Introduction
Diabetes is a complex metabolic disorder
characterized by abnormalities in glucose homeostasis and insulin
sensitivity (1). The increasing
prevalence of metabolic syndrome indications, including
dyslipidemia, obesity and insulin resistance, has stimulated rising
interest in the role environmental pollutants serve in such
diseases (2). Environmental
exposure to persistent organic pollutants is associated with
diabetes (3). Perfluorooctanoic
acid (PFOA) is one of the most common perfluoroalkyl acids (PFAAs),
which has been detected in humans and wildlife due to environmental
exposure. Substantial progress has been made in the toxicological
research of these compounds, particularly in the areas of
developmental toxicity, immunotoxicity, hepatotoxicity and the
potential associated modes of action (4); PFOA is not metabolized and has an
estimated human half-life of 2.3–3.4 years (5). Notably, PFOA is water-soluble and
migrates readily from soil to groundwater, and human exposure can
occur via contaminated drinking water, with other sources of
exposure including food, food packaging, treated fabrics, house
dust and air (4). Previous
research has suggested that PFOA is associated with risk factors
for type 2 diabetes, including glucose homeostasis and metabolic
disorder (6). Furthermore,
PFOA-exposed employees at manufacturing facilities presented an
increased risk of mortality from type 2 diabetes (7), and in a longitudinal prospective
cohort study of pregnant women followed from pre-conception to
delivery, Zhang et al (8)
revealed a significant positive association between serum PFOA
concentrations and the risk of gestational diabetes. Although
several epidemiological studies identified no association between
PFOA and other PFAAs and the incidence of diabetes, the evidence
suggested that these compounds influenced glucose metabolism
(2,9,10).
Notably, experimental studies have reported that PFOA is able to
induce oxidative stress and apoptosis in primary cultured tilapia
hepatocytes (11), and in human
hepatoma HepG2 cells (12).
Oxidative stress is considered to be a key risk
factor for the development and progression of various chronic
degenerative diseases, including diabetes (13). Oxidative stress in pancreatic
β-cells reduces the mitochondrial membrane potential (MMP), which
promotes the release of apoptogenic factors that activate
downstream death programs (14).
Although the exact mechanism underlying pancreatic β-cell
dysfunction is not currently known, the enhanced production of
mitochondrial reactive oxygen species (ROS) that occurs under
hyperglycemic and hyperlipidemic conditions is a major contributing
factor to the disruption of β-cell function in type 2 diabetes
(15). Pancreatic β-cells are
particularly sensitive to oxidative damage; therefore mitochondrial
oxidative damage may underlie the marked loss of β-cell function
(16). Apoptosis is the
predominant mode of pancreatic β-cell death in diabetes (17) and serves a crucial role in the
pathogenesis of diabetes (18).
The apoptotic process occurs via two pathways: The ‘extrinsic’ or
death receptor-initiated pathway, and the ‘intrinsic’ or
mitochondrial-initiated pathway (19). The intrinsic apoptotic pathway
involves fatal alterations to mitochondrial homeostasis, this
occurs via the loss of outer mitochondrial membrane integrity and
the subsequent release of cytochrome c. Mitochondrial
pore-formation and the release of cytochrome c are believed
to irreversibly commit a cell to apoptosis (20).
Proinflammatory cytokines, including interleukin-1β
(IL-1β) and tumor necrosis factor α (TNF-α), moderate the
expression and activity of antioxidant enzymes, further stimulating
an imbalance in the redox status of insulin-producing cells
(21). In addition,
proinflammatory cytokines stimulate inducible nitric oxide synthase
(iNOS) expression, which promotes nitric oxide (NO) formation
(21). NO has been widely
implicated in nitrosative stress, an event that is linked to cell
damage. Therefore, NO and ROS are considered crucial elements in
proinflammatory cytokine-mediated pancreatic β-cell death (22). During apoptosis, pancreatic β-cell
mitochondria may be influenced by proinflammatory cytokines, and
this occurs via activation of the intrinsic or extrinsic pathways
(23). Furthermore, cytokines may
impair the MMP and disturb adenosine triphosphate (ATP)
homeostasis, leading to the release of cytochrome c and
activation of β-cell apoptosis. The present study therefore
investigated the in vitro cytotoxic effects of PFOA on
pancreatic β-cells and its underlying mechanisms, using the RIN-m5F
rat pancreatic β-cell line.
Materials and methods
Materials
PFOA (Fig. 1) was
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Fetal bovine serum (FBS) and RPMI-1,640 medium were purchased from
Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Other
reagents were of the highest commercial grade available and were
purchased from Sigma-Aldrich (Merck KGaA).
Cell culture
RIN-m5F cells derived from rat pancreatic β-cells
were purchased from the American Type Culture Collection (Manassas,
VA, USA) and maintained in RPMI-1,640 supplemented with 10% FBS and
1% penicillin/streptomycin, at 37°C and saturated humidity with 5%
CO2. Cells were seeded into a 24-well plate (2×104
cells/well) and cultured for 48 h, and subsequently treated with
0–500 µM PFOA at 37°C for 48 h.
Cell viability
Cell viability was measured using an EZ-Cytox kit
(Daeil Lab Service Co. Ltd., Seoul, South Korea). Cells were
treated with water-soluble tetrazolium (WST) reagent and incubated
at 37°C for 2 h. Live cells metabolized the WST reagent, resulting
in an orange-colored product, the intensity of which was measured
at 450 nm using a Zenyth 3100 multimode detector spectrofluorometer
(Anthos Labtec Instruments GmbH, Salzburg, Austria). The data were
expressed as a percentage of the control [% control=100x
(absorbance of experimental group/absorbance of control
group)].
Measurement of apoptosis
Apoptosis was assessed using a Cell Death Detection
ELISAPLUS kit (Roche Diagnostics GmbH, Mannheim, Germany),
according to the manufacturer's protocol. The assay was based on
the quantitative sandwich-enzyme-immunoassay-principle, using DNA-
and histone-directed mouse monoclonal antibodies, and allowed the
specific determination of mono- and oligonucleosomes in the
cytoplasmic fraction of cell lysates (24).
Measurement of intracellular ROS
ROS was measured using 2,7-dichlorofluorescin
diacetate (DCFH-DA). Oxidation of the non-fluorescent DCFH-DA
yields dichlorofluorescin (DCF), a highly fluorescent product that
detects reactive oxygen intermediates in intact cells (25). Cells were treated with 10 µM
DCFH-DA at 37°C for 1 h. Following washing with Dulbecco's PBS, ROS
levels were determined by measuring the fluorescence intensity at
an excitation wavelength of 485 nm and an emission wavelength of
530 nm, using a Zenyth 3100 multimode detector
spectrofluorometer.
Measurement of mitochondrial
superoxide
Mitochondrial superoxide levels were detected using
MitoSOX™ Red mitochondrial superoxide indicator (Invitrogen; Thermo
Fisher Scientific Inc.), a fluorogenic dye used for the highly
selective detection of mitochondrial superoxide (26). Briefly, cells were incubated with 5
µM MitoSOX™ Red at 37°C for 20 min, according to the manufacturer's
protocol. The cells were subsequently washed by Dulbecco's PBS, and
then MitoSOX™ Red fluorescence was measured at an excitation
wavelength of 510 nm, and an emission wavelength of 580 nm.
