Introduction
A previous study reported that individuals who
regularly consume cruciferous vegetables demonstrate a lower risk
of developing cancer, including breast, prostate and hepatic cancer
(1). Cruciferous vegetables are a
rich source of isothiocyanates (ITCs), which have been demonstrated
to exhibit chemopreventive effects in animal cancer models
(1,2). Among ITCs, sulforaphane (SFN;
C6H11S2NO) is abundant in broccoli
and has been demonstrated to induce growth arrest and apoptosis of
tumor cells in a variety of experimental cancer models (2,3). SFN
exerts its anticancer effects on tumor cells by regulating a number
of cell signaling pathways including Kelch-like ECH-associated
protein 1-nuclear factor (NF)-erythroid 2-related factor 2
signaling, mitogen-activated protein kinase (MAPK) and NF-κB
signaling (4–6). Therefore, SFN is considered to be a
conceptually promising agent for cancer therapy. However, the
detailed molecular mechanisms underlying the effects of SFN in
colon cancer have yet to be fully elucidated.
Colorectal tumors first arise in the large intestine
or the rectum, and is the third and second most common cancer in
males and females, respectively (7,8).
This progressive cancer arises from the accumulation of mutations
in tumor suppressor genes and oncogenes, which disrupt the
homeostatic balance between cell proliferation and apoptosis
(9). The p53 tumor suppressor gene
serves important roles in the regulation of cell proliferation and
apoptosis (10–12). p53 is activated by a number of
intracellular and extracellular stressors, including irradiation,
DNA damaging agents or reactive oxygen species (ROS) (13–15).
Deficiencies in the function of p53 are critical events during the
development of tumors and resistance to anticancer therapies
(16,17). It has been estimated that ~29% of
colorectal cancers harbor p53 gene mutations (18). Deficiencies in p53 function
attenuate p53-mediated cell apoptosis and may lead to multi-drug
resistance (MDR) (19,20). An increasing body of evidence has
indicated that p53-mutant or p53-null cancer cells are more likely
to be resistant to different cytotoxic drugs; particularly to
chemotherapy and radiation therapy in the clinic, as these
therapies target cells primarily by inducing apoptosis (19,21).
Therefore, anticancer agents that induce cancer cell death in a
p53-independent manner may be promising candidates for preventing
MDR induced by p53 deficiency.
ROS serve important roles in the induction of
apoptosis under normal physiological and abnormal pathological
conditions (22,23). Tumor cells exhibit increased ROS
generation when compared to normal cells due to their vigorous
metabolic activities (24–26). Increased ROS production in tumor
cells increases their susceptibility to oxidative stress-induced
apoptosis, which may be exploited by selective anticancer agents
(27). A number of known natural
compounds, including Aloin, curcumin and resveratrol, increase ROS
levels, which leads to cancer cell death (28). A previous study demonstrated that
SFN generate ROS, which subsequently induces apoptosis in a number
of cancer cells. However, the specific role of ROS generation in
response to SFN exposure in p53-deficient human colon cancer cells
has not been fully elucidated (29).
In addition to p53, MAPK family members are
activated by ROS (30–32). MAPKs, including extracellular
signal-regulated kinase (Erk), c-Jun N-terminal kinase (JNK) and
p38, are activated in response to diverse stimuli, and serve
important roles in mammalian cells by regulating cell apoptosis,
differentiation, migration and proliferation (33). JNK, also known as the
stress-activated protein kinase, is predominantly activated by
different stressors via the phosphorylation of the
NH2-terminus of the c-Jun transcription factor. In the
majority of cases, p38 MAPK signaling has been demonstrated to
promote apoptosis or facilitate cell survival depending on the cell
type and stimulus (28). However,
Erk activation commonly induces cell proliferation or
differentiation in response to cell stressors (34,35).
A previous study revealed that SFN increases the
generation of intracellular ROS, leading to mitochondrial
dysfunction and human cancer cell apoptosis (36); however, the effects of SFN on
p53-deficient colon cancer cells is currently unknown. In the
present study, it was hypothesized that SFN induces apoptosis in
association with ROS generation and mitochondrial dysfunction. To
verify this hypothesis, the effect of SFN on the mitochondrial
membrane potential (MMP), and ROS and apoptosis levels was
determined using p53-deficient SW480 cells. The results
demonstrated that, SFN increased ROS generation, which subsequently
lead to activation of Erk and p38 MAPKs and apoptosis induction via
the mitochondrial-dependent apoptotic pathway.
Materials and methods
Reagents and antibodies
Dulbecco's modified Eagle's medium (DMEM), RPMI-1640
medium and fetal bovine serum (FBS) were supplied by Hyclone; GE
Healthcare Life Sciences (Logan, UT, USA). MTT, SFN and Cisplatin
(CPDD; cat. no. P4394) were obtained from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). The ROS, phosphorylated (p)-p38 and Erk
inhibitors [ammonium pyrrolidinedithiocarbamate (APDC), SB203580
and SCH772984], the caspase 3, 7, 8 and 9 activity assay kits (cat.
nos. C1116, C1136, C1151 and C1157), ROS Assay kit (cat. no.
S0033), Lipid Peroxidation Malondialdehyde (MDA) Assay kit (cat.
no. S0131), Total Superoxide Dismutase (SOD) Assay kit with
water-soluble tetrazolium salt (WST-8; cat. no. S0101) and Cellular
Glutathione Peroxidase (GSH-Px) Assay kit (cat. no. S0056), were
all supplied by Beyotime Institute of Biotechnology (Nanjing,
China). Protein isolation and bicinchoninic acid (BCA) protein
quantification kits were supplied by Bio-Rad Laboratories, Inc.
(Hercules, CA, USA). The annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) apoptosis detection kit was obtained
from Multi Sciences (Lianke) Biotech Co, Ltd. (Hangzhou, China).
Primary antibodies against B-cell lymphoma (Bcl-2; cat. no.
BS4024), Bcl-2-associated X protein (Bax; cat. no. MB9013), p73
(cat. no. BS2452), p53 upregulated modulator of apoptosis (PUMA;
cat. no. BS2922), phorbol-12-myristate-13-acetate-induced protein 1
(NOXA; cat. no. BS6214) and γH2AX (cat. no. BS4760) were obtained
from Bioworld Technology, Inc. (St. Louis Park, MN, USA). Primary
antibodies against p-Erk 1/2 (cat. no. sc-23759-R), p-p38 (cat. no.
sc-7973) and p-JNK (cat. no. sc-135642) were obtained from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA). The primary antibody
against β-actin (cat. no. BA2359) was supplied by Wuhan Boster
Biological Technology, Ltd. (Wuhan, China). IRDye-conjugated
anti-rabbit (cat. no. 611-645-122) and anti-mouse (cat. no.
610-142-002) secondary antibodies were obtained from Rockland
Immunochemicals, Inc. (Limerick, PA, USA).
Cell culture
The human colon adenocarcinoma cell line, SW480, and
the human colon cancer cell line, HCT-116, were obtained from The
Cell Bank of Type Culture Collection of Chinese Academy of Sciences
(Shanghai, China). SW480 and HCT-116 cells were cultured in DMEM
and RPMI-1640 medium, respectively, supplemented with 10% (v/v)
FBS, and incubated at 37°C in a humidified 5% CO2
incubator.
Cell viability assay
Cell viability was determined using an MTT assay.
Briefly, SW480 or HCT-116 cells were plated into 96-well culture
plates at an optimal density of 5×103 cells/well. SW480
cells were incubated for 24 h until cells had reached 90%
confluence, and were pretreated with or without 0.1 µM of the
specific inhibitors against p38, Erk and ROS for 1 h, before they
were treated with 0, 1, 5, 10, 15 and 20 µM SFN for 3, 6, 12, 24 or
48 h, respectively. HCT-116 cells were incubated for 24 h until
cells had reached 90% confluence before they were treated with 0,
1, 5, 10 or 15 µM SFN, together with 0 or 10 µM CPDD, for 24 h. The
supernatant was replaced with 100 µl (500 µg/ml) MTT solution and
incubated for a further 4 h. The supernatant was removed and 150 µl
dimethyl sulfoxide was added to dissolve the formazan crystals. The
optical density was measured at 570 nm for each sample using a
microtiter plate reader (BioTek Instruments, Inc, Winooski, VT,
USA). The results are expressed as the percentage viability of the
control at each time point (100%).
Determination of cell apoptosis
Cell apoptosis was analyzed using an annexin V-FITC
and PI double-staining method. Briefly, SW480 cells were seeded in
6-well plates at 3×105 cells/well and incubated for 24 h
until they had reached 90% confluence. Cells were then treated with
0, 1, 5, 10, 15 and 20 µM SFN for 24 h, and adherent cells were
washed with PBS, collected and re-suspended in a binding buffer
containing 1% (v:v) annexin V-FITC and 2% (v:v) PI. Cells were then
incubated in the dark at room temperature for 20 min. Apoptotic
cells were quantified using a flow cytometer (BD Biosciences,
Franklin Lakes, NJ, USA). A minimum of 1×105 cells for
each sample were collected for flow cytometry analysis, recorded on
a dot plot, and the percentage of apoptotic cells in the population
was calculated using ModFit software (version 2.0.0; Verity
Software House, Inc, Topsham, ME, USA).
