Introduction
Monosomy 8p is a rare chromosomal disorder
characterized by deletion of a part of the eighth chromosome. The
incidence of the 8p23.1 deletion was estimated at 1:18,542 in
amniotic fluid samples and 1:5,072 in postnatal samples (1). Since the first report of an 8p23.1
deletion by Fagan and Morris (2),
>50 cases have been reported (3). The majority of the cases are not
studied with high resolution molecular techniques or characterized
at the molecular level (4).
Interstitial deletions of the sub-band 8p23.1 have primarily been
associated with facial and other phenotypic abnormalities, whereas
terminal deletions are associated with heart defects (3,5).
Notably, distal deletion of 8p23.2-pter has additionally been
observed in apparently healthy individuals (1).
In the majority of cases, monosomy 8p appears to
result from de novo errors in early embryonic development
that occur for unknown reasons. Associated symptoms and findings
differ between cases (6). However,
in most cases clinical manifestations including growth deficiency,
mental retardation, post-natal growth retardation, developmental
delay and speech problems are observed. Furthermore, patients
present with common signs of body and craniofacial dysmophisms, in
addition to behavioral difficulties (1,3,5,6).
Facial dysmorphisms, which are more remarkable in early years,
include microcephaly, malformed or low set ears, arched eyebrows,
depressed nasal bridge, epicanthus, strabismus, hypermetropia
and/or myopia, serrated teeth, short neck and retrognathia. In
addition, vertebral abnormalities are frequently observed (7–11).
It has additionally been reported that children with
this chromosomal disorder present with behavioral difficulties,
including aggressiveness and attention deficit disorder, and
problems associated with cardiovascular and central nervous system
(5,9,12).
Furthermore, genito-urinary anomalies, in particular cryptorcidism
and hypospadias, are observed in boys (6).
In contrast to 8p deletion syndrome, partial
trisomies of the terminal 16qter are rare (1). A total of nine cases of partial
distal chromosome 16 trisomy have been reported but only a few were
studied with high resolution molecular techniques. Only one patient
presented a pure partial trisomy 16q24.1q24.3, whereas all the
others corresponded to unbalanced translocations where 16q24 was
rearranged with other chromosome regions (7,8,10,11,13–15).
For two of the patients, there was no detailed phenotypic
information (10,15). A number of clinical characteristic
features were common in all patients (low birth weight, growth
retardation, intellectual disability, muscular hypotonia, small
palpebral fissures, long philtrum, low set/dysplastic ears and
osteochondroma), so it is difficult to characterize with precision
the 16q24 trisomy phenotype or to establish a genotype-phenotype
correlation (11) (Table I).
 | Table I.Clinical characteristics associated
with 8p23 and 16q24 regions in the literature. |
Table I.
Clinical characteristics associated
with 8p23 and 16q24 regions in the literature.
| Clinical
characteristics | 8p23.1 →pter | 8p23.2 →pter | 16q24.1 →qter | 16q24.1 →qter | Patient 1 | Patient 2 | Other studies | Total |
|---|
| Prematurity |
|
|
|
| − | + |
|
|
| Post-natal growth
retardation | + | − | − | + | − | − | 7–12 | 7/15 |
| Low birth
weight | − | − | − | + | − | − | 7,8,11 | 3/3 |
| Developmental
delay | + | − | − | − | + | + | 5,9,12 | 15/20 |
| Mental
retardation | + | + | − | + | − | − | 5,6,9,10 | 14/20 |
|
Behavioral/neurodevelopmental
(hyperactivity, aggressiveness, no self- confidence, attention
deficit disorder, anxious) | + | − | − | − | + | + | 5,9,12 | 25/58 |
| Dysmorphic
craniofacial features |
|
|
|
|
|
|
|
|
| Microcephaly | + | + | − | − | + | − | 5,6,9,12 | 13/21 |
| Hypertelorism |
|
|
|
| + | + |
|
|
| Epicanthus | − | − | − | + | + | + | 7,12,14 | 4/5 |
| Broad forehead |
|
|
|
| − | + |
|
|
| Arched
eyebrows | + | + | − | − | − | + | 6 | 1/1 |
| Diffuse
depigmentation of retina |
|
|
|
| − | + |
|
|
| Alternating
esotropia |
|
|
|
| − | + |
|
|
| Long philtrum | − | − | − | + | − | + | 10,11,14 | 3/5 |
| Thin face |
|
|
|
| − | + |
|
|
| Thin lips | + | − | − | + | − | + | 9,14 | 5/9 |
| Small mouth |
|
|
|
| − | + |
|
|
| Retrognathia | + | − | − | − | − | + | 9 | 8/8 |
| Depressed nasal
bridge | + | + | − | − | − | + | 6,9 | 2/9 |
| Dysplastics/low set
ears | + | − | − | + | − | + | 5,7,8,9,11,12 | 12/23 |
| Major
malformations |
|
|
|
|
|
|
|
|
| Clinodactyly |
|
|
|
| + | − |
|
|
| Laryngeal
stridor/laryngomalacia | + | + | − | − | − | + | 6 | 1/1 |
| Cardiovascular
system problems | + | + | − | − | + | − | 6;16 | 3/3 |
| Abdominal
distension |
|
|
|
| − | + |
|
|
| Necrotizing
enterocolitis |
|
|
|
| − | + |
|
|
| Genito-urinary
anomalies | + | + | − | − | + | − | 5,6,9 | 9/28 |
| Central nervous
system |
|
|
|
|
|
|
|
|
| Speech
problems | + | − | − | − | + | − | 5,9,12 | 5/20 |
| Dystonic
posturing |
|
|
|
| − | + |
|
|
| Myelination
delay |
|
|
|
| − | + |
|
|
In the present study, two cases of liveborn
unrelated children with an unbalanced 8;16 translocation resulting
in partial monosomy of chromosome 8 and partial trisomy of
chromosome 16 were reported. The effect on the phenotype of
monosomy 8 seems to be more prominent than that of trisomy 16.