Measurement of intracellular NO
production
A sensitive fluorescent indicator of NO,
2,7-difluorofluorescein (DAF-FM) diacetate, was used to detect
intracellular NO production. DAF-FM diacetate is a cell-permeable
derivative of DAF-FM, which is converted to the less permeable
DAF-FM by cellular esterases when it enters the cell, thus
preventing signal loss due to diffusion of the molecule from the
cell. In the presence of oxygen, DAF-FM reacts with NO to yield the
highly fluorescent triazolofluorescein. Cells were incubated with 5
µM DAF-FM diacetate (Invitrogen; Thermo Fisher Scientific, Inc.)
for 2 h at 37°C (27). Following
the removal of excess probe, DAF-fluorescence intensity was
measured at an excitation wavelength of 495 nm and an emission
wavelength of 515 nm.
Cytokine (TNF-α and IL-1β)
immunoassay
Cell extracts were prepared using cell lysis buffer
(Cell Signaling Technology, Inc., Danvers, MA, USA) and centrifuged
at 10,000 × g for 15 min at 4°C. The cytosolic concentrations of
TNF-α (cat. no. RTA00) and IL-1β (cat. no. RBL00) were measured
using ELISA kits (R&D System Inc., Minneapolis, MN, USA)
according to the manufacturer's protocol. Briefly, cytoplasmic
cytokines were bound to antibodies immobilized on a pre-coated
microplate. Unbound substances were removed by washing, and a
cytokine-specific enzyme-linked polyclonal antibody was added to
each well. Unbound antibody-enzyme reagent was removed by washing,
and the provided substrate was added to each well. The enzyme
reaction yielded a blue product, which was converted to a yellow
product following addition of the stop solution. The absorbance was
measured at 450 nm; the measured color intensity was proportional
to the amount of bound cytokine.
Measurement of ATP concentration
Cells were homogenized in ATP assay buffer
(BioVision, Inc., Milpitas, CA, USA) Intracellular ATP
concentrations were determined using an ATP Colorimetric Assay kit
(BioVision, Inc.), which allows rapid measurement of intracellular
ATP. The assay was performed according to the manufacturer's
protocol. ATP concentrations were normalized to protein content in
the samples, protein concentrations were determined using the
Bradford Protein assay kit (Bio-Rad Laboratories, Inc., Hercules,
CA, USA) (25). A standard curve
using known ATP concentrations was plotted to allow the calculation
of nmoles ATP/mg protein. Results are presented as a percentage of
the control.
Measurement of the MMP
MMP was measured fluorometrically using the
fluorescent probe rhodamine 123 (28). Cells (1×104) were cultured in black
96-well plates and allowed to adhere overnight. Following adhesion,
cells were treated for a further 48 h with PFOA, as aforementioned.
The cells were then washed twice with PBS and incubated with 10 µM
rhodamine 123 solution, at 37°C in the dark, for 30 min. Following
a further 2 washes with PBS, the fluorescence intensity was
measured using a spectrofluorometer at an excitation wavelength of
505 nm and an emission wavelength of 534 nm. The data were analyzed
by GraphPad Prism software 4.0 (GraphPad Software, Inc., San Diego,
CA, USA).
Cytochrome c release assay
Cells were cultured at 2×104 cells/well onto 24-well
plates. Cell extracts were prepared using cell lysis buffer and
centrifuged at 10,000 × g for 15 min at 4°C. The
supernatants (200 µl) were detected using a cytochrome c
ELISA kit (cat. no. ab110172; Abcam, Cambridge, MA, USA). The assay
was performed according to the manufacturer's protocol. Cytochrome
c from the conditioned medium was immunocaptured within the
wells, and the concentration was determined by adding a cytochrome
c-specific antibody conjugated to horseradish peroxidase.
Following addition of the provided colorless substrate, the
peroxidase converted the substrate to a blue end-product. This
reaction occurred in a time-dependent manner, which was
proportional to the amount of protein captured in the wells. The
rate of blue color development was detected at 600 nm. The change
in absorbance was expressed as change in milliOD/min.
Measurement of cardiolipin
peroxidation
Cardiolipin peroxidation was assessed using
10-N-nonyl-Acridine Orange (NAO) (Molecular Probes; Thermo Fisher
Scientific, Inc.), which binds to mitochondria-specific
cardiolipin. NAO loses its affinity for peroxidized cardiolipin;
therefore, a decrease in NAO fluorescence reflects the peroxidation
of intracellular cardiolipin (29). Cells were labeled with NAO (5 µM,
at 37°C for 20 min), washed twicewithDulbecco's PBS, and the
fluorescence was measured at an excitation wavelength of 485 nm and
an emission wavelength of 530 nm.
Statistical analysis
The results were expressed as the mean ± standard
error of the mean, for triplicate experiments. One-way analysis of
variance was followed by Dunnett's t-test. P<0.05 was considered
to indicate a statistically significant difference. Data was
analyzed using SAS statistical software (version 9.1.3; SAS
Institute, Inc., Cary, NC, USA).
Results
Cytotoxicity of PFOA in RIN-m5F
cells
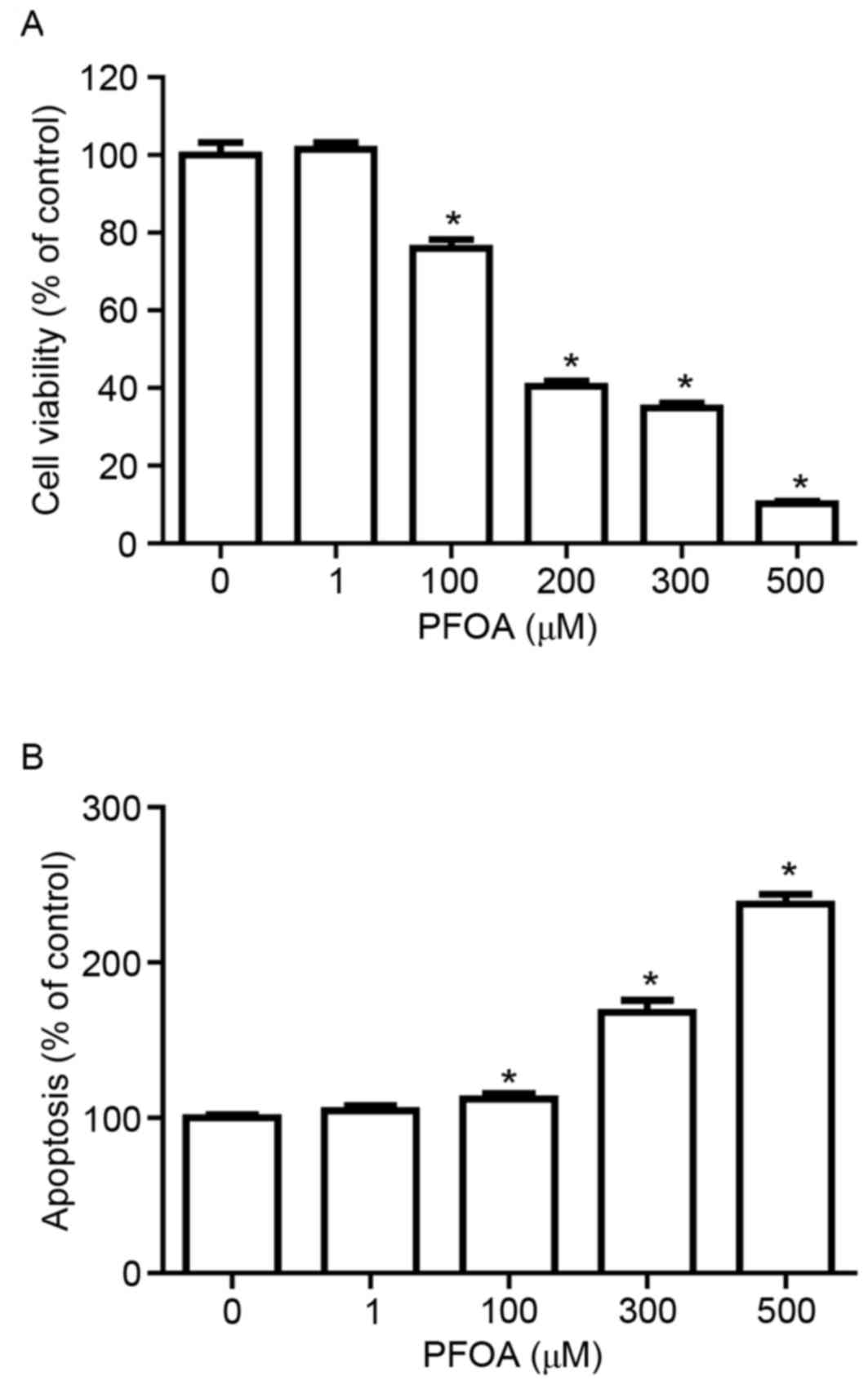
To determine whether PFOA induced cytotoxicity and
apoptosis in RIN-m5F cells, cells were treated with 0–500 µM PFOA
for 48 h. PFOA significantly decreased RIN-m5F cell viability in a
concentration-dependent manner (≥100 µM, Fig. 2A), with cells treated with 100 µM
PFOA exhibiting 76% cell viability, compared with control.