Western blot analysis
The protein expression levels of p73, PUMA, NOXA,
p-p38, p-Erk, p-JNK, Bcl-2 and Bax were measured in SW480 cells,
and the expression levels of γH2AX were measured in HCT-116 cells
by western blot analysis. β-actin was used as a loading control for
these cell lines. Briefly, SW480 or HCT-116 cells were seeded in 25
cm2 cell culture dishes (1×106 cells/dish)
and incubated for 24 h until they reached 90% confluence. SW480 and
HCT-116 cells were treated with 0, 1, 5, 10, 15 and/or 20 µM SFN,
together with 0 or 10 µM CPDD, for 24 h, and the adherent cells
were washed with PBS, collected and lysed using a protein isolation
kit (Bio-Rad Laboratories, Inc.) according to the manufacturer's
instructions. Protein concentrations were quantified using a BCA
protein quantification kit (Bio-Rad Laboratories, Inc.) according
to the manufacturer's instructions. Whole cell lysates were mixed
with a loading buffer (cat. no. P0015; Beyotime Institute of
Biotechnology), and a total of 40 µg cell lysate was boiled at 95°C
for 5 min and then separated by 8–12% SDS-PAGE. The protein bands
were transferred to polyvinylidene fluoride (PVDF) membranes
according to manufacturer's instructions (EMD Millipore, Billerica,
MA, USA). The PVDF membranes were blocked in Tris-buffered saline
with 0.1% Tween 20 (TBST) containing 5% non-fat skim milk at 4°Cfor
1 h, rinsed with TBST and probed with primary antibodies at
4°Covernight (dilution, 1:500). Membranes were then incubated with
the corresponding IRDye-conjugated secondary antibodies (dilution,
1:2,000) at room temperature for 1 h. Membranes were visualized
using an Odyssey Infrared Imaging System (LI-COR Biosciences,
Lincoln, NE, USA). The relative optical densities of the target
proteins were analyzed using Quantity One software (v4.62, Bio-Rad
Laboratories, Inc.). The expression levels of target proteins were
normalized to that of β-actin internal controls.
Caspase activity assay
The activities of caspase-3, −7, −8 and −9 were
determined using caspase activity kits (Beyotime Institute of
Biotechnology) according to the manufacturer's instructions.
Briefly, SW480 cells were seeded in 25 cm2 cell culture
dishes (1×106/dish) and incubated for 24 h until they
reached 90% confluence. Cells were then treated with 0, 1, 5, 10,
15 and 20 µM SFN for 24 h. Adherent cells were washed with PBS,
collected and lysed with Radioimmunoprecipitation (RIPA) Lysis
Buffer (cat. no. P0013C; Beyotime Institute of Biotechnology). The
protein concentration was determined using a BCA protein
quantification kit (Bio-Rad Laboratories, Inc.). Equal quantities
(4 µg/ml) of whole cell lysates were then incubated with the
corresponding caspase substrates to a final concentration of 2 mM
at 37°Cfor 2 h. Samples were measured using a EL800 Microplate
Reader (BioTek Instruments, Inc.) at 488 nm excitation and 525 nm
emission.
Measurement of MMP
The MMP was analyzed by staining SW480 cells using a
JC-1 probe (C2005, Beyotime Institute of Biotechnology), according
to the manufacturer's instructions. Briefly, SW480 cells were
seeded in 96-well plates at 3×103 cells/well and
incubated for 24 h until reaching 90% confluence. The cells were
subsequently treated with 0, 1, 5, 10, 15 and 20 µM SFN for 24 h.
The cells were then incubated with 100 µl JC-1 staining solution (5
µg/ml) at 37°C for 20 min, before cells were rinsed twice with
phosphate-buffered saline. The MMP was measured by determining the
relative quantity of dual emissions from mitochondrial JC-1
monomers or aggregates using a fluorescence EL800 Microplate Reader
(BioTek Instruments, Inc.) at excitation and emission wavelengths
of 488 and 525 nm, respectively. Mitochondrial depolarization is
expressed as the red/green fluorescence intensity ratio, according
to manufacturer's recommendations.
Measurement of intracellular ROS and
MDA generation
The production of ROS was analyzed by staining SW480
cells with a dichlorodihydrofluorescein diacetate
(H2DCF-DA) probe. Briefly, the SW480 cells were seeded
in 96-well plates at 3×103 cells/well and incubated for
24 h until they reached 90% confluence. The cells were then treated
with 0, 1, 5, 10, 15 and 20 µM SFN for 24 h, before the supernatant
was removed. Cells were subsequently incubated with
H2DCF-DA solution (2 µM) for 30 min at room temperature
and rinsed three times using FBS-free medium. The fluorescence
intensity was measured using a fluorescent ELISA reader (BioTek
Instruments, Inc.) at excitation and emission wavelengths of 488
and 525 nm, respectively.
MDA generation was detected using a lipid
peroxidation MDA assay kit based on thiobarbituric acid (TBA)
method. SW480 cells were seeded, cultured and treated with SFN
using the same aforementioned methods, before the supernatant was
subsequently collected and incubated with TBA (at a dilution of
1:200) at 100°C for 15 min. The remaining procedures were performed
according to the manufacturer's instructions (Beyotime Institute of
Biotechnology). The MDA content in the supernatant was measured at
532 nm using an EL800 Microplate Reader (BioTek Instruments,
Inc.).
Antioxidant activity assay
SOD activities in total cell lysates were examined
using a total SOD assay kit based on the WST-8 method (Beyotime
Institute of Biotechnology), according to the manufacturer's
instructions. Briefly, SW480 cells were seeded in 25 cm2
cell culture dishes (1×106/dish) and incubated for 24 h
until they reached 90% confluence. Cells were then treated with 0,
1, 5, 10, 15 and 20 µM SFN for 24 h. Subsequently, the adherent
cells were washed with PBS, collected and lysed with RIPA Lysis
Buffer (Beyotime Institute of Biotechnology) according to the
buffer manufacturer's instructions. The protein concentration was
determined using a BCA protein quantification kit (Bio-Rad
Laboratories, Inc.). Equal quantities of whole cell lysates (4
µg/µl) were incubated with WST-8 solution (at a dilution of 1:200)
at 37°C for 30 min, and the remaining procedures were performed
according to the manufacturer's instructions (Beyotime Institute of
Biotechnology). SOD activity in total cell lysates was measured at
550 nm using the EL800 Microplate Reader (BioTek Instruments,
Inc.).
GSH-Px activities in total cell lysates were
examined using a cellular GSH-Px assay kit. Briefly, SW480 cells
were seeded in 25 cm2 cell culture dishes and incubated
for 24 h until they reached 90% confluence. The cells were then
treated with 0, 1, 5, 10, 15 and 20 µM SFN for 24 h before the
adherent cells were washed with PBS, collected and lysed with the
RIPA Lysis Buffer (Beyotime Institute of Biotechnology), according
to the buffer manufacturer's instructions. Protein concentrations
were quantified using a BCA protein quantification kit. Equal
quantities of whole cell lysates (2 µg/µl) were incubated with the
assay kit buffer (GPx detection working buffer, at a 1:1 dilution)
at 37°C for 30 min, and the remaining procedures were performed
according to the manufacturer's instructions. The GSH-Px activity
in total cell lysates was measured at 412 nm using the EL800
Microplate Reader (BioTek Instruments, Inc.).
Statistical analysis
The results are presented as the mean ± standard
deviation of three independent experiments and the statistical
significance was analyzed by one-way analysis of variance followed
by a post-hoc Dunnett's test. Analysis was performed using SPSS
16.0 (SPSS, Inc, Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
SFN inhibits SW480 cell viability
To investigate the effects of SFN on SW480 cell
growth, the cells were treated with increasing concentrations (0,
1, 5, 10, 15 and 20 µM) of SFN for 3, 6, 12, 24 and 48 h, and an
MTT assay was performed to assess cell viability. As shown in
Fig. 1A, SFN inhibited SW480 cell
proliferation in a dose- and time-dependent manner. When compared
with the untreated control group at each time point, the viability
of cells treated with 5, 10, 15 and 20 µM SFN were significantly
inhibited (P<0.05 and P<0.01). In addition, the viability of
cells treated with 20 µM SFN at 3, 6, 12, 24 and 48 h were
significantly reduced when compared with controls, with 70.2, 66.3,
51.5, 38.6 and 20.1% viabilities, respectively (P<0.01; Fig. 1A). However, compared with the
untreated controls, no significant differences in the viability of
cells treated with 1 µM SFN for 3, 6, 12, 24 and 48 h were
observed. Therefore, for all subsequent assays, cells were exposed
to SFN for 24 h as it exhibited an appropriate inhibiting ratio for
cell viability.