However, this phenotype may result from the rearranged architecture
of the region, the structure and function of the genes and regions
involved, and their interactions.
Patients and methods
Ethical approval
The present study was approved by the Ethics
Committee of the P. & A. Kyriakou Children's Hospital (Athens,
Greece) and was performed with respect to the ethical standards of
the Declaration of Helsinki, as revised in 2008. Written, informed
consent was obtained from the patient's families.
Patient 1
Patient 1 was a 10- and a half-year-old boy, and the
second child of healthy, unrelated parents. The first child of the
family is a 16-year-old healthy boy. Patient 1 was referred for
developmental assessment for speech and language delay. The patient
was born following an uncomplicated full term pregnancy with birth
weight 3.350 kg, height 51 cm and head circumference (HC) 35 cm.
The perinatal history was non-significant. At the age of 8 months
the patient was diagnosed with a urinary tract infection and an
X-ray investigation revealed urinary reflux (V degree), and a
kidney dimercaptosuccinic acid scan revealed 20% decreased left
kidney function. The developmental milestones of the patient were
slightly delayed as he sat independently at the age of 9 months and
walked unaided at the age of 18 months.
On developmental examination at 3 years old the
patient was a sociable child, with mild dysmorphic facial and body
features including microcephaly, hypertelorism, epicanthus, and
clinodactyly. The patient demonstrated good ability for symbolic
play and his comprehension ability was limited to one concept per
sentence. His speech was limited to 3–4 simple words. His overall
developmental level was equivalent to 18 months. According to the
Bailey's Scales of Infant Development 2nd edition (16), his mental score was 51 and motor
score was 95. Heart auscultation revealed a mild systolic murmur.
On neurological examination, the patient was revealed to be
slightly hypertonic with borderline microcephaly (HC=48 cm;
3%).
Echocardiography revealed a small ventricular septal
defect without hemodynamic alterations. Metabolic screening
revealed a mild elevation of glutamate in blood amino acids and
small proteinuria involving lysine, arginin and cystin. His bone
age was increased (equivalent to 6-year-old boy). Thyroid function,
brain magnetic resonance imaging scans, visual and audiological
examinations, urine amino acids and blood lactic acid levels were
healthy.
The patient attended mainstream kindergarten and
received early intervention services twice a week based on a
Portage Scheme. His development was followed up at regular
intervals in the Developmental Unit and was monitored according to
his needs.
At the age of 3 years and 9 months, his cognitive
and language skills were equivalent to the level of a 20-month-old,
with severe behavioral difficulties characterized by frequent
temper tantrums. At the age of 4 years, 3 months, the cognitive
abilities of the patient increased to the level of a 30-month-old
while his language skills remained at a 26-month level. His
behavior had improved but he remained a difficult child, presenting
with hyperactivity, aggressiveness and impulsiveness.
He was additionally observed at the age of 5 years
and 2 months. He had made significant developmental progress and
his cognitive skills were equivalent to a 4 year and 6 month level,
with language skills equivalent to a 2 year and 9 month level.
According to Griffiths Scales (17), his performance developmental
subquotient (DQ) was 87 and his language DQ was 51. His weight was
19 kg (50th centile), his height was 107 cm (20th centile) and his
HC was 49.5 cm (3rd centile). The dysmorphia of his facial features
remained mild and passed unnoticed. He was well integrated in
mainstream kindergarten and his parents were planning to place him
in a mainstream school with extra educational help.
The patient was re-evaluated at the age of 7 years.