Furthermore, apoptosis was significantly increased in PFOA-treated
cells (100–500 µM; Fig. 2B);
following treatment with 500 µM PFOA apoptosis was increased
2.4-fold. These results indicated that PFOA may reduce cell
viability by inducing apoptosis.
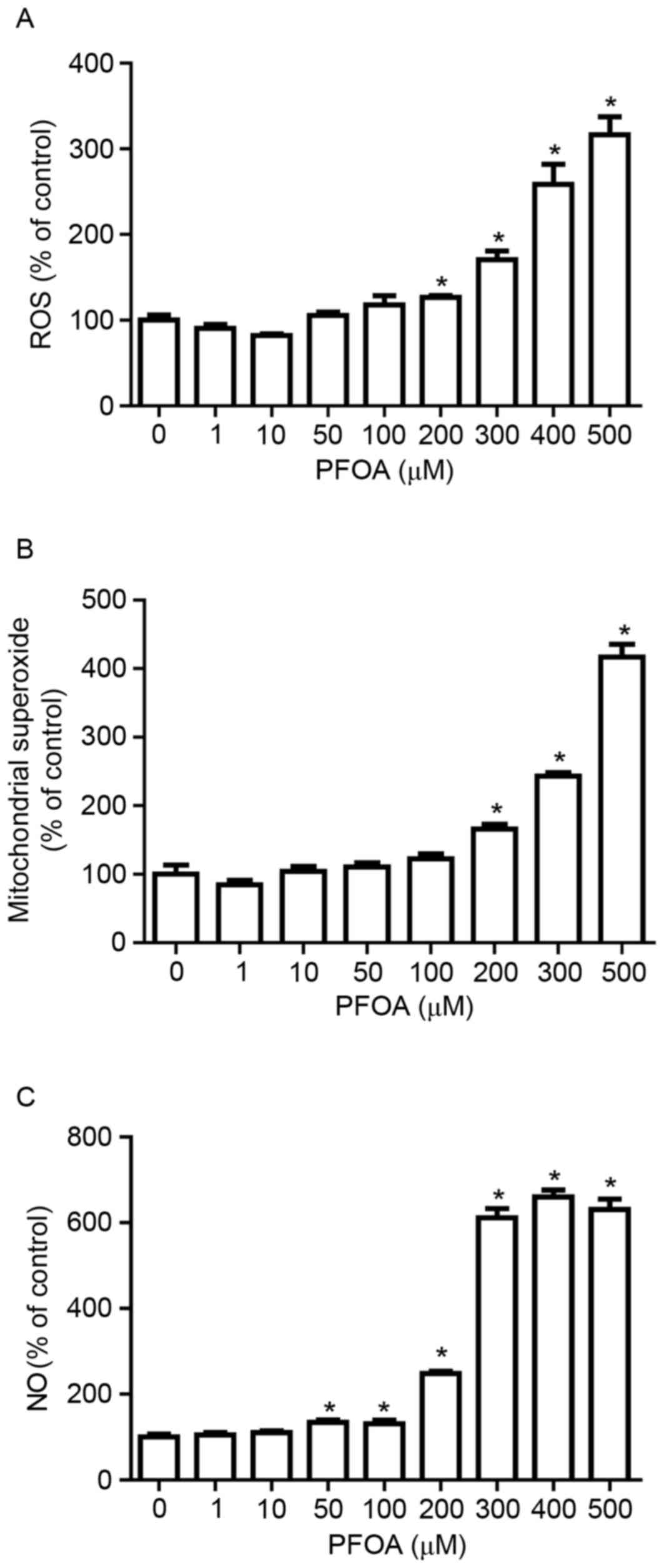
PFOA induces oxidative/nitrosative
stress in RIN-m5F cells
To investigate whether PFOA-induced apoptosis in
RIN-m5F cells was associated with the accumulation of ROS, RIN-m5F
cells were treated with DCFH-DA, a fluorescent probe for ROS. PFOA
(≥200 µM) significantly increased DCF fluorescence, which was
3.2-fold greater following treatment with 500 mM PFOA (Fig. 3A).
Mitochondria are a major source of ROS (25). To determine whether PFOA regulated
mitochondrial ROS accumulation in RIN-m5F cells, cells were
incubated with MitoSOX Red, a fluorogenic dye that specifically
detects superoxide in the mitochondria of live cells (30). Cells treated with PFOA (≥200 µM)
demonstrated significantly higher levels of MitoSOX™ Red
fluorescence, compared with control cells, which was 4-fold greater
following treatment with 500 µM PFOA. This result indicated that
high concentrations of PFOA may increase superoxide accumulation in
the mitochondria of RIN-m5F cells (Fig. 3B).
NO overproduction induces oxidative/nitrosative
stress, which results in cell apoptosis or necrosis. The
NO-specific DAF-FM probe was employed to investigate PFOA-induced
NO production. NO production was greater in RIN-m5F cells incubated
with PFOA for 48 h (≥50 µM) compared with control cells, up to
6.6-fold following treatment with 400 µM PFOA (Fig. 3C). This result suggested that PFOA
may induce oxidative/nitrosative stress in pancreatic β-cells, via
stimulation of NO overproduction.