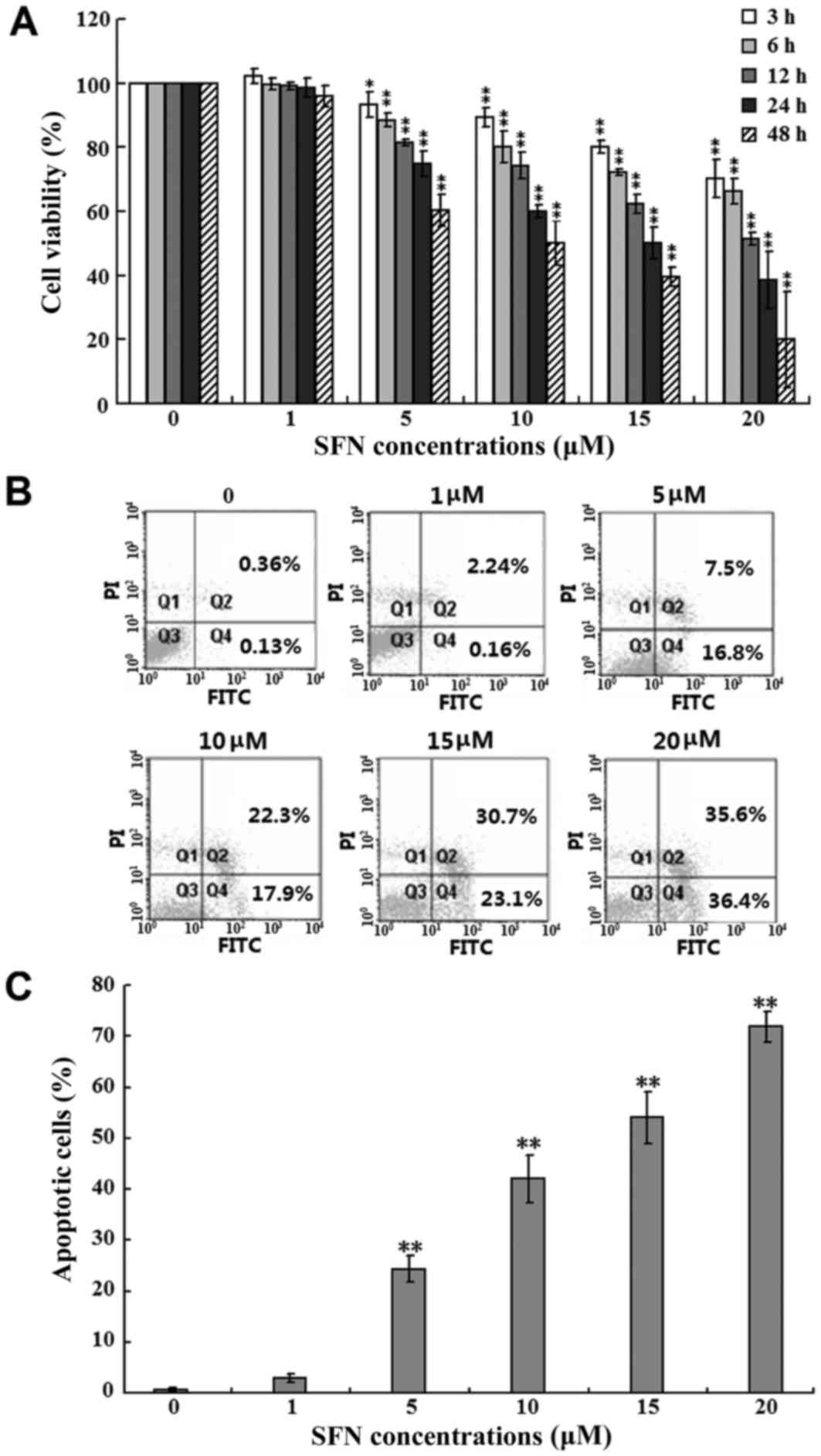 | Figure 1.Effects of SFN on SW480 cell
viability and apoptosis. (A) SW480 cells were treated with 0, 1, 5,
10, 15 and 20 µM SFN for 3, 6, 12, 24 and 48 h, and cell viability
was assessed using an MTT assay. The viability of cells without SFN
treatment was considered as 100%, and the results from three
independent experiments are shown (*P<0.05 and **P<0.01 vs. 0
µM SFN at each time point). (B) SW480 cells were treated with 0, 1,
5, 10, 15 and 20 µM SFN for 24 h. The percentage of apoptotic cells
was then examined by flow cytometry analysis. The percentage of
apoptotic cells was calculated as the sum of Q2 and Q4. (C)
Quantification of the percentage of apoptotic cells from three
independent experiments are shown (**P<0.01 vs. 0 µM SFN). SFN,
sulforaphane; Q, quadrant. |
In order to determine whether the SFN-induced
inhibition of SW480 cell viability was due to the induction of
apoptosis, the proportion of apoptotic cells in the population was
analyzed by flow cytometry using FITC and PI double-staining. As
shown in Fig. 1B and summarized in
Fig. 1C, exposure of SW480 cells
to 5, 10, 15 and 20 µM SFN for 24 h, significantly increased the
percentage of apoptotic cells when compared with the untreated
controls (P<0.01); the percentage of apoptotic cells in the 20
µM SFN-treated group reached 71.9%. However, 1 µM SFN failed to
induce a significant difference in the proportion of apoptotic
cells (Fig. 1B and C). These
results indicated that SW480 cells may undergo apoptosis following
treatment with SFN, and suggested an association between the
induction of apoptosis and the inhibition of cell viability.
SFN-induced apoptosis is
p73-independent and is associated with the intrinsic apoptotic
signaling pathway
To determine whether SFN-induced SW480 cell
apoptosis is p73-dependent, the protein expression levels of p73,
as well as its downstream effectors, PUMA and NOXA, were detected.
As shown in Fig. 2A, p73 protein
was not detected in the untreated control group nor in the
SFN-treated groups. By contrast, PUMA and NOXA were expressed in
the SW480 cells; however, no significant differences were observed
between the untreated and SFN-treated groups (Fig. 2A).
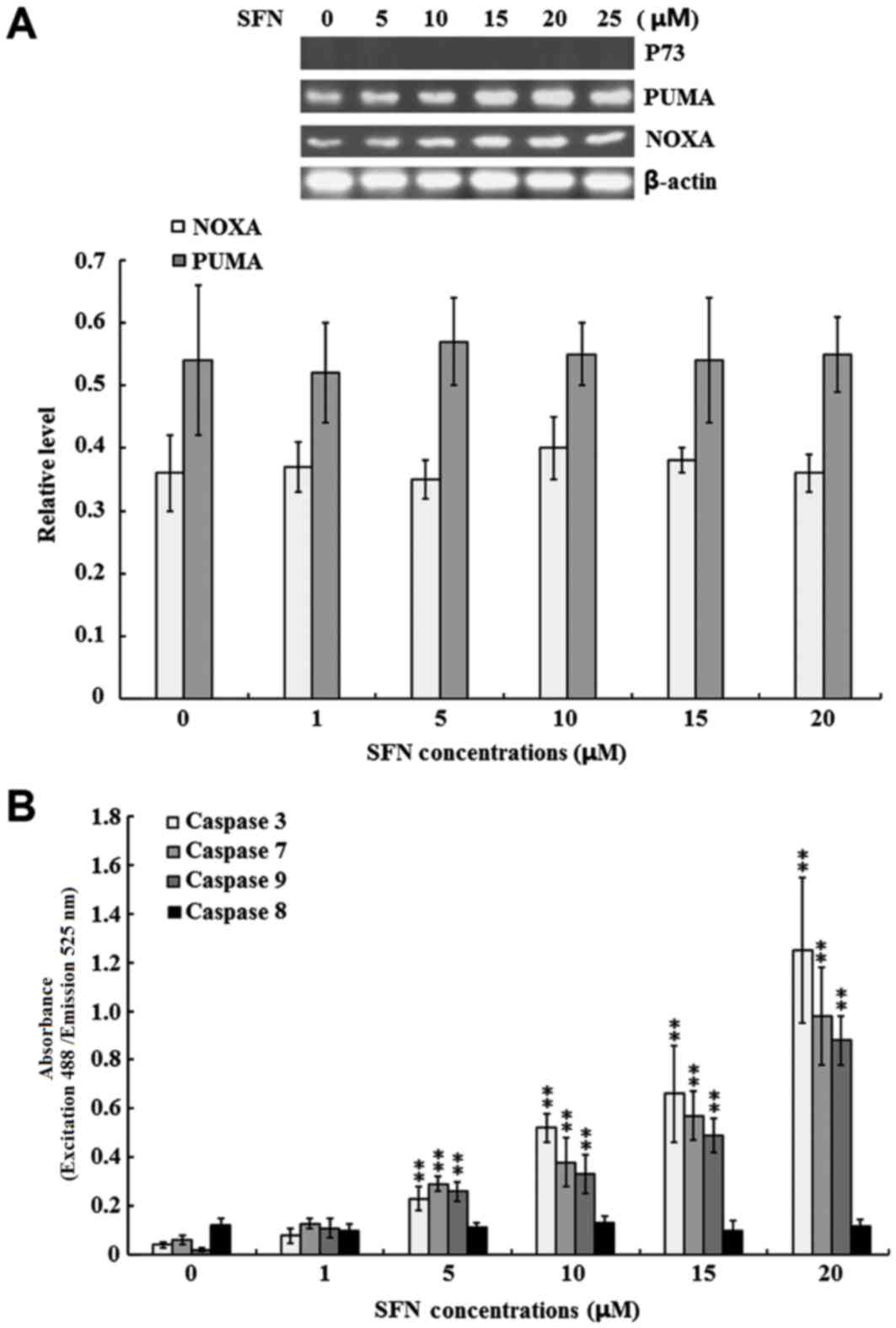 | Figure 2.Effect of SFN on p73 expression and
caspase enzyme activities. SW480 cells were treated with 0, 1, 5,
10, 15 and 20 µM SFN for 24 h. (A) The protein expression levels of
p73, PUMA and NOXA were examined by western blotting. Quantified
band densities are summarized below the images of the bands, and
target protein expression levels were normalized to that of
b-actin. (B) The activity of caspase-3, −7, −8 and −9 was
determined using specific ELISA kits. The results of three
independent experiments are shown. **P<0.01 vs. 0 µM SFN. SFN,
sulforaphane; PUMA, p53 upregulated modulator of apoptosis; NOXA,
phorbol-12-myristate-13-acetate-induced protein 1; ELISA,
enzyme-linked immunosorbent assay. |
The present study further investigated the role of
caspase-3, −7, −8, and −9 in SFN-induced SW480 cell apoptosis. As
shown in Fig. 2B, the activity of
the intrinsic apoptotic executioners, caspase-3, −7 and −9, were
significantly increased in the 5, 10, 15 and 20 µM SFN-treated
groups when compared with the untreated control group (P<0.01).
