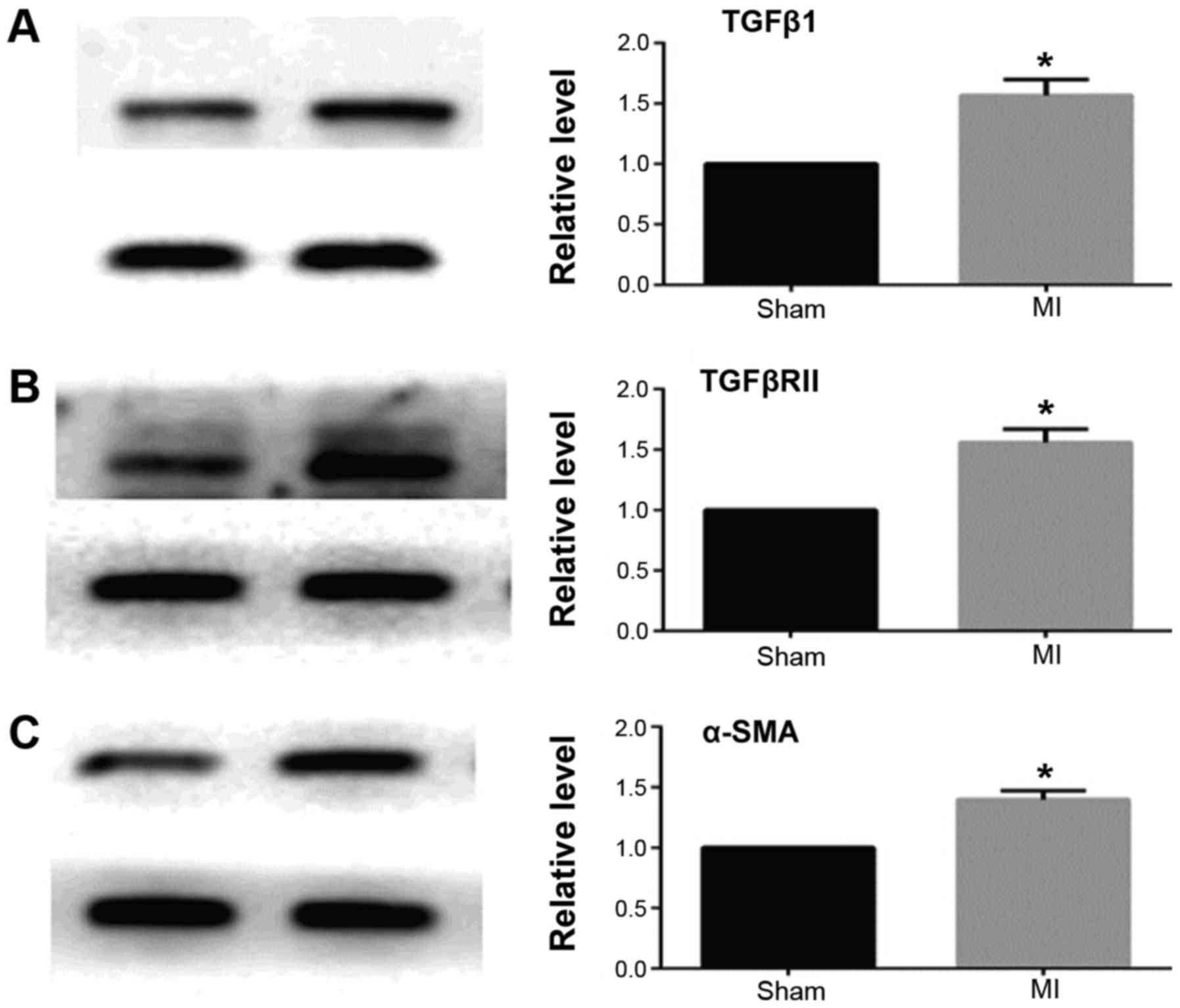
MASSON staining and qRT-PCR results showed that 2
weeks after myocardial infarction, the collagen deposition in the
infarct border area was significantly increased in the myocardial
infarction group when compared with the sham operation group
(Fig. 1). The expression of TGFβ1
and TGFβRII was significantly increased and miR-370 expression
decreased in the border area of the heart infarction. Western
blotting results showed that the expression of α-SMA was also
significantly increased in the border area, which suggested that
CFbs in the infarct border area partially differentiated into
myofibroblasts (Figs. 2 and
3).