He was well integrated into the 1st grade of mainstream primary
school with special educational provision. His behavior had
significantly improved and he was sociable and co-operative. His
cognitive abilities were increased, with a developmental level of 5
years and 8 months with a general DQ of 86. His weight was 24 kg
(25th centile), his height was 119 cm (25th centile) and his HC was
49.5 cm (below 3rd centile). Dysmorphia of his body and facial
features remained mild. On neurological examination, he was
revealed to be slightly hypertonic. His thyroid functions and
detailed endocrinological examination (GH, IGF1, prolactin, LH,
FSH, 17-OH prog., cortisol, insulin) proved normal. Previous
echo-triplex results additionally proved normal.
The patient was last observed at the age of 10 years
and 6 months. He was attending the 3rd grade of the same mainstream
primary school with special educational support. He remained
sociable with severe attention deficit disorder, impulsivity and
lack of self-confidence. His cognitive deficits were more evident
in reading and mathematics. His developmental level was equivalent
to that of a healthy 6-year-old, with mild phonological and
morphological language problems. His general DQ was 78. The
dysmorphia of his body and facial features (microcephaly,
hypertelorism, epicanthus and clinodactyly) was more evident. On
neurological examination, he remained slightly hypertonic with
brisk reflexes but without focal neurological signs. His weight was
34 kg (25th centile), his height was 140 cm (25th centile) and his
HC was 49.5 cm (<3rd centile).
Patient 2
Patient 2 was a girl was born to non-consanguineous
healthy parents at 36 weeks of gestational age, following a normal
pregnancy and an uncomplicated delivery. Prenatal karyotype was
performed due to advanced maternal age, and it was normal. The
family history was unremarkable and there was no previous history
of infertility or spontaneous abortion prior to this pregnancy. The
birth weight was 2,400 g (25th centile), height 48 cm (75th-90th
centile), and HC 30.5 cm (2nd-10th centile). Apgar scores were 9
and 10 at 1 and 5 min, respectively.
Two days following birth, the patient presented with
abdominal distension and bloody stools. An X-ray revealed the
presence of air outside the intestines in the abdominal cavity.
Necrotizing enterocolitis with perforation was diagnosed and
surgical removal of the caecum was performed, and the ileocecal
valve was perforated. However, three months following surgery, she
presented with intestinal obstruction caused by narrowing of the
previously diseased bowel, requiring further surgical intervention.
In addition, the neonatal period was complicated by laryngeal
stridor due to laryngomalacia. Some dysmorphic features and
dystonic posturing were noticed in early infancy. The patient
acquired head control at the age of 6 months, trunk control at the
age of 9 months, and autonomous deambulation at the age of 12
months. The patient started to speak at two years of age, but then
stopped any further development of verbal language and developed a
preference for gestural communication. Verbal comprehension was
good.
Extensive studies for metabolic diseases (including
blood and urine amino acids, urine organic acids, blood lactate,
pyruvate and ammonia) gave normal results. Electroencephalogram,
audiometric examination, cardiological evaluation including
echocardiogram, X-rays of the thorax and renal ultrasound returned
normal results. Ophthalmologic assessment (at 3 months of age)
revealed diffuse depigmentation of the retina. Brain magnetic
resonance (at 6 months of age) revealed myelination delay. At 7
months of age, the patient's height was 62 cm (10th centile),
weight was 5.035 g (<3rd centile) and HC was 39.5 cm (<2nd
centile). Morphological evaluation evidenced a thin face, broad
forehead, low-set and posteriorly rotated ears, bilateral pits
above the tragus, arched eyebrows, hypertelorism, epicanthus
inversus, depressed nasal bridge, long philtrum, thin lips, small
mouth with down-turned corners, and retrognathia. Neurological
examination revealed developmental delay, with gross motor
milestones limited to uncompleted head control. Dystonic axial
posturing and fluctuating muscular tone of the four limbs was
present. Alternating esotropia was additionally observed.
Cytogenetic and fluorescence in situ
hybridization (FISH) analyses
Chromosome analysis was performed from 2–2.5 ml
cultured blood lymphocytes using Giemsa banding and high resolution
banding techniques obtained following cell culture synchronization
and thymidine incorporation. FISH studies were performed using a
set of probes specific for 8p (TelVysion 8p SpectrumGreen D8S504)
and 16q (TelVysion 16q SpectrumOrange 16qTEL013) subtelomeres
according to the manufacturer's protocol (Vysis; Abbott Molecular,
Des Plaines, Illinois, USA) (18).
The slides were washed and counterstained with
4′,6-diamidino-2-phenylindole, and cells were examined under a
Zeiss Axioplan II, Imager.M1/Imager.Z1 fluorescence microscope
equipped with a triple-bandpass filter (Zeiss GmbH, Jena, Germany).
Digital images were captured and stored with Isis software version
3.4.0 (MetaSystems, Altlussheim, Germany).