By contrast, the activity of the extrinsic apoptotic executioner,
caspase 8, was not significantly induced under the same conditions
(Fig. 2B).
SFN-induced apoptosis is associated
with p38 and Erk activation
In addition to p53 and p73, the MAPK signaling
pathway regulates cellular proliferation and apoptosis under cell
stress conditions (30).
Therefore, the present study investigated whether SFN-induced
apoptosis was regulated by the activation of Erk, JNK and p38. As
shown in Fig. 3A, the protein
expression levels of p-Erk and p-p38 were significantly increased
in 5, 10, 15 and 20 µM SFN-treated groups when compared with the
untreated control group (P<0.05 and P<0.01).
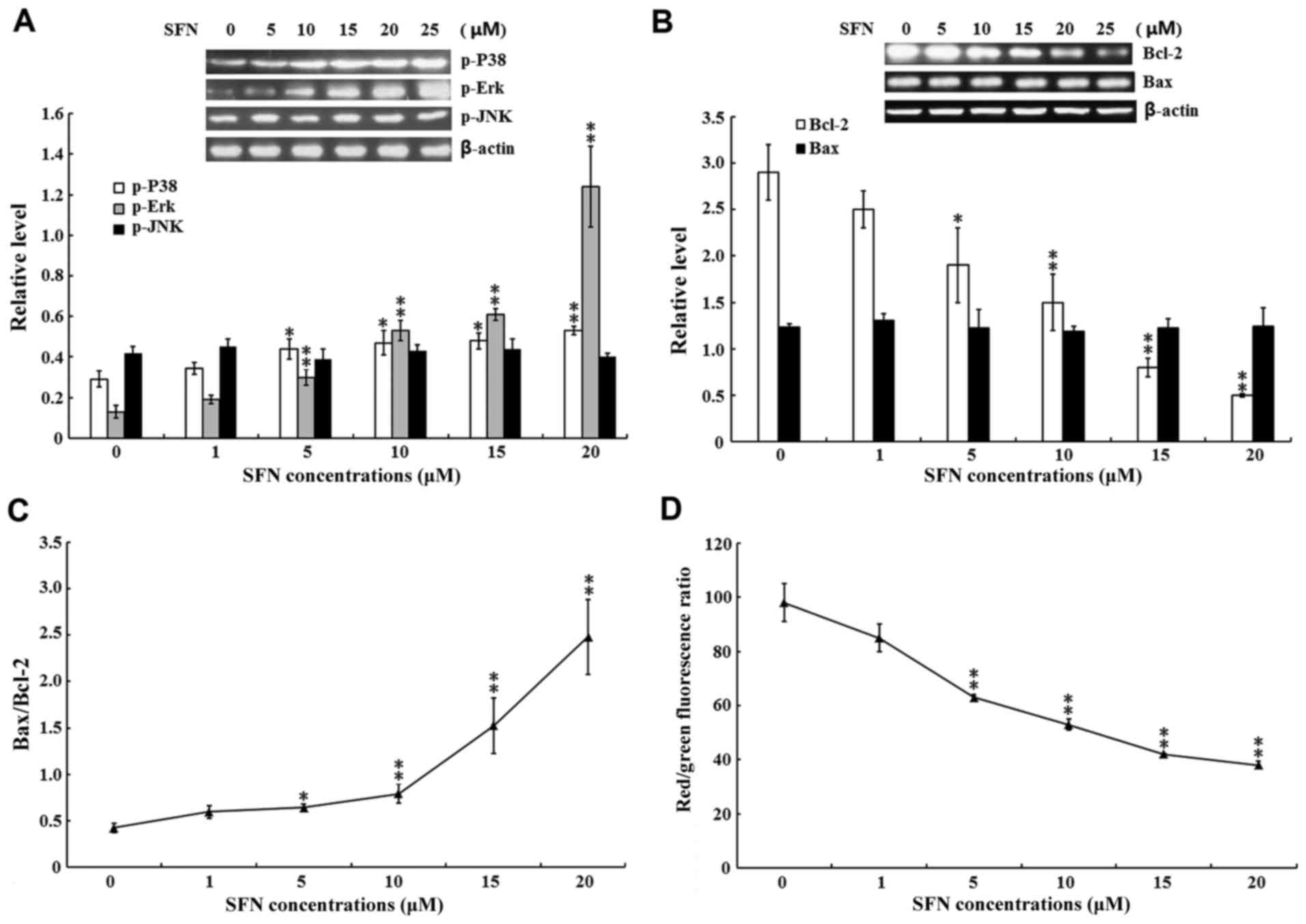 | Figure 3.Effect of SFN on MAPKs and intrinsic
apoptotic signaling pathways. SW480 cells were treated with 0, 1,
5, 10, 15 and 20 µM SFN for 24 h. (A) The protein expression levels
of (A) p-p38, p-Erk and p-JNK and (B) Bcl-2 and Bax were examined
by western blotting, and the quantified results are summarized
below the images of the protein bands. Target protein expression
levels were normalized to that of b-actin. (C) The Bax/Bcl-2 ratio.
(D) The MMP was examined using a MMP assay kit with a JC-1 probe.
The results of three independent experiments are shown. *P<0.05
and **P<0.01 vs. 0 µM SFN. SFN, sulforaphane; MAPK,
mitogen-activated protein kinases; p-, phosphorylated; Erk,
extracellular signal-regulated kinases; JNK, c-Jun N-terminal
kinases; Bcl-2, B-cell lymphoma-2; Bax, Bcl-2-associated protein X;
MMP, mitochondrial membrane potential. |
A previous study indicated that the anti-apoptotic
functions of Bcl-2 and the pro-apoptotic functions of Bax are
regulated by MAPKs (37).
Therefore, the present study examined the protein expression levels
of Bax and Bcl-2 in SFN-treated SW480 cells. As shown in Fig. 3B, the protein expression levels of
Bcl-2 were significantly decreased in the 5, 10, 15 and 20 µM
SFN-treated groups when compared with the untreated control group
(P<0.05 and P<0.01); however, no significant differences in
the level of Bax protein were detected in the SFN-treated groups
compared with the controls. Accordingly, a significant increase in
the Bax/Bcl-2 ratio was observed when compared with the untreated
controls (P<0.05 and P<0.01; Fig. 3C). Previous studies have
demonstrated that an increased Bax/Bcl-2 ratio induces alterations
in the mitochondrial permeability transition; an event which
subsequently leads to disruption of the MMP (38,39).
Therefore, the present study investigated whether SFN exposure
altered the MMAs shown in Fig. 3D,
exposure to 5, 10, 15 and 20 µM SFN induced significant
mitochondrial depolarization when compared with the untreated
group, as indicated by the decreased red/green fluorescence ratio
(P<0.01).
SFN induces oxidative stress in SW480
cells
ROS are known to function as messengers that
activate MAPKs (26). Therefore,
the present study investigated whether SFN induced SW480 cell
apoptosis via the generation of ROS. As shown in Fig. 4A, exposure to 5, 10, 15 and 20 µM
SFN significantly increased ROS production when compared with the
untreated group (P<0.01). This result was further confirmed by
the significant increase in MDA production following exposure of
cells to 1, 5, 10, 15 and 20 µM SFN when compared with untreated
controls (Fig. 4B). MDA production
is the direct consequence of ROS production (40). The activity of the anti-oxidative
enzymes, SOD and GSH-Px, were not significantly altered following
exposure to increasing concentrations of SFN (Fig. 4C and D).