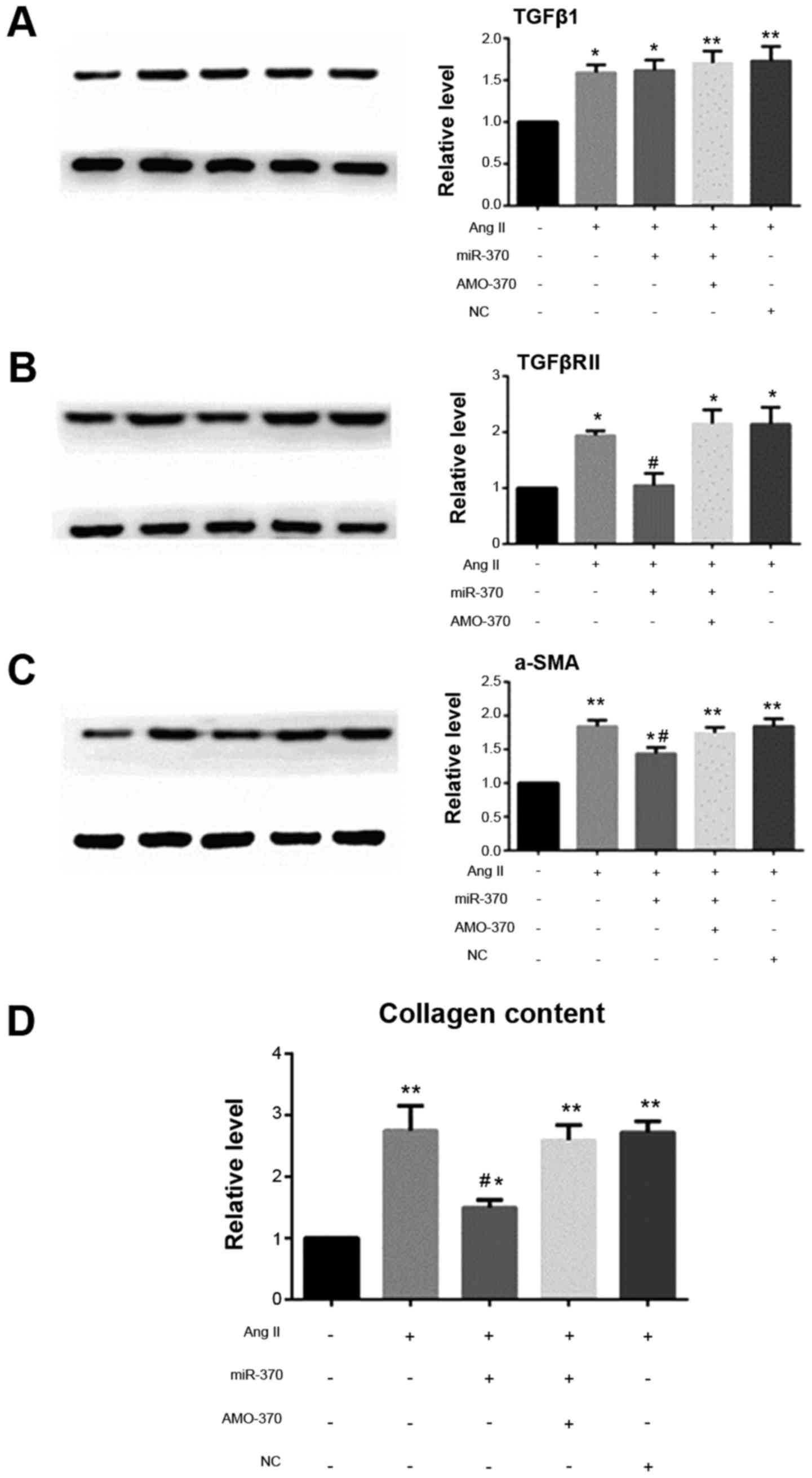
AngII promotes the differentiation of
CFbs and collagen secretion and inhibits the expression of
miR-370
When SD rat CFbs were passaged for the third
generation and cells were fused to 85%, the cells were synchronized
with serum-free DMEM for 24 h and stimulated with AngII for 24 h,
and then the indicators were detected. It showed that TGFβ1, and
TGFβRII mRNA and protein levels were increased in fibroblasts under
the induction of AngII. The increase of α-SMA promoted the
transformation of fibroblasts into myofibroblasts and increased the
expression of collagen. The results also showed that AngII
significantly inhibited the expression of CFb miR-370, which
indicated that the decrease of miR-370 may be related to the
increase of TGFβRII.