Array comparative genomic
hybridization (aCGH), polymerase chain reaction (PCR) and
microsatellite analysis
High molecular weight genomic DNA was extracted from
the patient's blood lymphocytes using aQiamp DNA Blood Midi kit
(Qiagen, Inc., Valencia, CA, USA). aCGH analysis was performed with
DNA from cultured amniocytes in order to characterize the extent of
the deletion in Patient 1 and to justify the clinical findings in
Patient 2. Molecular karyotyping was performed via oligonucleotide
aCGH platforms using an 100 kb resolution array kit 44K (Agilent
Technologies, Inc., Santa Clara, CA, USA). Gene dosage for 9
sequence tagged sites (STSs) from chromosome 8 was performed by PCR
using the LightCycler FastStart DNA Master SYBR Green 1 Kit (Roche
Diagnostics, Monza Italy), according to the manufacturer's
instructions, on a Roche LightCycler 1.5 instrument (Roche
Diagnostics). Primer sequences for the telomeric STSs amplified,
including the genesceroid-lipofuscinosis, neuronal 8 (CLN8),
CUB and Sushi multiple domains 1 (CSMD1), microcephalin 1
primary autosomal recessive 1 (MCPH1) and GATA binding
protein 4 (GATA4), are listed in Table II. Altogether, the analyzed region
covered ~11 Mb of DNA of the telomeric 8p region. PCR was performed
using the following program: 95°C for 10 min, followed by 40 cycles
of 95°C for 10 sec, 55°C for 10 sec, and 72°C for 25 sec.
Copy-number/genome of each STS was evaluated by a relative
quantification method using the software RelQuant (Roche
Diagnostics). A 156 bp fragment of the human beta-globin gene (HBB)
was used as reference DNA for normalization and amplified in
separate capillaries simultaneously to the STS targets. Primer
sequences for HBB were as follows: forward
5′-CAGCTCACTCAGTGTGGCAAAG-3′ and reverse
5′-AGGTTCTTTGAGTCCTTTGGGG-3. Relative standard curves were produced
using 5 control DNA samples to correct for differences in
efficiency of amplification between STS target and reference DNA.
For each locus the test was replicated three times.
| Table II.Genotypic information of Patient 1 at
the chromosome 8 STS markers obtained by quantitative polymerase
chain reaction and gene dosage assay. |
Table II.
Genotypic information of Patient 1 at
the chromosome 8 STS markers obtained by quantitative polymerase
chain reaction and gene dosage assay.
| STS name | Gene | Position (bp) | Deletion | Primer | Sequence
(‘5-3’) | size |
|---|
| STS-N21307 | LOC286161 | 427685–427914 | Yes | F |
CAGGTTGGCAAGTGAAATAC | 230 |
|
|
|
|
| R |
GCAGTAGTGGCATGAAGC |
|
| SHGC-149177 | DLGAP2 | 952948–953243 | Yes | F |
GCCTCCTGGGATAAAAATCCTTT | 296 |
|
|
|
|
| R |
GGTTTGCTCTCCTGATTTAGGGT |
|
| SHGC-149177 | CLN8 |
1728163–1728478 | Yes | F |
AAGAGCAAGAGGAGCAGGAAAAC | 316 |
|
|
|
|
| R |
GTGAAACATGTGAATCATCAGCC |
|
| SHGC-105022 | CSMD1 |
4126904–4127196 | Yesa | F |
TTTTATTTTGGATCAGGCAACCT | 293 |
|
|
|
|
| R |
TGTGCTTTGAACCACACTCCTAA |
|
| RH119760 |
CSMD1 |
4950952–4951296 | Yes | F |
TATCCAGTCTCTGCATTTGATGG | 345 |
|
|
|
|
| R |
AGAATCCCAAAGGAGTTACCGAA |
|
| A004X20 | MCPH1 |
6302850–6303049 | Yes | F |
TAAGTTTTCCTTCTCTTCTGTAG | 216 |
|
|
|
|
| R |
AAGGACATGATGATGATT |
|
| SHGC-77726 | MCPH1 |
6478893–6479173 | Yesa | F |
GAAGTAAACTGCAACAGTTCGCC | 281 |
|
|
|
|
| R |
TCTTCTTTCCGCTGTAGGGC |
|
| RH120376 | TDH |
11224233–11224519 | No | F |
AAAATCCACGCTTTGACCTAACA | 287 |
|
|
|
|
| R |
TGGTAAGGGAATGAGTGTGTTCA |
|
| RH11694 | GATA4 |
11617203–11617417 | No | F |
TGCACATTGCTGTTTCTGCC | 234 |
|
|
|
|
| R |
GTTTGTGGGTTAGGGAGGGT |
|
Bioinformatic analyses
Sequence features of 8p and 16q regions were
analysed in the University of California Santa Cruz (UCSC) Genome
Browser (19) using data from the
International Standards for Cytogenomic Arrays Consortium (ISCA;
www.iscaconsortium.org/) database
(20,21) and the corresponding data track for
UCSC genes. The Basic Local Alignment Search Tool (BLAST) algorithm
(http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used for
analysis (22). The results were
used to study the nucleotide sequence similarity between the
breakpoint regions.