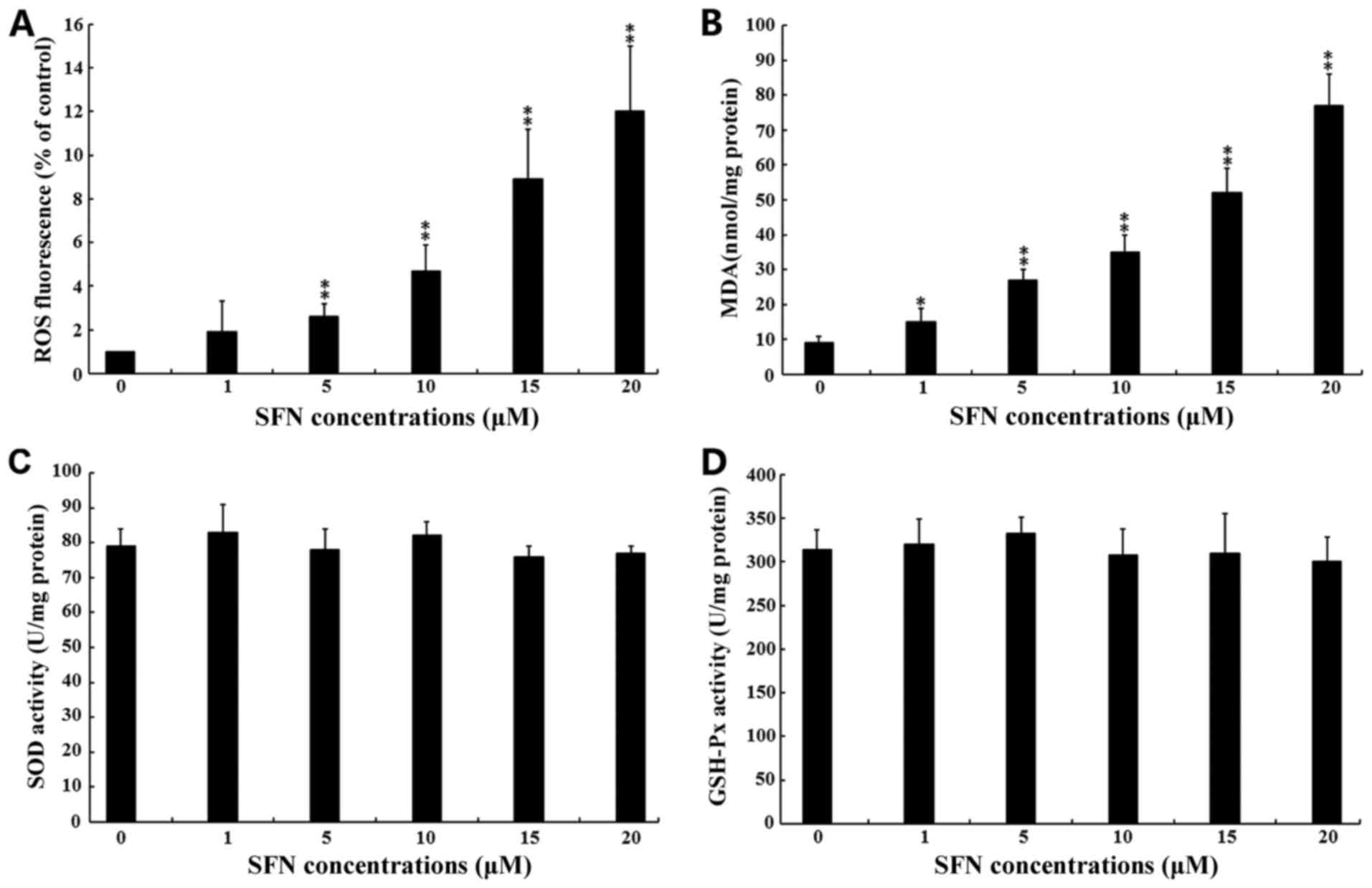 | Figure 4.Effect of SFN on oxidative stress
levels. SW480 cells were treated with 0, 1, 5, 10, 15 and 20 µM SFN
for 24. (A) Intracellular ROS production was examined using a ROS
assay kit. (B) Intracellular MDA levels were examined using a lipid
peroxidation MDA assay kit. (C) SOD activities in total cell
lysates were examined using a total SOD assay kit using
water-soluble tetrazolium salt-8. (D) GSH-Px activities in total
cell lysates were examined using a cellular GSH-Px assay kit. For
all experiments, the results of three independent experiments are
shown. *P<0.05 and **P<0.01 vs. 0 µM SFN. SFN, sulforaphane;
ROS, reactive oxygen species; MDA, malondialdehyde; SOD, superoxide
dismutase; GSH-Px, glutathione peroxidase. |
SFN-induced apoptosis is ROS dependent
and involves p38 and Erk cell signaling pathways
To discriminate between the different roles of ROS,
p38 and Erk in SFN-induced apoptosis, SFN-treated SW480 cells were
treated with specific inhibitors against p38, Erk and ROS. First,
the effects of the inhibitors on ROS generation were investigated.
As shown in Fig. 5A, the ROS
inhibitor, APDC, completely attenuated the SFN-induced production
of ROS. By contrast, treatment with the p38 and Erk inhibitors
(SB203580 and SCH772984, respectively), demonstrated no marked
effects on the SFN-induced production of ROS. The effects of the
inhibitors on cell viability were then evaluated. As presented in
Fig. 5B, treatment of cells with
the ROS inhibitor, APDC, completely attenuated the SFN-mediated
inhibition of SW480 cell viability. By contrast, treatment with the
p38 inhibitor (SB203580), and the Erk inhibitor (SCH772984)
partially attenuated the SFN-mediated inhibition of SW480 cell
proliferation. These results indicated that SFN increased ROS
generation and induced cell apoptosis via the p38 and Erk cell
signaling pathways.
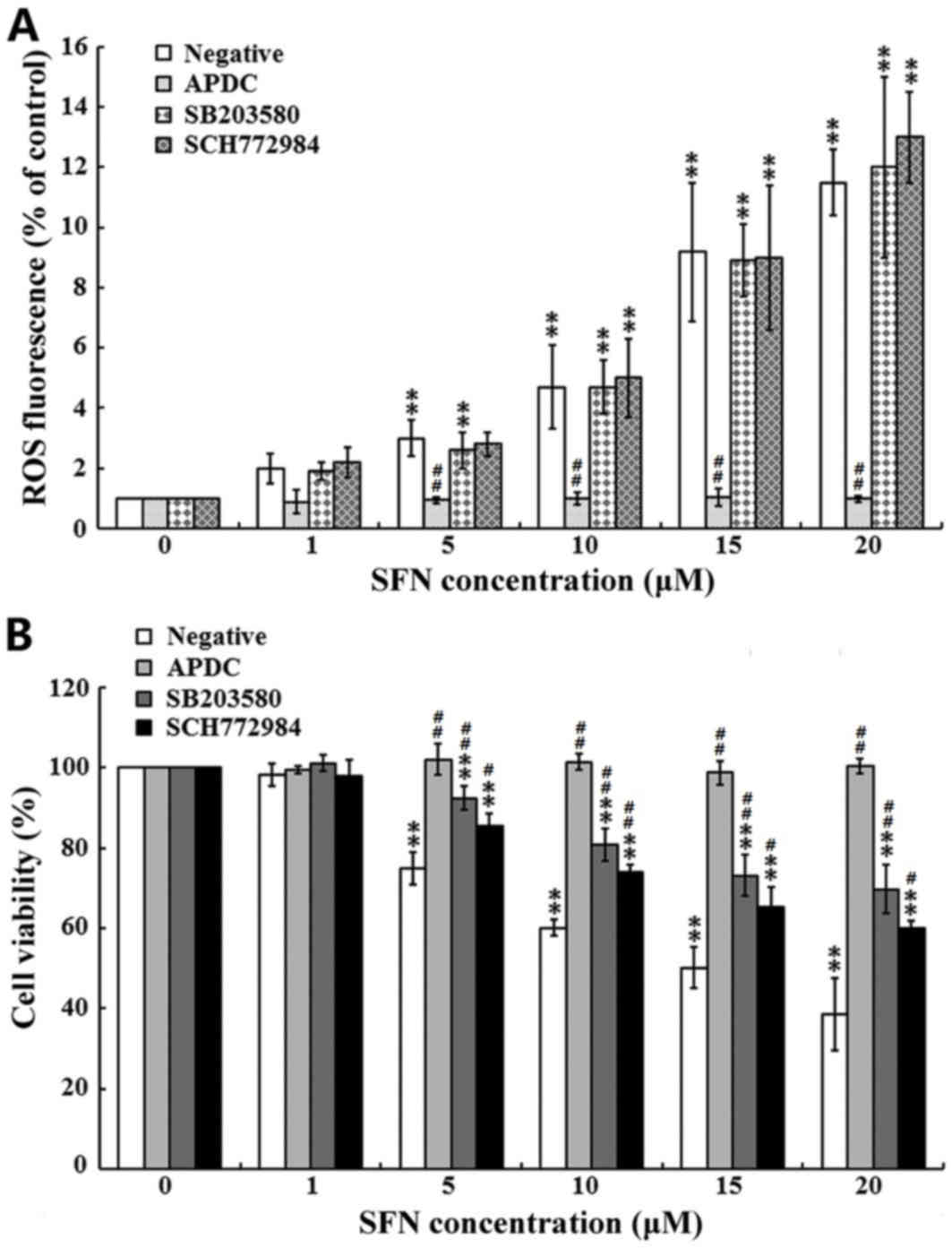 | Figure 5.Role of MAPKs and ROS in SFN-induced
apoptosis. SW480 cells were pre-treated with or without specific
inhibitors against p38, Erk and ROS for 1 h, before cells were
treated with 0, 1, 5, 10, 15 and 20 µM SFN for 24 h. (A)
Intracellular ROS production was examined using an ROS assay kit.