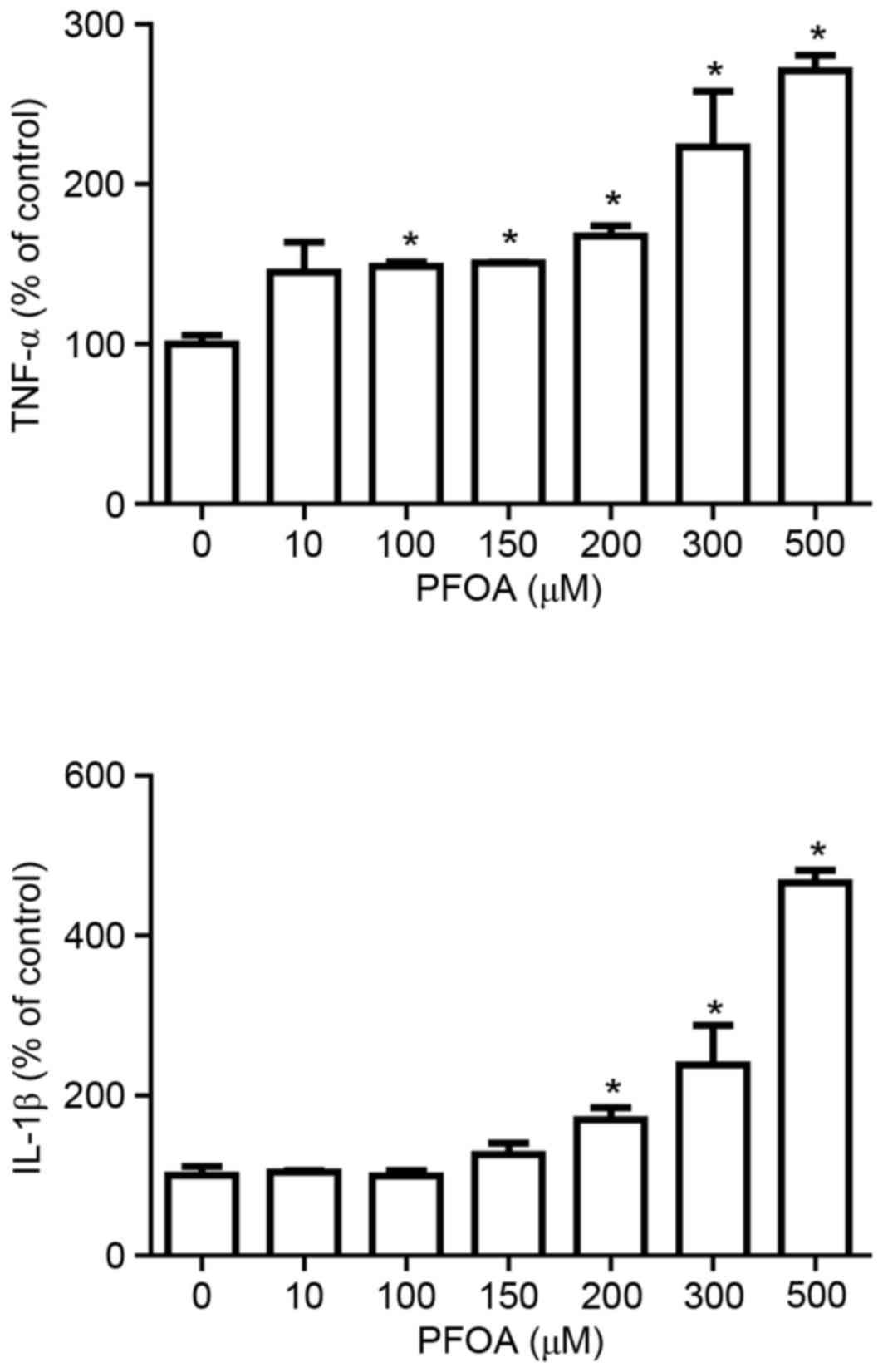
PFOA increases cytosolic TNF-α and
IL-1β levels in RIN-m5F cells
Proinflammatory cytokines activate various metabolic
pathways in pancreatic β-cells that result in cell death (31). Therefore, the present study
investigated whether PFOA modulates the production of the cytokines
TNF-α and IL-1β. The production of TNF-α and IL-1β was
significantly increased at PFOA concentrations ≥100 and ≥200 µM
respectively; TNF-α was increased by 2.7-fold and IL-1β was
increased by 4.6-fold following treatment with 500 µM PFOA
(Fig. 4).
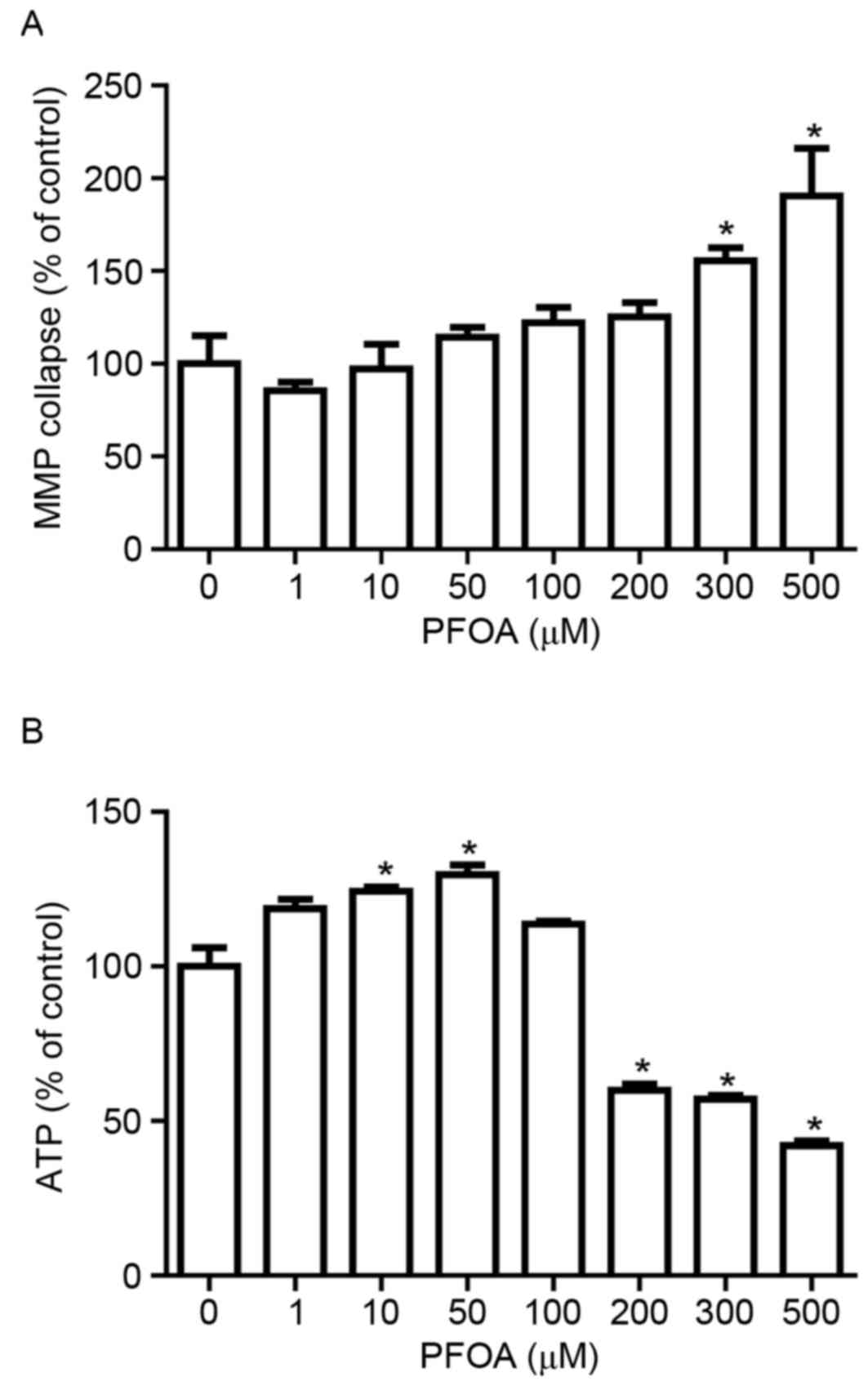
Effects of PFOA on MMP and ATP
production in RIN-m5F cells
The irreversible loss of mitochondrial function is a
prerequisite for apoptosis (32).