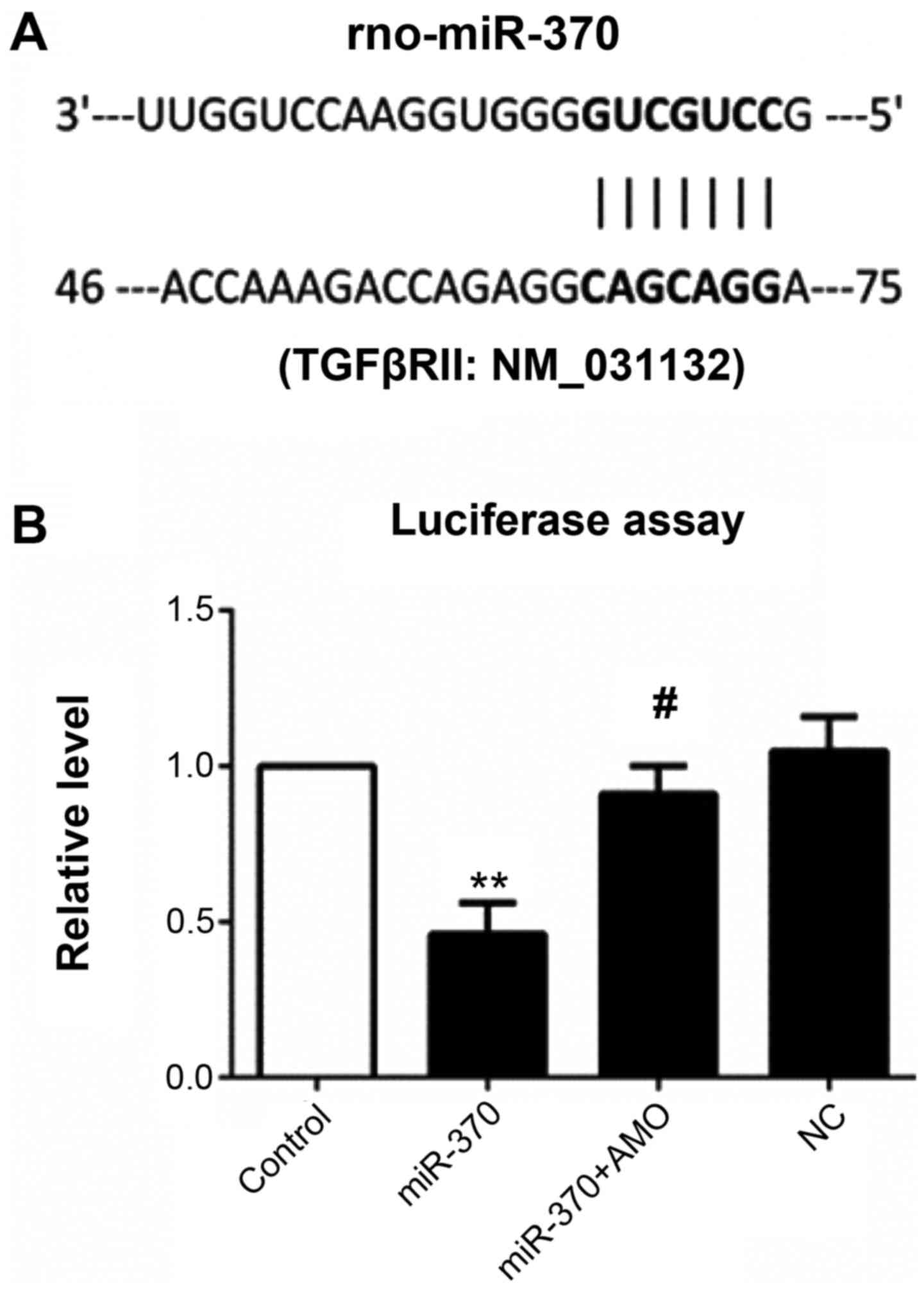
miR-370 inhibits TGFBRII expression
and inhibits myocardial fibrosis induced by AngII
Dual luciferase reporter gene assay showed that
luciferase activity was significantly decreased in the plasmid
group co-transfected with miR-370 mimics and contained the 3′UTR
gene sequence of TGFβRII when compared with the control group.
miR-370 mimics and inhibitor co-transfection significantly
inhibited decreased luciferase activity induced by miR-370 mimics,
while there were no effects in the NC transfection group (Fig. 4).
qRT-PCR results showed that the levels of TGFβRII,
ColI1a and ColIII1a mRNAs in the miR-370 mimics + AngII group were
significantly decreased compared with the AngII group. There were
no significant differences in the levels of the above indexes in
the miR-370 inhibitor + AngII, NC + AngII and AngII groups. Level
of TGFβ1 mRNA did not differ in the above four groups. Western blot
analysis revealed that, compared with the other three groups,
TGFβRII and α-SMA protein levels and collagen secretion decreased
in the miR-370 mimics + AngII group, however, miR-370 mimics did
not inhibit the increase in TGFβ1 induced by AngII (Figs. 5 and 6).