(B) Cell viability was assessed using an MTT assay, and the
viability of SFN-untreated cells was considered as 100%. For all
experiments, the results of three independent experiments are
shown. **P<0.01 vs. 0 µM SFN; #P<0.05 and
##P<0.01 vs. the negative control. MAPKs,
mitogen-activated protein kinases; ROS, reactive oxygen species;
SFN, sulforaphane; Erk, extracellular signal-regulated kinases;
APDC, ammonium pyrrolidinedithiocarbamate; SB203580, inhibitor of
p38 MAPK; SCH772984, inhibitor of Erk. |
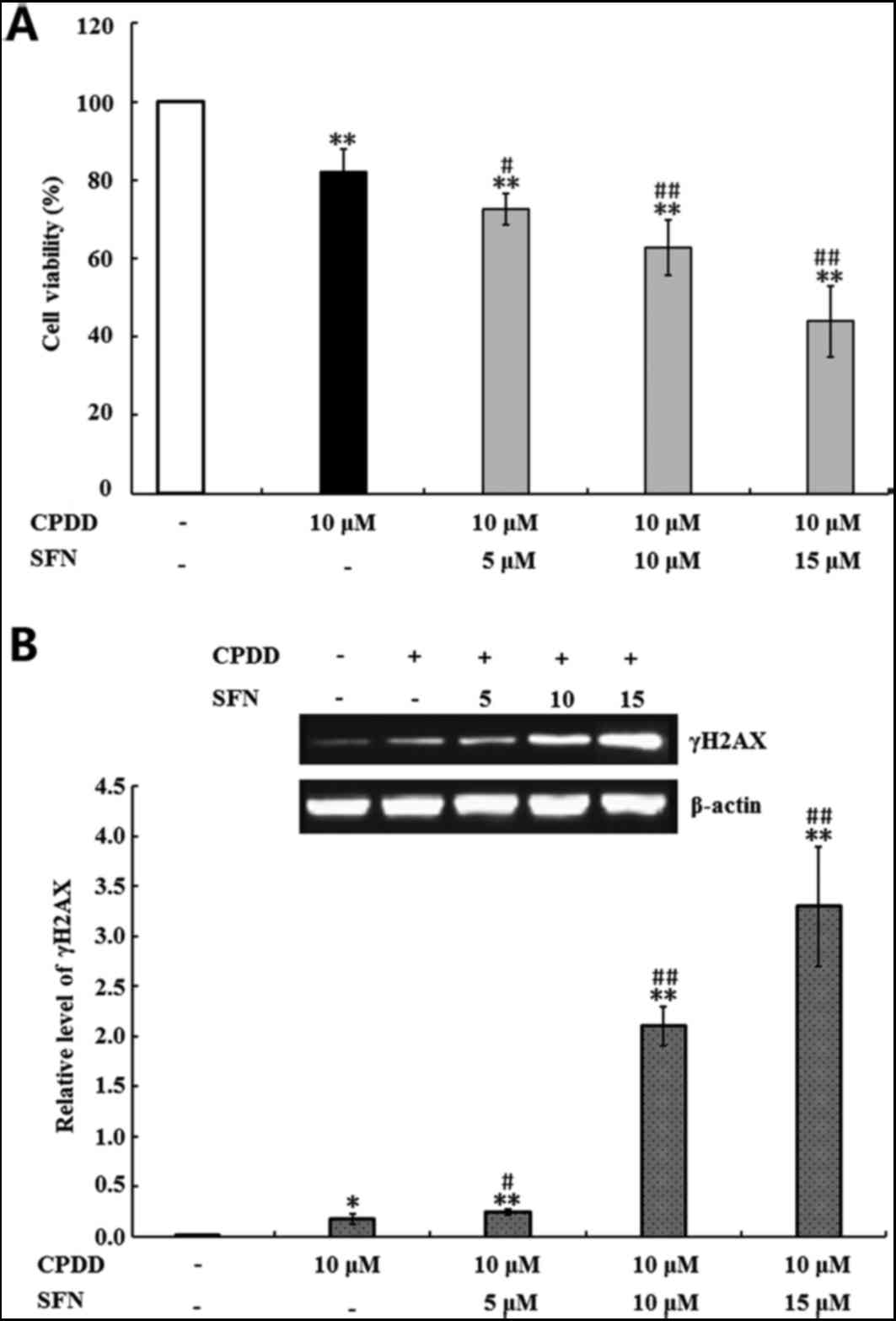
SFN sensitizes p53 proficient HCT-116
cells to CPDD-induced apoptosis by increasing DNA damage
Finally, the present study evaluated the ability of
SFN to potentiate the inhibitory action of CPDD against the
proliferation of p53-proficient HCT-116 colon cancer cells. The
effects of SFN on CPDD-mediated inhibition of HCT-116 cell
viability were first determined. As shown in Fig. 6A, treatment with 10 µM CPDD alone,
and 10 µM CPDD combined with 5, 10 and 15 µM SFN, significantly
inhibited HCT-116 cell viability when compared with the untreated
control group (P<0.01). In addition, when compared with 10 µM
CPDD treatment alone, combined SFN and CDDP treatment significantly
reduced the viability of HCT-116 cells (P<0.05 and P<0.01;
Fig. 6A). The anti-cancer activity
of CPDD is based on its interactions with DNA and the subsequent
inhibition of DNA replication, which leads to DNA damage (41). Therefore, the present study
investigated the effect of SFN on CPDD-induced DNA damage by
detecting the levels of γH2AX, a biomarker of DNA damage (42). As shown in Fig. 6B, treatment with 10 µM CPDD alone,
or 10 µM CPDD combined with 5, 10 and 15 µM SFN, significantly
increased the level of γH2AX when compared with the untreated group
(P<0.05 and P<0.01). In addition, combined treatment with SFN
and CDDP induced a significant increase in γH2AX expression when
compared with CPDD treatment alone (P<0.05 and P<0.01;
Fig. 6B), which suggested that SFN
may increase the cytotoxic effects of CDDP by increasing the
production of γH2AX.
Discussion
SFN is one of the most commonly studied ITCs, and
has been reported to inhibit the proliferation of different types
of cancer cells by inducing apoptosis (43,44).
In agreement with previous studies, the results of the present
study revealed that SFN at concentrations between 5 and 20 µM
significantly inhibited the viability of SW480 cells following
exposure for 3, 6, 12, 24 and 48 h. These results were confirmed by
flow cytometry analysis, which demonstrated that SFN at
concentrations between 5 and 20 µM increased apoptosis in SW480
cells following treatment for 24 h. In addition, SFN was observed
to induce apoptosis in SW480 cells by increasing ROS generation and
the activation of Erk and p38 MAPKs.
ROS are byproducts of aerobic metabolic events that
include superoxide O2−, peroxide
H2O2 and hydroxyl OH− radicals
(43). In the present study, SFN
disturbed the homeostatic balance between the generation and
elimination of ROS, leading to the accumulation of ROS. MDA, SOD
and GSH-Px are important biomarkers of oxidative damage (45). Exposure to SFN increased MDA
production in a dose-dependent manner; however, it did not alter
the activities of the anti-oxidative enzymes, SOD and GSH-Px. The
observed alterations in the levels of these biological parameters
indicated that severe oxidative damage was induced by SFN
exposure.
In mammalian cells, ROS serve as second messengers
to mediate diverse redox-sensitive signaling pathways, such as the
MAPK and p53 pathways (46). MAPKs
are a family of serine/threonine kinases that produce intracellular
signals in response to various stimuli (47). The oxidative stress-dependent
activation of MAPKs has been demonstrated to be involved in the
induction of cell apoptosis (35).