To determine whether PFOA treatment disrupts the MMP in RIN-m5F
cells, MMP was evaluated using the fluorescent dye rhodamine 123,
which accumulates in the mitochondrial compartment in an
MMP-dependent manner. PFOA treatment (≥300 µM) for 48 h
significantly disrupted the MMP in RIN-m5F cells, up to 1.9-fold
following treatment with 500 µM PFOA (Fig. 5A). Living cells require a
continuous supply of ATP to support the complex biological
functions that are essential for survival, and ATP concentration is
an important indicator of mitochondrial function. Intracellular ATP
was measured in cells exposed to increasing concentrations of PFOA.
ATP levels were significantly increased in response to low PFOA
concentrations (10–50 µM), up to 1.3-fold following treatment with
50 µM PFOA (Fig. 5B). However,
intracellular ATP decreased following exposure to high
concentrations (≥200 µM) of PFOA, and had decreased to 42%,
compared with the control, in cells treated with 500 µM PFOA.
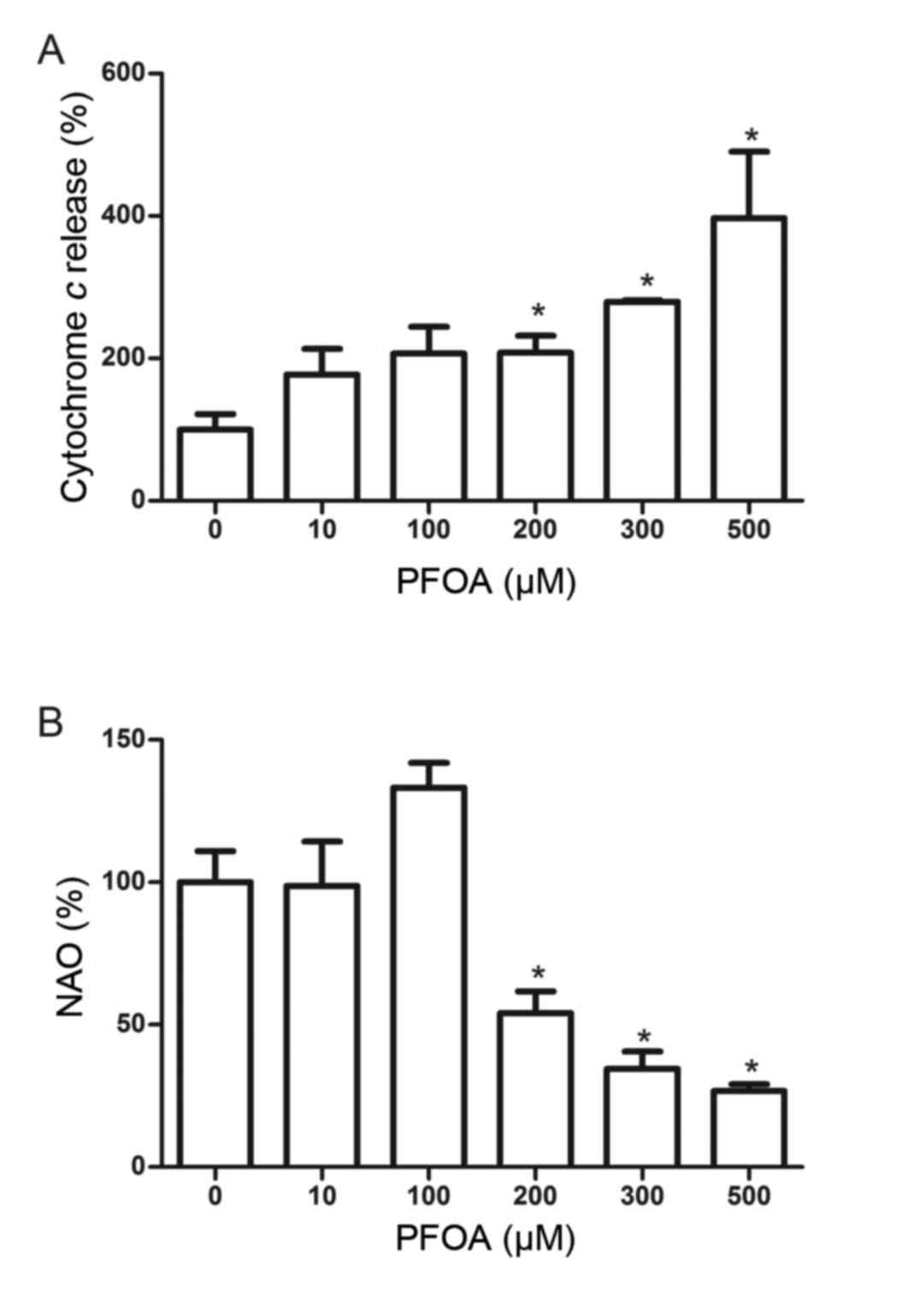
Effects of PFOA on cytochrome c
release and cardiolipin peroxidation in RIN-m5F cells
Mitochondrial cytochrome c release is a key
event in apoptotic initiation. PFOA treatment (≥200 µM) stimulated
a significant increase in cytochrome c release into the
medium, and was increased 4-fold following treatment with 500 µM
PFOA (Fig. 6A). Cardiolipin, which
is a phospholipid found almost exclusively at the inner
mitochondrial membrane, is an early target of oxygen-free radical
attack due to its high content of unsaturated fatty acids. An
oxidation-induced decrease in cardiolipin may facilitate the
release of cytochrome c into the cytosol. The fluorescent
probe NAO binds to cardiolipin, and this was used to determine
whether PFOA induced cardiolipin peroxidation in RIN-m5F cells. NAO
binding was reduced in cells treated with PFOA (≥200 µM), and was
reduced to 27%, compared with the control, in cells treated with
500 µM PFOA, thus indicating the occurrence of mitochondrial
PFOA-induced cardiolipin peroxidation (Fig. 6B).