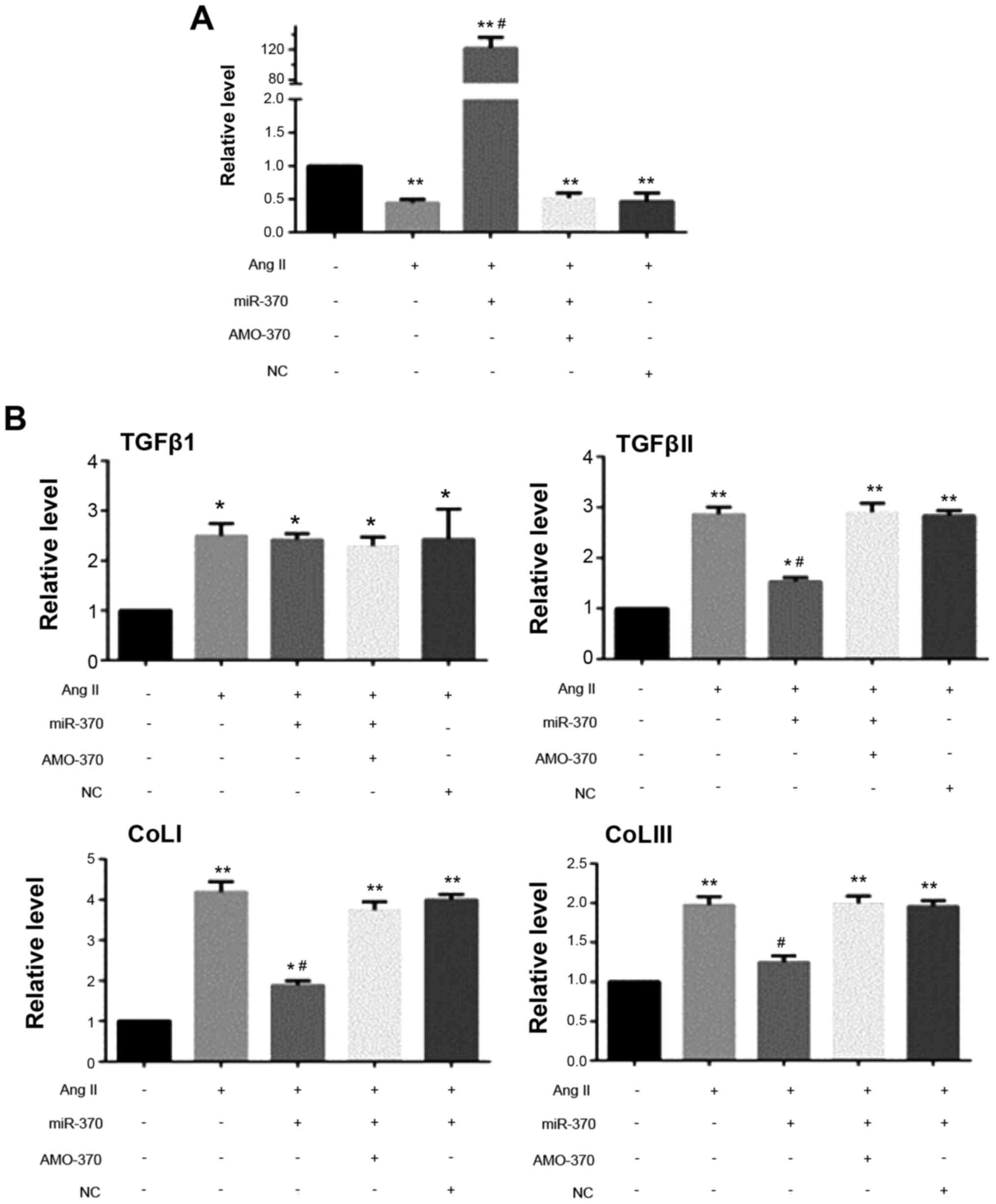 | Figure 5.qRT-PCR results. (A) Detection of
AngII (100 nM); transfection of miR mimics (50 nM) + AngII;
transfection of miR inhibitor (100 nM) + AngII/transfection of miR
NC (50 nM) + AngII stimulating CFbs for 24 h, the level of miR-370
was detected. (B) Detection of AngII (50 nM); transfection of miR
mimics (50 nM) + AngII; transfection of miR inhibitor (100 nM) +
AngII/transfection of miR NC (50 nM) + AngII stimulating CFbs for
24 h, levels of TGFβ1, TGFβRII, ColIa1, ColIIIa1, GAPDH mRNAs were
detected (*P<0.05 vs. Sham group; **P<0.01 vs. sham group;
#P<0.05 vs. AngII group). N=3 in each group; The data
are expressed as mean ± SEM. AMO-370 AMO, miR-370 inhibitor;
miR-370, miR-370 mimics; NC, negative control; CFbs, cardiac
fibroblasts. |
Discussion
The present study showed that the expression of
miR-370 in the myocardium was decreased after myocardial
infarction. It can inhibit the myocardial remodeling induced by
AngII after myocardial infarction. The underlying mechanism is that
miR-370 reduces the expression of TGFβRII, inhibits the TGFβ1-SMAD
signal transduction pathway so as to exert the effects of
inhibiting myocardial fibrosis.