Results
Patient 1
The conventional karyotype of Patient 1 revealed
‘additional’ material in the short arm of chromosome 8 (46, XY,
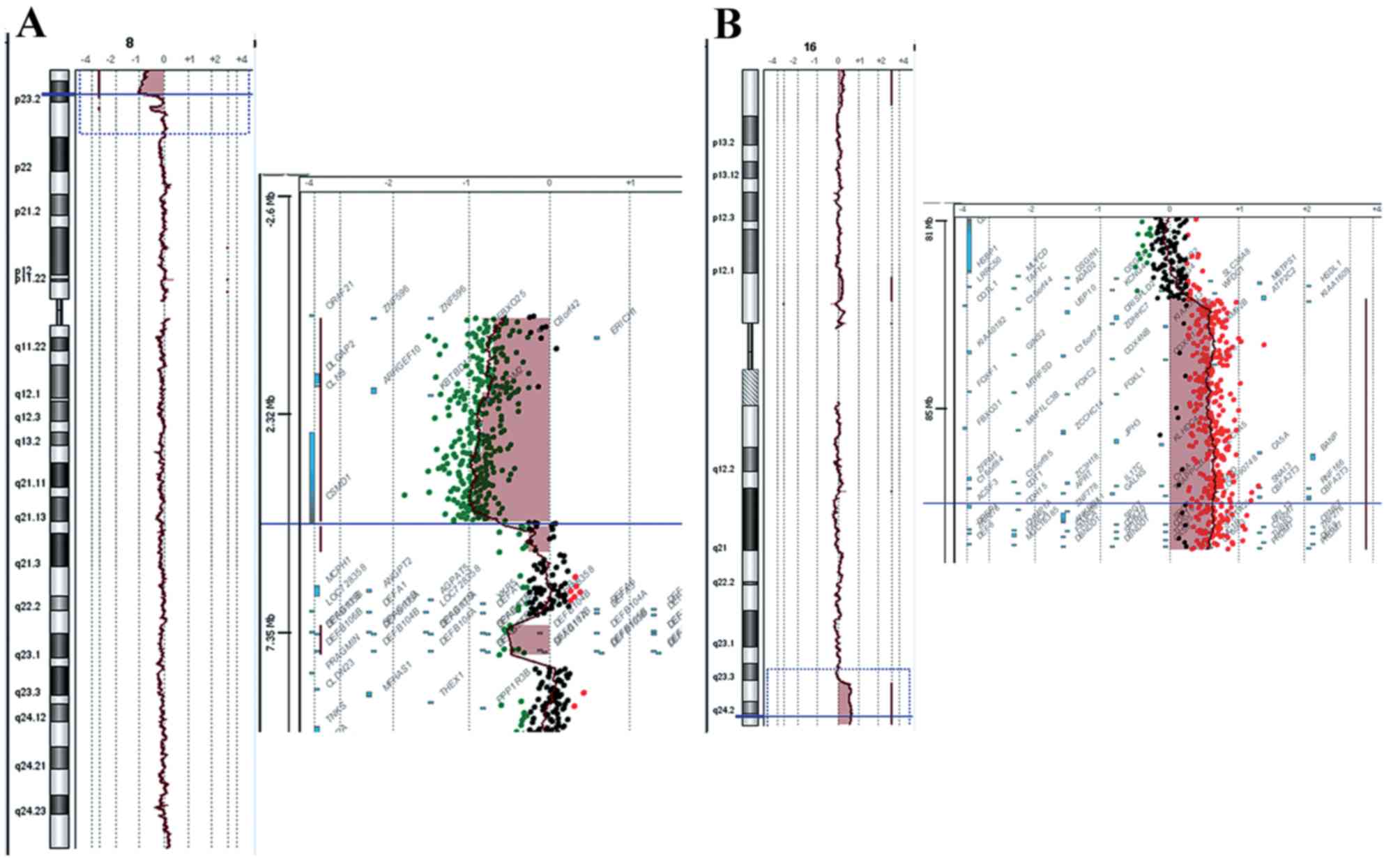
8p+). aCGH analysis revealed the chromosomal origin of the
additional material and the exact position of the breakpoints,
namely a deletion of 8p and a duplication of 16q. The 8p deletion
was a 4,8 Mb deletion of the distal short arm of chromosome 8 with
the proximal breakpoints between 4,814,649 bp (last deleted oligo)
and 4,833,351 bp (first normal oligo), with the last
oligonucleotide present in the array at 8p position 161,472 kb
being deleted (Fig. 1A). The
deleted region of the CSMD1 gene began at the first intron.