Discussion
The present study demonstrated that PFOA induces
apoptosis and impairs the viability of the rat pancreatic β-cell
line RIN-m5F. Furthermore, PFOA damages β-cells by mediating the
accumulation of reactive nitrogen species (RNS) and ROS, and
through mitochondrial dysfunction. To the best of our knowledge,
the present study is the first to demonstrate a direct cytotoxic
effect of PFOA on pancreatic β-cells. Treatment with PFOA
independently disrupted MMP and reduced intracellular ATP levels in
β-cells, thus suggesting that inhibition of mitochondrial
metabolism may mediate this cytotoxic effect. Mitochondria are the
main source of intracellular ROS; superoxide is produced at
complexes I and III in the mitochondrial matrix and is converted to
hydrogen peroxide (H2O2) by manganese
superoxide dismutase (1).
Pancreatic β-cells are particularly sensitive to destruction by
mitochondrial ROS because the expression of antioxidant enzymes is
relatively low in these cells (33). PFOA-induced alterations in
mitochondrial membrane integrity result in the subsequent leakage
of ions, which may affect the proton gradient and, subsequently,
the oxidative phosphorylation required to produce ATP. Therefore,
it may be hypothesized that in β-cells exposed to PFOA,
RNS/ROS-induced oxidative stress damages the cell membrane, and the
subsequent oxidative stimulus induces the cytosolic signaling
pathway of the cell, disrupts mitochondrial function and initiates
apoptosis.
The mitochondrial-mediated apoptotic pathway serves
a major role in pancreatic β-cell death. Previous research has
demonstrated that a common effector phase of mitochondrial
permeability transition (MPT) may be shared by several types of
apoptosis and necrosis (34). MPT
is caused by the opening of a multiprotein complex pore positioned
at a connecting point between the inner and outer mitochondrial
membranes leading to MMP disruption, uncoupling of the respiratory
chain and termination of ATP production, hyperproduction of ROS,
Ca2+ release and depletion of reduced-glutathione, some
of which may provoke necrosis (34). Furthermore, MPT is associated with
the mitochondrial release of apoptosis-inducing factor and
cytochrome c, which may activate caspases involved in the
degradation phase of apoptosis (35). A decrease in cardiolipin levels
induces cytochrome c detachment from the inner mitochondrial
membrane, and facilitates the subsequent release into the cytosol
(36). Once released into the
cytoplasm, cytochrome c promotes assembly of the apoptosome
in response to the cell-death stimulus, and activated caspase-9
subsequently induces the processing and activation of effector
caspases, ultimately culminating in apoptosis (37). The discovery that PFOA treatment
results in the release of mitochondrial cytochrome c
indicates that this mechanism may serve an important role in the
promotion of oxidative damage and pancreatic β-cell damage.
Mitochondrial ROS stimulate lipid peroxidation and
reactive aldehyde formation in pancreatic β-cells, leading to the
development of type 2 diabetes (38). The present study demonstrated that
PFOA-induced apoptosis is associated with increased levels of
mitochondrial ROS production and cardiolipin peroxidation.
Cardiolipin is a unique dimeric phospholipid found almost
exclusively in the inner mitochondrial membrane. This lipid
contains a high percentage of unsaturated fatty acids and is
readily oxidized by ROS, a step that is considered essential for
the release of cytochrome c (39). Following mitochondrial damage,
cardiolipin is repositioned to the outer membrane of the
mitochondria where it functions as a recognition signal for
dysfunctional mitochondria. Cardiolipin and cytochrome c
interact at two sites on cytochrome c (40); cytochrome c can accept a
hydrogen proton and it can oxidize cardiolipin with an extra oxygen
molecule (41). Korytowski et
al (42) demonstrated that
oxidized cardiolipin species were significantly increased in the
mitochondria, following exposure to apoptotic stress.
The outcomes of the present study are consistent
with previous investigations into the toxicity of fluorochemicals.
Panaretakis et al (43)
observed MMP dissipation in HepG2 cells following incubation with
PFOA. Starkov and Wallace (44)
reported that treatment with perfluorinated derivatives induced
collapse of the MMP and subsequent swelling of rat liver
mitochondria. The study by Starkov and Wallace (44) indicated that PFOA may induce
peroxisome proliferation and interfere with mitochondrial metabolic
pathways. Free acid PFOA has previously been reported to induce a
small increase in the intrinsic proton leak of the mitochondrial
inner membrane, and the resulting alteration in membrane fluidity
was similar to that induced by a surfactant (44). Peroxisome proliferators can
slightly increase the steady-state level of
H2O2 in rodents, perhaps due to an
upregulation of acyl-CoA oxidase combined with a small increase in
catalase activity (45), and it is
well known that long-chain fatty acids are capable of inducing MPT
in vitro (46).
The present study indicated that PFOA induces the
production of high levels of NO. Increased generation of NO during
insulitis may contribute to pancreatic β-cell destruction (47). NO can diffuse through the
mitochondrial membrane and react with H2O2 in
an environment rich in free-iron, which promotes the formation of
hydroxyl radicals. The mitochondria are therefore the predominant
site of hydroxyl radical formation in pancreatic β-cells, and the
primary site of ROS toxicity, suggesting that mitochondrial damage
may be responsible for PFOA-induced cell death. Catalase enzyme
activity is reduced by the induction of iNOS and the accompanying
production of NO, which binds to the iron moiety in the catalase
heme groups (48). Furthermore, a
reaction of NO with superoxide results in the production of
peroxynitrite (49), a highly
reactive oxidant species that is associated the development of
autoimmune diabetes (50).
Although the precise role of NO in the development of diabetes is
not fully understood, synthesis of the NO radical contributes
significantly to β-cell dysfunction and apoptosis. Previous
research demonstrated that iNOS serves an important role in reduced
mitochondrial function in β-cells and islets (51). Hirst and Robson (52) reported that NO initiated
alterations in the MMP, and the subsequent release of cytochrome
c induced apoptosis. Notably, cytokine-induced production of
NO inhibits the mitochondrial enzyme aconitase, thus resulting in a
subsequent reduction in ATP production, which may contribute to the
promotion of necrotic cell death (53). The reaction of NO with superoxide
mediates physiological processes, another method through which this
may occur involves NO interaction with a metal at enzymatic active
sites, particularly in the Krebs cycle, which ultimately results in
significantly decreased glucose metabolism and ATP production
(54). Therefore, it is possible
that PFOA may exert its toxic effect through the damaging effects
of NO on mitochondrial function.
Pancreatic β-cell damage-initiated diabetes is a
complex process that is mediated, at least in part, by interactions
among cytokines, NO and oxygen-free radicals with the target
β-cells (55). The present study
revealed that PFOA increased the release of TNF-α and IL-1β. These
inflammatory cytokines are cytotoxic to β-cells in vitro,
and they have demonstrated an ability to induce apoptosis in
primary human (56) and mouse
(57) pancreatic β-cells, possibly
by stimulating NO synthesis (58).
These cytokines increase mitochondrial ROS production in several
cell types (59). Therefore,
mitochondrial ROS serves an important role in cytokine toxicity.