Similar to the results of previous studies, the present study
demonstrated that SFN exposure significantly increased the
expression levels of p-p38 and p-ERK; however, exposure did not
alter the expression levels of the p53 family member, p73. These
results suggested that MAPKs were activated by SFN treatment. The
roles of ROS, p-Erk and p-p38 in SFN-mediated inhibition of SW480
cell viability were then determined. SW480 cells were pretreated
with APDC, SB203580 or SCH772984, which are selective inhibitors of
ROS, p38 and Erk, respectively, prior to treatment with SFN. It was
revealed that SB203580 and SCH772984 partially alleviated the
inhibitory effects of SFN on cell viability. Notably, the ROS
inhibitor, APDC, completely attenuated the SFN-mediated reduction
in SW480 cell viability. These results indicated that a positive
interaction between ROS generation and cell apoptosis may exist,
and that this ROS-mediated cell apoptosis may be involved in the
activation of p-Erk and p-p38. However, the detailed interactions
between ROS production and p-Erk/p-P38 activation requires further
investigation in future studies.
Bcl-2 family members are known to be important
gatekeepers of apoptotic responses (48–50).
Bcl-2 family members are classified into the following three
functional groups: Anti-apoptotic factors, such as Bcl-2;
pro-apoptotic factors, such as Bax; and pro-apoptotic activators,
such as NOXA and PUMA (50,51).
Bcl-2 binds and interacts with Bax to prevent mitochondrial pore
formation, which subsequently inhibits the execution of cell
apoptosis. Therefore, the Bax/Bcl-2 ratio may be used to determine
response to therapy and apoptosis (52). The pro-apoptotic activators, which
contain a single BH3 domain, are downstream mediators of p53- and
p73-dependent apoptosis pathways (53). In the present study, exposure of
SW480 cells to SFN did not increase NOXA and PUMA expression, which
may be attributed to the deficiencies in p53 and p73 function in
SW480 cells. However, SFN treatment significantly inhibited Bcl-2
expression, which led to an increase in the Bax/Bcl-2 ratio and
cell apoptosis. These results were further confirmed by the
observed decrease in MM However, the detailed mechanisms by which
these cell death signals produced during SFN-induced apoptosis
increase the expression of Bcl-2 remains to be elucidated. Previous
studies have suggested that the anti-apoptotic functions of Bcl-2
are inhibited by p38 MAPK via phosphorylation (54,55).
Therefore, it is possible that Bcl-2 is a mediator that connects
the activation of MAPKs to SFN-induced apoptosis.
CPDD is a typical alkylating agent that demonstrates
cytotoxic activities against human cancers in vitro.
CPDD-based chemotherapy is one of the most important
chemotherapeutic treatments available for patients with colon
cancer. However, CPDD often produces severe side effects, which
limit its efficacy (56).
Therefore, there is an urgent requirement to identify more
effective agents as cancer therapies. Combination therapy with
multiple drugs or modalities is a common treatment strategy for
patients with cancer, as it may achieve greater therapeutic
benefits when compared with a single drug or modality. In the
present study, the effects of SFN exposure on p53-proficient
HCT-166 cells in response to CPDD-induced apoptosis were further
examined. It was demonstrated that exposure to SFN, even at a low
concentration (5 µM), increased the sensitivity of HCT-116 cells to
the 10 µM CPDD-induced reduction in cell viability, potentially via
an increase in γH2AX expression. These results suggest that SFN may
be effective as an adjuvant therapy for patients with colon cancer
undergoing treatment with CPDD.
In conclusion, the present study successfully
established an in vitro colon cancer model of
p53-independent, SFN-induced apoptosis. The preliminary results
demonstrated that SFN-induced apoptosis in this model may be
associated with activation of an intrinsic apoptotic signaling
pathway, and that accumulation of ROS is a key event. These
observations provide a mechanistic insight into the therapeutic
potential of SFN as a clinical treatment for patients with
p53-deficient colon cancers. However, further studies are required
to confirm the effects of SFN-induced apoptosis in vivo.
Glossary
Abbreviations
Abbreviations:
|
SFN
|
sulforaphane
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
Erk
|
extracellular signal-regulated
kinases
|
|
FBS
|
fetal bovine serum
|
|
ITCs
|
isothiocyanates
|
|
FITC
|
fluorescein isothiocyanate
|
|
PI
|
propidium iodide
|
|
MMP
|
mitochondrial membrane potential
|
|
ROS
|
reactive oxygen species
|
|
H2DCF-DA
|
dichlorodihydrofluorescein
diacetate
|
|
MDR
|
multi-drug resistance
|
|
JNK
|
c-Jun N-terminal kinase
|
|
SAPK
|
stress-activated protein kinase
|
|
CPDD
|
cisplatin
|
References
|
1
|
Bauman JE, Zang Y, Sen M, Li C, Wang L,
Egner PA, Fahey JW, Normolle DP, Grandis JR, Kensler TW and Johnson
DE: Prevention of carcinogen-induced oral cancer by sulforaphane.
Cancer Prev Res (Phila). 9:547–557. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Amjad AI, Parikh RA, Appleman LJ, Hahm ER,
Singh K and Singh SV: Broccoli-derived sulforaphane and
chemoprevention of prostate cancer: From bench to bedside. Curr
Pharmacol Rep. 1:382–390. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Traka MH, Melchini A and Mithen RF:
Sulforaphane and prostate cancer interception. Drug Discov Today.
19:1488–1492. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Folkard DL, Marlow G, Mithen RF and
Ferguson LR: Effect of Sulforaphane on NOD2 via NF-κB: Implications
for Crohn's disease. J Inflamm (Lond). 12:62015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun CC, Li SJ, Yang CL, Xue RL, Xi YY,
Wang L, Zhao QL and Li DJ: Sulforaphane attenuates muscle
inflammation in dystrophin-deficient mdx mice via NF-E2-related
factor 2 (Nrf2)-mediated Inhibition of NF-κB signaling pathway. J
Biol Chem. 290:17784–17795. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jang M and Cho IH: Sulforaphane
ameliorates 3-nitropropionic acid-induced striatal toxicity by
activating the keap1-Nrf2-ARE pathway and Inhibiting the MAPKs and
NF-κB pathways. Mol Neurobiol. 53:2619–2635. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tammali R, Reddy AB, Saxena A, Rychahou
PG, Evers BM, Qiu S, Awasthi S, Ramana KV and Srivastava SK:
Inhibition of aldose reductase prevents colon cancer metastasis.
Carcinogenesis. 32:1259–1267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Coriat R, Marut W, Leconte M, Ba LB,
Vienne A, Chéreau C, Alexandre J, Weill B, Doering M, Jacob C, et
al: The organotelluride catalyst LAB027 prevents colon cancer
growth in the mice. Cell Death Dis. 2:e1912011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu T, Wang L, Zhang L, Lu L, Shen J, Chan
RL, Li M, Wu WK, To KK and Cho CH: Sensitivity of
apoptosis-resistant colon cancer cells to tanshinones is mediated
by autophagic cell death and p53-independent cytotoxicity.
Phytomedicine. 22:536–544. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Acedo P and Zawacka-Pankau J: p53 family
members-important messengers in cell death signaling in
photodynamic therapy of cancer? Photochem Photobiol Sci.
14:1390–1396. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Basu S and Murphy ME: p53 family members
regulate cancer stem cells. Cell Cycle. 15:1403–1404. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pflaum J, Schlosser S and Müller M: p53
family and cellular stress responses in cancer. Front Oncol.
4:2852014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tu HC, Ren D, Wang GX, Chen DY, Westergard
TD, Kim H, Sasagawa S, Hsieh JJ and Cheng EH: The p53-cathepsin
axis cooperates with ROS to activate programmed necrotic death upon
DNA damage. Proc Natl Acad Sci USA. 106:1093–1098. 2009; View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wiseman A: p53 protein or BID protein
select the route to either apoptosis (programmed cell death) or to
cell cycle arrest opposing carcinogenesis after DNA damage by ROS.
Med Hypotheses. 67:296–299. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Y, Jiang L, She Y, Chen M, Li Q, Yang
G, Geng C, Tang L, Zhong L, Jiang L and Liu X: Olaquindox induces
DNA damage via the lysosomal and mitochondrial pathway involving
ROS production and p53 activation in HEK293 cells. Environ Toxicol
Pharmacol. 40:792–799. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dueñas M, Santos M, Aranda JF, Bielza C,
Martínez-Cruz AB, Lorz C, Taron M, Ciruelos EM, Rodríguez-Peralto
JL, Martín M, et al: Mouse p53-deficient cancer models as platforms
for obtaining genomic predictors of human cancer clinical outcomes.
PLoS One. 7:e424942012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou S, Kachhap S and Singh KK:
Mitochondrial impairment in p53-deficient human cancer cells.