The 16q duplication was a 5.6 Mb duplication of the long arm of
chromosome 16 with the proximal breakpoint between 84,468,454 bp
(normal) and 84,511,640 bp (duplicated), and the last
oligonucleotide present in the array at 16q position 88,690,571 bp
being duplicated (Fig. 1B). The
size of the breakpoint intervals were 18,702 bp for 8p and 43,186
bp for 16q. The analysis revealed an unbalanced translocation, and
the aCGH karyotype was
46,XY,der(8)t(8;16)(p23.2;q23.3)dn.arr[hg18]8p23.3p23.2(151,472–4814649)x1,16q23.3q24.3
(84,511,640–88,690,571)x3.
A list of Online Mendelian Inheritance in Man (OMIM;
https://www.omim.org/) genes deleted and
duplicated is presented in Table
III. FISH analysis was performed to confirm the aCGH data.
Three signals were detected; one on chromosome 8 and two on
chromosome 16 of the 16q subtelomeric probe. FISH analysis
performed in the parents revealed a normal result, indicating a
de novo rearrangement.
| Table III.List of OMIM genes deleted and
duplicated in both patients. |
Table III.
List of OMIM genes deleted and
duplicated in both patients.
| Duplication |
|---|
|
|---|
| Patient 1 | Patient 2 |
|---|
|
|
|---|
| Gene | OMIM | Gene | OMIM |
|---|
| FBX025 | 609098 | FBX025 | 609098 |
| DLGAP2 | 605438 | DLGAP2 | 605438 |
| CLN8 | 607837 | CLN8 | 607837 |
|
ARHGEF10 | 608136 |
ARHGEF10 | 608136 |
| MYOM2 | 603509 | MYOM2 | 603509 |
| CSMD1 | 608397 | CSMD1 | 608397 |
|
|
| MCPH1 | 607117 |
|
|
| ANGPT2 | 601922 |
|
|
| AGPAT5 | 614796 |
|
|
| DEFB1 | 602056 |
|
|
| DEFA6 | 600471 |
|
|
| DEFA4 | 601157 |
|
|
| DEFA1 | 125220 |
|
|
| DEFA3 | 604522 |
|
|
| DEFA5 | 600472 |
|
|
|
DEFB103B | 606611 |
|
|
| SPAG11B | 606560 |
|
|
|
FAM90A7P | 613044 |
|
|
|
FAM90A10P | 613047 |
|
|
| DEFB4A | 602215 |
|
|
| CLDN23 | 609203 |
|
|
| MFHAS1 | 605352 |
|
|
| ERI1 | 608739 |
|
|
| PPP1R3B | 610541 |
|
|
| TNKS | 603303 |
|
| Duplication |
|
|
| Patient 1 | Patient 2 |
|
|
|
|
| Gene | OMIM | Gene | OMIM |
|
| ATP2C2 | 613082 | WWOX | 605131 |
| COTL1 | 606748 | MAF | 177075 |
| USP10 | 609818 | MAFTRR | 616264 |
|
CRISPLD2 | 612434 | DYNLRB2 | 607168 |
| ZDHHC7 | 614604 | CENPN | 611509 |
|
KIAA0513 | 611675 | ATMIN | 614693 |
| FAM92B | 617274 | GCSH | 238330 |
| GSE1 | 616886 | PKD1L2 | 607894 |
| GINS2 | 610609 | BCO1 | 605748 |
| EMC8 | 604886 | GAN | 605379 |