The mechanism that underlies mitochondrial ROS signaling in
cytokine-induced apoptosis remains unknown, however it has been
suggested that the destructive effects include cardiolipin
peroxidation, MPT facilitation and inhibition of mitochondrial
metabolism (60). Previous
research has indicated that various inhibitors of NO generation may
protect insulin-secreting cells against cytokine-mediated toxicity,
however this occurs with variable efficiency depending on the
chemical inhibitor involved and the cytokine combination (61).
In conclusion, the present study demonstrated that
PFOA induced an increase in the production of RNS, ROS and
inflammatory cytokines, and reduced ATP levels. Furthermore, PFOA
induced MMP collapse and the release of cytochrome c from
RIN-m5F rat pancreatic β-cell mitochondria. Therefore, these
results indicated that PFOA may exert its cytotoxic effect on
RIN-m5F cells through the increased oxidative stress and
mitochondrial dysfunction associated with the induction of
apoptosis.
Acknowledgements
The present study was supported by a grant of the
Korea Health Technology R&D Project through the Korea Health
Industry Development Institute (KHIDI), funded by the Ministry of
Health & Welfare, Republic of Korea (grant no.
HI14C-2700-020014).
References
|
1
|
Hsu HC, Chiou JF, Wang YH and Chen CH, Mau
SY, Ho CT, Chang PJ, Liu TZ and Chen CH: Folate deficiency triggers
an oxidative-nitrosative stress-mediated apoptotic cell death and
impedes insulin biosynthesis in RINm5F pancreatic islet β-cells:
Relevant to the pathogenesis of diabetes. PLoS One. 8:e779312013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nelson JW, Hatch EE and Webster TF:
Exposure to polyfluoroalkyl chemicals and cholesterol, body weight
and insulin resistance in the general U.S. population. Environ.
Health Perspect. 118:197–202. 2010. View Article : Google Scholar
|
|
3
|
Taylor KW, Novak RF, Anderson HA, Birnbaum
LS, Blystone C, Devito M, Jacobs D, Köhrle J, Lee DH, Rylander L,
et al: Evaluation of the association between persistent organic
pollutants (POPs) and diabetes in epidemiological studies: A
national toxicology program workshop review. Environ Health
Perspect. 121:774–783. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lau C, Anitole K, Hodes C, Lai D,
Pfahles-Hutchens A and Seed J: Perfluoroalkyl acids: A review of
monitoring and toxicological findings. Toxicol Sci. 99:366–394.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartell SM, Calafat AM, Lyu C, Kato K,
Ryan PB and Steenland K: Rate of decline in serum PFOA
concentrations after granular activated carbon filtration at two
public water systems in Ohio and West Virginia. Environ Health
Perspect. 118:222–228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin CY, Chen PC, Lin YC and Lin LY:
Association among serum perfluoroalkyl chemicals, glucose
homeostasis, and metabolic syndrome in adolescents and adults.
Diabetes Care. 32:702–707. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leonard RC, Kreckmann KH, Sakr CJ and
Symons JM: Retrospective cohort mortality study of workers in a
polymer production plant including a reference population of
regional workers. Ann Epidemiol. 18:15–22. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang C, Sundaram R, Maisog J, Calafat AM,
Barr DB and Louis GM Buck: A prospective study of prepregnancy
serum concentrations of perfluorochemicals and the risk of
gestational diabetes. Fertil Steril. 103:184–189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kames C, Winquist A and Steenland K:
Incidence of type II diabetes in a cohort with substantial exposure
to perfluorooctanoic acid. Environ Res. 128:78–83. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lind L, Zethelius B, Salihovic S, van
Bavel B and Lind PM: Circulating levels of perfluoroalkyl
substances and prevalent diabetes in the elderly. Diabetologia.
57:473–479. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu C, Yu K, Shi X, Wang J, Lam PK, Wu RS
and Zhou B: Induction of oxidative stress and apoptosis by PFOS and
PFOA in primary cultured hepatocytes of freshwater tilapia
(Oreochromis niloticus). Aquat Toxicol. 82:135–143. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eriksen KT, Raaschou-Nielsen O, Sørensen
M, Roursgaard M, Loft S and Møller P: Genotoxic potential of the
perfluorinated chemicals PFOA PFOS, PFBS, PFNA and PFHxA in human
HepG2 cells. Mutat Res. 700:39–43. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rains JL and Jain SK: Oxidative stress,
insulin signaling, and diabetes. Free Radic Biol Med. 50:567–575.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsujimoto Y: Cell death regulation by the
Bcl-2 protein family in the mitochondria. J Cell Physiol.
195:158–167. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nishikawa T and Araki E: Impact of
mitochondrial ROS production in the pathogenesis of diabetes
mellitus and its complications. Antioxid Redox Signal. 9:343–353.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Poitout V and Robertson RP:
Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr
Rev. 29:351–366. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mathis D, Vence L and Benoist C: beta-Cell
death during progression to diabetes. Nature. 414:792–798. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Atkinson MA: ADA outstanding scientific
achievement lecture 2004. Thirty years of investigating the
autoimmune basis for type 1 diabetes: Why can't we prevent or
reverse this disease? Diabetes. 54:1253–1263. 2005.
|
|
19
|
Millan A and Huerta S: Apoptosis-inducing
factor and colon cancer. J Surg Res. 151:163–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jin Z and El-Deiry WS: Overview of cell
death signaling pathways. Cancer Biol Ther. 4:139–163. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Souza KL, Gurgul-Convey E, Elsner M and
Lenzen S: Interaction between pro-inflammatory and
anti-inflammatory cytokines in insulin-producing cells. J
Endocrinol. 197:139–150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lenzen S: Oxidative stress: The vulnerable
beta-cell. Biochem Soc Trans. 36:343–347. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barbu A, Welsh N and Saldeen J:
Cytokine-induced apoptosis and necrosis are preceded by disruption
of the mitochondrial membrane potential (Deltapsi(m)) in pancreatic
RINm5F cells: Prevention by Bcl-2. Mol Cell Endocrinol. 190:75–82.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shi B, De Girolami U, He J, Wang S,
Lorenzo A, Busciglio J and Gabuzda D: Apoptosis induced by HIV-1
infection of the central nervous system. J Clin Invest.
98:1979–1990. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
López-Sánchez N, Rodriguez JR and Frade
JM: Mitochondrial c-Jun NH2-terminal kinase prevents the
accumulation of reactive oxygen species and reduces necrotic damage
in neural tumor cells that lack trophic support. Mol Cancer Res.
5:47–60. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schroeder P, Pohl C, Calles C, Marks C,
Wild S and Krutmann J: Cellular response to infrared radiation
involves retrograde mitochondrial signaling. Free Radic Biol Med.
43:128–135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang H, Meng QH, Chang T and Wu L:
Fructose-induced peroxynitrite production is mediated by
methylglyoxal in vascular smooth muscle cells. Life Sci.
79:2448–2454. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baracca A, Sgarbi G, Solaini G and Lenaz
G: Rhodamine 123 as a probe of mitochondrial membrane potential:
Evaluation of proton flux through F(0) during ATP synthesis.