It has been confirmed that the expression of miR-370
in peripheral blood leukocytes was higher in patients with acute
coronary syndrome than that in normal patients (9). But the expression of miR-370 in the
myocardium after myocardial infarction and the corresponding
mechanisms have not been studied. The present study showed that in
myocardial remodeling, the expression of miR-370 in the infarct
border area was decreased after myocardial infarction. Fibroblasts
differentiated into myofibroblasts and collagen secretion was
increased. AngII promoted the expression of TGFβ1 and TGFβRII in
CFbs and inhibited the expression of miR-370 at the same time.
miR-370 can be combined with the 3-UTR in TGFβRII mRNA and inhibit
the expression of TGFβRII, which result in the inhibition of the
biological effects of AngII on CFbs, including differentiation of
CFbs and the increase of collagen expression. However, miR-370 did
not block the increase in TGFβ1 induced by AngII. The results
suggested that the biological effects of miR-370 on AngII may be
mediated by inhibiting TGFβRII expression and blocking downstream
signal transduction pathways, but not inhibiting the generation of
TGFβ1.
Myocardial remodeling in myocardial infarction is
divided into two stages. The first stage is the physiological
repair after infarction, which belongs to the protective repair.
The second stage is the pathological remodeling caused by the
continuous accumulation of extracellular matrix (9). In pathological remodeling after
myocardial infarction, CFbs transform into α-SMA positive
myofibroblasts under the continuous action of multiple biological
effector molecules, such as AngII, TNF-α and TGF-β. The ability of
myofibroblasts to migrate, proliferate and secrete collagen is
obviously stronger. Hence, the transformation into myofibroblasts
is the key link of pathological remodeling after infarction
(10).
TGFβ1-SMAD signal transduction pathway plays an
important role in the process of cardiac remodeling. Previous
studies have indicated that many induced fibrosis factors, such as
AngII and ET1, take effect by promoting the expression of TGFβ1
(11). Indeed, TGFβ1 is the key
factor of fibrogenic effects. TGFβ1 has an effect on TGFβ receptor
complex on the cell surface and leads to the phosphorylation of
downstream transcription factor SMAD2/3, which is further combined
with SMAD4 to form the complex. The complex enters into the cell
and combines with cellular DNA, or it can form a special
transcription factor to regulate the target genes and target
proteins (4). Previous findings
have shown that nicotine can lead to atrial fibrosis in dogs by
increasing the expression of TGFβRII receptor in the atrial
fibroblasts, enhancing the downstream signal transduction and
increasing the expression of collagen (12).
An increasing number of microRNAs including Let-7,
mir-133 and −30c have been shown to exert an inhibitory effect on
organ fibrosis by blocking the TGFβ1-TGFβRI/TGFβRII-SMAD signal
transduction pathway (13,14). Of these, Let-7 has been shown to
inhibit the expression of TGFβRI and thus, play a protective role
in renal fibrosis (15). miR-24
was demonstrated to exert anti-fibrotic effects by inhibiting the
expression of TGFβ1 in CFbs (15).
miR-21, a small RNA that has been widely confirmed to have
fibrogenic effects, has also been shown that it leads to fibrosis
by inhibiting the expression of TGFβRIII, seen that TGFβRIII can
inhibit TGFβRI/II, thereby promoting TGFβRI/II-induced downstream
signal transduction (16,17). A recent study has showed that
nicotine in tobacco reduced mir-590 in dog atrial fibroblasts,
increased the expression of TGFβRII and then enhanced the secretion
of collagen and proliferation (12). This suggests that it is feasible to
affect the expression of TGFβR and to inhibit myocardial fibrosis
through the intervention of miRNAs levels.