| COX4I1 | 123864 | CMIP | 610112 |
| IRF8 | 601565 | PLCG2 | 600220 |
|
LINC01082 | 614978 | SDR42E1 | 616164 |
|
LINC01081 | 614977 | HSD17B2 | 109685 |
| FENDRR | 614975 |
MPHOSPH6 | 605500 |
| FOXF1 | 601089 | CDH13 | 601364 |
| MTHFSD | 616820 | HSBP1 | 604553 |
| FOXC2 | 602402 | MLYCD | 606761 |
| FOXL1 | 603252 | OSGIN1 | 607975 |
| FBXO31 | 609102 | SLC38A8 | 615585 |
|
MAP1LC3B | 609604 | MBTPS1 | 603355 |
| JPH3 | 605268 | DNAAF1 | 613190 |
| SLC7A5 | 600182 | TAF1C | 604905 |
| CA5A | 114761 | KCNG4 | 607603 |
| BANP | 611564 | WFDC1 | 605322 |
| ZNF469 | 612078 | ATP2C2 | 613082 |
| ZFPM1 | 601950 | COTL1 | 606748 |
| IL17C | 604628 | USP10 | 609818 |
| CYBA | 608508 |
CRISPLD2 | 612434 |
| MVD | 603236 | ZDHHC7 | 614604 |
| SNAI3 | 612741 |
KIAA0513 | 611675 |
| RNF166 | 617178 | FAM92B | 617274 |
| CTU2 | 617057 | GSE1 | 616886 |
| PIEZO1 | 611184 | GINS2 | 610609 |
| CDT1 | 605525 | EMC8 | 604886 |
| APRT | 102600 | COX4I1 | 123864 |
| GALNS | 612222 | IRF8 | 601565 |
|
TRAPPC2L | 610970 |
LINC01082 | 614978 |
| CBFA2T3 | 603870 |
LINC01081 | 614977 |
| ACSF3 | 614245 | FENDRR | 614975 |
| CDH15 | 114019 | FOXF1 | 601089 |
| ANKRD11 | 611192 | MTHFSD | 616820 |
| SPG7 | 602783 | FOXC2 | 602402 |
| RPL13 | 113703 | FOXL1 | 603252 |
| CPNE7 | 605689 | FBXO31 | 609102 |
| DPEP1 | 179780 |
MAP1LC3B | 609604 |
| CHMP1A | 164010 | JPH3 | 605268 |
| SPATA33 | 615409 | SLC7A5 | 600182 |
| CDK10 | 603464 | CA5A | 114761 |
| ZNF276 | 608460 | BANP | 611564 |
| FANCA | 607139 | ZNF469 | 612078 |
| SPIRE2 | 609217 | ZFPM1 | 601950 |
| TCF25 | 612326 | IL17C | 604628 |
| MC1R | 155555 | CYBA | 608508 |
| TUBB3 | 602661 | MVD | 603236 |
| AFG3L1P | 603020 | SNAI3 | 612741 |
| GAS8 | 605178 | RNF166 | 617178 |
|
GAS8-AS1 | 605179 | CTU2 | 617057 |
| URAHP | 615805 | PIEZO1 | 611184 |
| PRDM7 | 609759 | CDT1 | 605525 |
|
|
| APRT | 102600 |
|
|
| GALNS | 612222 |
|
|
|
TRAPPC2L | 610970 |
|
|
| CBFA2T3 | 603870 |
|
|
| ACSF3 | 614245 |
|
|
| CDH15 | 114019 |
|
|
| ANKRD11 | 611192 |
|
|
| SPG7 | 602783 |
|
|
| RPL13 | 113703 |
|
|
| CPNE7 | 605689 |
|
|
| DPEP1 | 179780 |
|
|
| CHMP1A | 164010 |
|
|
| SPATA33 | 615409 |
|
|
| CDK10 | 603464 |
|
|
| ZNF276 | 608460 |
|
|
| FANCA | 607139 |
|
|
| SPIRE2 | 609217 |
|
|
| TCF25 | 612326 |
|
|
| MC1R | 155555 |
|
|
| TUBB3 | 602661 |