Mutagenesis. 18:287–292. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Robles AI and Harris CC: Clinical outcomes
and correlates of TP53 mutations and cancer. Cold Spring Harb
Perspect Biol. 2:a0010162010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oren M, Tal P and Rotter V: Targeting
mutant p53 for cancer therapy. Aging (Albany NY). 8:1159–1160.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Q, Zeng SX and Lu H: Targeting
p53-MDM2-MDMX loop for cancer therapy. Subcell Biochem. 85:281–319.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xiang JF, Wang WQ, Liu L, Xu HX, Wu CT,
Yang JX, Qi ZH, Wang YQ, Xu J, Liu C, et al: Mutant p53 determines
pancreatic cancer poor prognosis to pancreatectomy through
upregulation of cavin-1 in patients with preoperative serum CA19-9
≥1,000 U/ml. Sci Rep. 6:192222016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi Y, Nikulenkov F, Zawacka-Pankau J, Li
H, Gabdoulline R, Xu J, Eriksson S, Hedström E, Issaeva N, Kel A,
et al: ROS-dependent activation of JNK converts p53 into an
efficient inhibitor of oncogenes leading to robust apoptosis. Cell
Death Differ. 21:612–623. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu B, Yuan B, Zhang L, Mu W and Wang C:
ROS/p38/p53/Puma signaling pathway is involved in emodin-induced
apoptosis of human colorectal cancer cells. Int J Clin Exp Med.
8:15413–15422. 2015.PubMed/NCBI
|
|
24
|
Panieri E and Santoro MM: ROS homeostasis
and metabolism: A dangerous liason in cancer cells. Cell Death Dis.
7:e22532016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Prasad S, Gupta SC and Tyagi AK: Reactive
oxygen species (ROS) and cancer: Role of antioxidative
nutraceuticals. Cancer Lett. 387:95–105. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Walton EL: The dual role of ROS,
antioxidants and autophagy in cancer. Biomed J. 39:89–92. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nogueira V and Hay N: Molecular pathways:
Reactive oxygen species homeostasis in cancer cells and
implications for cancer therapy. Clin Cancer Res. 19:4309–4314.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Santabárbara-Ruiz P, López-Santillán M,
Martínez-Rodríguez I, Binagui-Casas A, Pérez L, Milán M, Corominas
M and Serras F: ROS-Induced JNK and p38 signaling is required for
unpaired cytokine activation during Drosophila regeneration. PLoS
Genet. 11:e10055952015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Amin PJ and Shankar BS: Sulforaphane
induces ROS mediated induction of NKG2D ligands in human cancer
cell lines and enhances susceptibility to NK cell mediated lysis.
Life Sci. 126:19–27. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhai JW, Gao C, Ma WD, Wang W, Yao LP, Xia
XX, Luo M, Zu YG and Fu YJ: Geraniin induces apoptosis of human
breast cancer cells MCF-7 via ROS-mediated stimulation of p38 MAPK.
Toxicol Mech Methods. 26:311–318. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Youn GS, Lee KW, Choi SY and Park J:
Overexpression of HDAC6 induces pro-inflammatory responses by
regulating ROS-MAPK-NF-κB/AP-1 signaling pathways in macrophages.
Free Radic Biol Med. 97:14–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang H, Li D, Hu Z, Zhao S, Zheng Z and Li
W: Protective effects of green tea polyphenol against renal injury
through ROS-Mediated JNK-MAPK pathway in lead exposed rats. Mol
Cells. 39:508–513. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang S, Xu R, Luo X, Jiang Z and Shu H:
Genome-wide identification and expression analysis of MAPK and
MAPKK gene family in Malus domestica. Gene. 531:377–387. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rhim JH, Luo X, Gao D, Xu X, Zhou T, Li F,
Wang P, Wong ST and Xia X: Cell type-dependent Erk-Akt pathway
crosstalk regulates the proliferation of fetal neural progenitor
cells. Sci Rep. 6:265472016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu E, Li J, Shi S, Wang X, Liang T, Wu B
and Li Q: Sustained ERK activation-mediated proliferation
inhibition of farrerol on human gastric carcinoma cell line by
G0/G1-phase cell-cycle arrest. Eur J Cancer Prev. 25:490–499. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hsu YC, Chang SJ, Wang MY, Chen YL and
Huang TY: Growth inhibition and apoptosis of neuroblastoma cells
through ROS-independent MEK/ERK activation by sulforaphane. Cell
Biochem Biophys. 66:765–774. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen YJ, Liu WH, Kao PH, Wang JJ and Chang
LS: Involvement of p38 MAPK- and JNK-modulated expression of Bcl-2
and Bax in Naja nigricollis CMS-9-induced apoptosis of human
leukemia K562 cells. Toxicon. 55:1306–1316. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Smaili SS, Hsu YT, Sanders KM, Russell JT
and Youle RJ: Bax translocation to mitochondria subsequent to a
rapid loss of mitochondrial membrane potential. Cell Death Differ.
8:909–920. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Benadiba M, Dos Santos RR, Dde O Silva and
Colquhoun A: Inhibition of C6 rat glioma proliferation by
[Ru2Cl(Ibp)4] depends on changes in p21, p27, Bax/Bcl2 ratio and
mitochondrial membrane potential. J Inorg Biochem. 104:928–935.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ray G, Batra S, Shukla NK, Deo S, Raina V,
Ashok S and Husain SA: Lipid peroxidation, free radical production
and antioxidant status in breast cancer. Breast Cancer Res Treat.
59:163–170. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rebillard A, Lagadic-Gossmann D and
Dimanche-Boitrel MT: Cisplatin cytotoxicity: DNA and plasma
membrane targets. Curr Med Chem. 5:2656–2663. 2008. View Article : Google Scholar
|
|
42
|
Redon CE, Dickey JS, Bonner WM and
Sedelnikova OA: γ-H2AX as a biomarker of DNA damage induced by
ionizing radiation in human peripheral blood lymphocytes and
artificial skin. Adv Space Res. 43:1171–1178. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Buenz EJ: Aloin induces apoptosis in
Jurkat cells. Toxicol In Vitro. 22:422–429. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tabolacci C, Rossi S, Lentini A,
Provenzano B, Turcano L, Facchiano F and Beninati S: Aloin enhances
cisplatin antineoplastic activity in B16-F10 melanoma cells by
transglutaminase-induced differentiation. Amino Acids. 44:293–300.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jing S, Jiang WH and Sun W: Effects of
smoking on serum SOD and GSH-PX activities and MDA contents in rats
with gastric ulcer. Appl Mech Mater 675–677. 1–129. 2014.
|
|
46
|
Bragado P, Armesilla A, Silva A and Porras
A: Apoptosis by cisplatin requires p53 mediated p38alpha MAPK
activation through ROS generation. Apoptosis. 12:1733–1742. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hao W, Wang S and Zhou Z: Tubeimoside-1
(TBMS1) inhibits lung cancer cell growth and induces cells
apoptosis through activation of MAPK-JNK pathway. Int J Clin Exp
Pathol. 8:12075–12083. 2015.PubMed/NCBI
|
|
48
|
Bao FK: The bcl-2 gene family, an
important regulator of apoptosis. Sheng Li Ke Xue Jin Zhan.
27:67–69. 1996.(In Chinese). PubMed/NCBI
|
|
49
|
Wu Y and Tang L: Bcl-2 family proteins
regulate apoptosis and epithelial to mesenchymal transition by
calcium signals. Curr Pharm Des. 22:4700–4704. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Carpio MA, Michaud M, Zhou W, Fisher JK,
Walensky LD and Katz SG: BCL-2 family member BOK promotes apoptosis
in response to endoplasmic reticulum stress. Proc Natl Acad Sci
USA. 112:7201–7206. 2015; View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gallenne T, Gautier F, Oliver L, Hervouet
E, Noël B, Hickman JA, Geneste O, Cartron PF, Vallette FM, Manon S
and Juin P: Bax activation by the BH3-only protein Puma promotes
cell dependence on antiapoptotic Bcl-2 family members. J Cell Biol.
185:279–290. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cory S, Huang DC and Adams JM: The Bcl-2
family: Roles in cell survival and oncogenesis. Oncogene.
22:8590–8607. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Melino G, Bernassola F, Ranalli M, Yee K,
Zong WX, Corazzari M, Knight RA, Green DR, Thompson C and Vousden
KH: p73 Induces apoptosis via PUMA transactivation and Bax
mitochondrial translocation. J Biol Chem. 279:8076–8083. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Haddad JJ: The role of Bax/Bcl-2 and
pro-caspase peptides in hypoxia/reperfusion-dependent regulation of
MAPK(ERK): Discordant proteomic effect of MAPK(p38). Protein Pept
Lett. 14:361–371. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pan MH, Chiou YS, Cheng AC, Bai N, Lo CY,
Tan D and Ho CT: Involvement of MAPK, Bcl-2 family, cytochrome c,
and caspases in induction of apoptosis by
1,6-O,O-diacetylbritannilactone in human leukemia cells. Mol Nutr
Food Res. 51:229–238. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
He G, He G, Zhou R, Pi Z, Zhu T, Jiang L
and Xie Y: Enhancement of cisplatin-induced colon cancer cells
apoptosis by shikonin, a natural inducer of ROS in vitro and in
vivo. Biochem Biophys Res Commun. 469:1075–1082. 2016. View Article : Google Scholar : PubMed/NCBI
|