In total, our results suggest that miR-370 exerts
anti-fibrotic effects by decreasing the expression level of TGFβRII
and inhibiting TGFβ1-TGFβRI/II-SMAD signal transduction pathway.
However, a large number of studies are needed to validate the
specific mechanisms of miRNAs in myocardial remodeling after
myocardial infarction and the application of miRNAs in the
treatment of cardiovascular diseases.
References
|
1
|
Poole-Wilson P: The prevention of
cardiovascular disease worldwide: Whose task and WHO's task? Clin
Med (Lond). 5:379–384. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Porter KE and Turner NA: Cardiac
fibroblasts: At the heart of myocardial remodeling. Pharmacol Ther.
123:255–278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shinde AV and Frangogiannis NG:
Fibroblasts in myocardial infarction: A role in inflammation and
repair. J Mol Cell Cardiol. 70:74–82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Blobe GC, Schiemann WP and Lodish HF: Role
of transforming growth factor beta in human disease. N Engl J Med.
342:1350–1358. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
van Rooij E: The art of microRNA research.
Circ Res. 108:219–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Orenes-Piñero E, Montoro-García S, Patel
JV, Valdés M, Marín F and Lip GY: Role of microRNAs in cardiac
remodelling: New insights and future perspectives. Int J Cardiol.
167:1651–1659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang
F, Zhang Y, Shan H, Luo X, Bai Y, et al: MicroRNA-328 contributes
to adverse electrical remodeling in atrial fibrillation.
Circulation. 122:2378–2387. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hoekstra M, van der Lans CA, Halvorsen B,
Gullestad L, Kuiper J, Aukrust P, van Berkel TJ and Biessen EA: The
peripheral blood mononuclear cell microRNA signature of coronary
artery disease. Biochem Biophys Res Commun. 394:792–797. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Espira L and Czubryt MP: Emerging concepts
in cardiac matrix biology. Can J Physiol Pharmacol. 87:996–1008.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Adiarto S, Heiden S, Vignon-Zellweger N,
Nakayama K, Yagi K, Yanagisawa M and Emoto N: ET-1 from endothelial
cells is required for complete angiotensin II-induced cardiac
fibrosis and hypertrophy. Life Sci. 91:651–657. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shan H, Zhang Y, Lu Y, Zhang Y, Pan Z, Cai
B, Wang N, Li X, Feng T, Hong Y, et al: Downregulation of miR-133
and miR-590 contributes to nicotine-induced atrial remodelling in
canines. Cardiovasc Res. 83:465–472. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duisters RF, Tijsen AJ, Schroen B,
Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE,
Schellings MW, Barenbrug P, et al: miR-133 and miR-30 regulate
connective tissue growth factor: implications for a role of
microRNAs in myocardial matrix remodeling. Circ Res. 104:170–178.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang B, Jha JC, Hagiwara S, McClelland AD,
Jandeleit-Dahm K, Thomas MC, Cooper ME and Kantharidis P:
Transforming growth factor-β1-mediated renal fibrosis is dependent
on the regulation of transforming growth factor receptor 1
expression by let-7b. Kidney Int. 85:352–361. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang J, Huang W, Xu R, Nie Y, Cao X, Meng
J, Xu X, Hu S and Zheng Z: MicroRNA-24 regulates cardiac fibrosis
after myocardial infarction. J Cell Mol Med. 16:2150–2160. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang L, Tang J, Chen H, Ge D, Sui T, Que
J, Cao X and Ge Y: Taurine reduced epidural fibrosis in rat models
after laminectomy via downregulating EGR1. Cell Physiol Biochem.
38:2261–2271. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liang H, Zhang C, Ban T, Liu Y, Mei L,
Piao X, Zhao D, Lu Y, Chu W and Yang B: A novel reciprocal loop
between microRNA-21 and TGFβRIII is involved in cardiac fibrosis.
Int J Biochem Cell Biol. 44:2152–2160. 2012. View Article : Google Scholar : PubMed/NCBI
|