Biochim Biophys Acta. 1606:137–146. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nomura K, Imai T, Koumura T, Kobayashi T
and Nakagawa Y: Mitochondrial phospholipid hydroperoxide
glutathione peroxidase inhibits the release of cytochrome c from
mitochondria by suppressing the peroxidation of cardiolipin in
hypoglycaemia-induced apoptosis. Biochem J. 351:183–193. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Robinson KM, Janes MS and Beckman JS: The
selective detection of mitochondrial superoxide by live cell
imaging. Nat Protoc. 3:941–947. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cieślak M, Wojtczak A and Cieślak M: Role
of pro-inflammatory cytokines of pancreatic islets and prospects of
elaboration of new methods for the diabetes treatment. Acta Biochim
Pol. 62:15–21. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mathews CE and Leiter EH: Constitutive
differences in antioxidant defense status distinguish
alloxan-resistant and alloxan-susceptible mice. Free Radic Biol
Med. 27:449–455. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Green D and Kroemer G: The central
executioners of apoptosis: Caspases or mitochondria? Trends Cell
Biol. 8:267–271. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Reed JC: Cytochrome c: Can't live with
it-can't live without it. Cell. 91:559–562. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gogvadze V and Zhivotovsky B: Alteration
of mitochondrial function and cell sensitization to death. J
Bioenerg Biomembr. 39:23–30. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Susnow N, Zeng L, Margineantu D and
Hockenbery DM: Bcl-2 family proteins as regulators of oxidative
stress. Semin Cancer Biol. 19:42–49. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Miwa I, Ichimura N, Sugiura M, Hamada Y
and Taniguchi S: Inhibition of glucose-induced insulin secretion by
4-hydroxy-2-nonenal and other lipid peroxidation products.
Endocrinology. 141:2767–2772. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kagan VE, Tyurin VA, Jiang J, Tyurina YY,
Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V,
et al: Cytochrome c acts as a cardiolipin oxygenase required for
release of proapoptotic factors. Nat Chem Biol. 1:223–232. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kagan VE, Bayır HA, Belikova NA, Kapralov
O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek
PM, et al: Cytochrome c/cardiolipin relations in mitochondria: A
kiss of death. Free Radic Biol Med. 46:1439–1453. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kapralov AA, Yanamala N, Tyurina YY,
Castro L, Samhan-Arias A, Vladimirov YA, Maeda A, Weitz AA,
Peterson J, Mylnikov D, et al: Topography of tyrosine residues and
their involvement in peroxidation of polyunsaturated cardiolipin in
cytochrome c/cardiolipin peroxidase complexes. Biochim Biophys
Acta. 1808:2147–2155. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Korytowski W, Basova LV, Pilat A,
Kernstock RM and Girotti AW: Permeabilization of the mitochondrial
outer membrane by Bax/Truncated Bid (tbid) proteins as sensitized
by cardiolipin hydroperoxide translocation: mechanistic
implications for the intrinsic pathway of oxidative apoptosis. J
Biol Chem. 286:26334–26343. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Panaretakis T, Shabalina IG, Grandér D,
Shoshan MC and DePierre JW: Reactive oxygen species and
mitochondria mediate the induction of apoptosis in human hepatoma
HepG2 cells by the rodent peroxisome proliferator and
hepatocarcinogen, perfluorooctanoic acid. Toxicol Appl Pharmacol.
173:56–64. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Starkov AA and Wallace KB: Structural
determinants of fluorochemical-induced mitohcondrial dysfunction.
Toxicol Sci. 66:244–252. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yeldandi AV, Rao MS and Reddy JK: Hydrogen
peroxide generation in peroxisome proliferator-induced oncogenesis.
Mutat Res. 448:159–177. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hermesh O, Kalderon B, Berman B and
Bar-Tana J: Mitochondrial protonophoric activity induced by a
thyromimetic fatty acid analogue. Biochim Biophys Acta.
1457:166–174. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Eizirik DL and Darville MI: beta-cell
apoptosis and defense mechanisms: Lessons from type 1 diabetes.
Diabetes. 50 Suppl 1:S64–S69. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sigfrid LA, Cunningham JM, Beeharry N,
Lortz S, Tiedge M, Lenzen S, Carlsson C and Green IC: Cytokines and
nitric oxide inhibit the enzyme activity of catalase but not its
protein or mRNA expression in insulin-producing cells. J Mol
Endocrinol. 31:509–518. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Arteel GE and Sies H: Protection against
peroxynitrite by cocoa polyphenol oligomers. FEBS Lett.
462:167–170. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Suarez-Pinzon WL, Szabó C and Rabinovitch
A: Development of autoimmune diabetes in NOD mice is associated
with the formation of peroxynitrite in pancreatic islet beta-cells.
Diabetes. 46:907–911. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Størling J, Binzer J, Andersson AK, Züllig
RA, Tonnesen M, Lehmann R, Spinas GA, Sandler S, Billestrup N and
Mandrup-Poulsen T: Nitric oxide contributes to cytokine-induced
apoptosis in pancreatic beta cells via potentiation of JNK activity
and inhibition of Akt. Diabetologia. 48:2039–2050. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hirst DG and Robson T: Nitrosative stress
as a mediator of apoptosis: Implications for cancer therapy. Curr
Pharm Des. 16:45–55. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sandler S, Bendtzen K, Eizirik DL,
Strandell E, Welsh M and Welsh N: Metabolism and beta-cell function
of rat pancreatic islets exposed to human interleukin-1 beta in the
presence of a high glucose concentration. Immunol Lett. 26:245–251.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Welsh N, Eizirik DL, Bendtzen K and
Sandler S: Interleukin-1 beta-induced nitric oxide production in
isolated rat pancreatic islets requires gene transcription and may
lead to inhibition of the Krebs cycle enzyme aconitase.
Endocrinology. 129:3167–3173. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Eizirik DL and Mandrup-Poulsen T: A choice
of death-the signal-transduction of immune-mediated beta-cell
apoptosis. Diabetologia. 44:2115–2133. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Delaney CA, Pavlovic D, Hooren A,
Pipeleers DG and Eizirik DL: Cytokines induce deoxyribonucleic acid
strand breaks and apoptosis in human pancreatic islet cells.
Endocrinology. 138:2610–2614. 1997. View Article : Google Scholar
|
|
57
|
Pavlovic D, Chen MC, Gysemans CA, Mathieu
C and Eizirik DL: The role of interferon regulatory factor-1 in
cytokine-induced mRNA expression and cell death in murine
pancreatic beta-cells. Eur Cytokine Netw. 10:403–412.
1999.PubMed/NCBI
|
|
58
|
Heitmeier MR, Scarim AL and Corbett JA:
Interferon-gamma increases the sensitivity of islets of Langerhans
for inducible nitric-oxide synthase expression induced by
interleukin 1. J Biol Chem. 272:13697–13704. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang D, Elner SG, Bian ZM, Till GO, Petty
HR and Elner VM: Pro-inflammatory cytokines increase reactive
oxygen species through mitochondria and NADPH oxidase in cultured
RPE cells. Exp Eye Res. 85:462–472. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Orrenius S: Reactive oxygen species in
mitochondria-mediated cell death. Drug Metab Rev. 39:443–455. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Darville MI and Eizirik DL: Regulation by
cytokines of the inducible nitric oxide synthase promoter in
insulin-producing cells. Diabetologia. 41:1101–1108. 1998.
View Article : Google Scholar : PubMed/NCBI
|