|
|
| AFG3L1P | 603020 |
|
|
| GAS8 | 605178 |
|
|
|
GAS8-AS1 | 605179 |
|
|
| URAHP | 615805 |
|
|
| PRDM7 | 609759 |
Patient 2
Prenatal diagnosis due to elevated maternal age
revealed a normal karyotype of 46, XX. During the neonatal period
and due to dysmorphic features, hypotonia and clinical
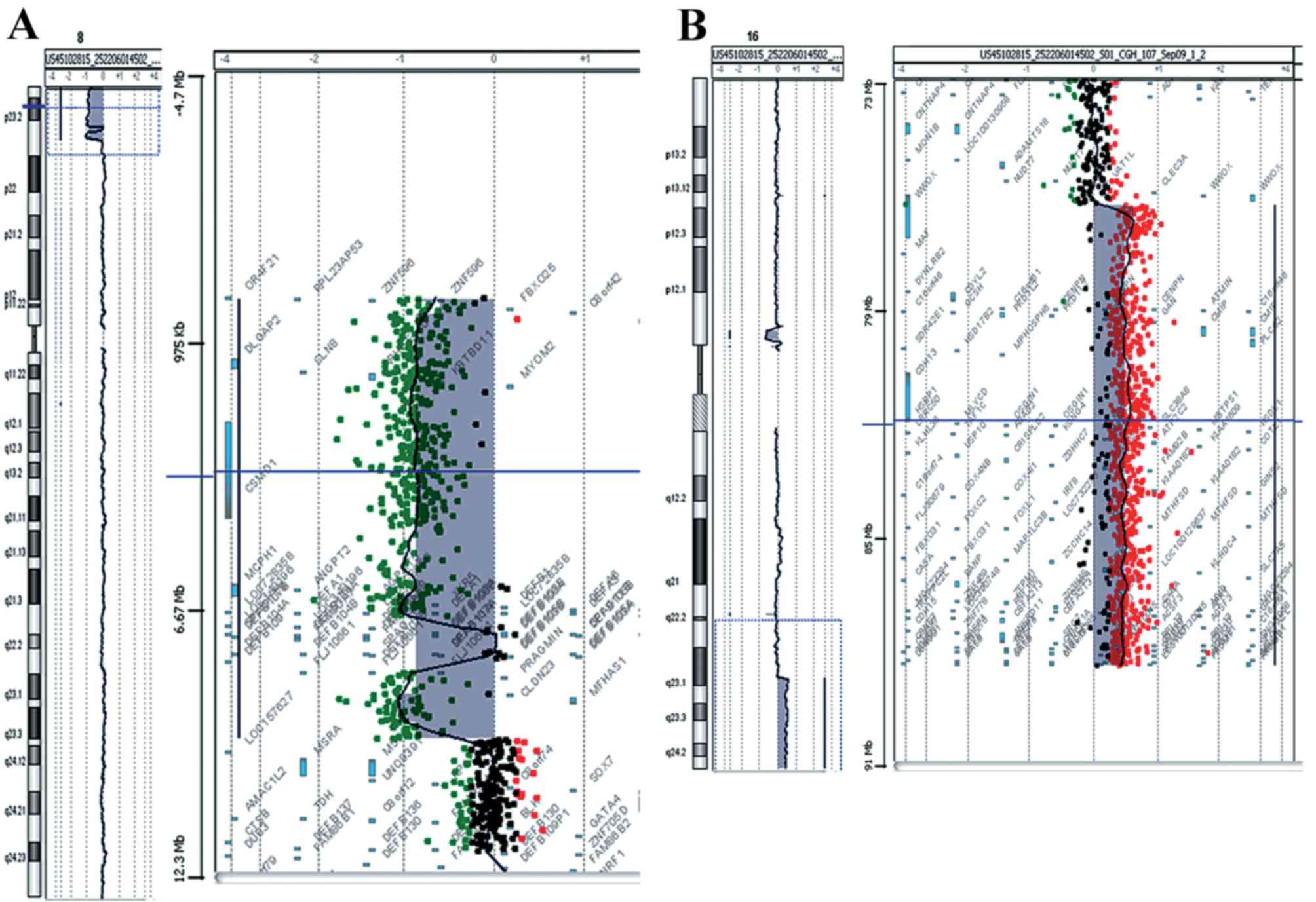
complications, aCGH analysis was performed. The analysis revealed a
deletion of 9.5 Mb of the distal short arm of chromosome 8 with the
proximal breakpoints between 95,48,146 bp (last deleted oligo) and
95,62,020 bp (first normal oligo), with the last oligonucleotide
present in the array at 8p position 151,472 kb being deleted
(Fig. 2A). The deleted region of
the tankyrase (TNKS) gene began at the fifth intron. A
duplication of 11.7 Mb of the long arm of chromosome 16 with the
proximal breakpoint between 76,961,103 bp (duplicated) and
76,938,723 bp (normal) was observed and the last oligonucleotide
present in the array at 16q position 88,690,571 bp was duplicated
(Fig. 2B). The size of the
breakpoint intervals were 13,874 bp for 8p and 22,380 bp for 16q.
The analysis revealed an unbalanced translocation and the aCGH
karyotype was 46,XX,der(8)t(8;16)(p23.1;q23.1).arr[hg18]8p23.3p23.1
(151,472–9548146)x1,16q23.1q24.3(76,961,103–88,690,571)x3.
A list of OMIM genes deleted and duplicated is
presented in Table III.
Microsatellite analysis of the trio revealed that deletion and
duplication occurred on maternally-derived chromosomes (Table IV).
| Table IV.Results from microsatellite analysis
on Patient 2. |
Table IV.
Results from microsatellite analysis
on Patient 2.
| Sample | 253-10 Proband | 254-10 father | 255-10 mother | Origin |
|---|
| D8S201 | 259.5 | 255.5/259.5 | 259.5/267.2 | Uninformative |
| D8S504 | 200.7 | 200.7/202.8 | 198.1/202.7 | Maternal |
| D8S264 | 138.2 | 138.1/138.1 | 126.4/126.4 | Maternal |
| D8S1781 | 259.4 | 259.4/263.2 | 251.1/263.1 | Maternal |
| D8S351 | 119.2 | 119/119 | 105/105 | Maternal |
| D8S1706 | 228/234.2 | 228/234.2 | 228/234.2 | Uninformative |
| D16S3023 | 79.5/83.5 | 83.5/83.5 | 79.5/83.6 | Uninformative |
| D16S413 | 128/132 | 130/132 | 128/132 | Uninformative |
| STS1 (chr16) | 210.8*/214.9 | 214.9/214.9 | 210.9/210.9 | Maternal |
| STS2 (chr16) | 125.2/125.2 | 125.2/125.2 | 125.2/125.2 | Uninformative |
| STS3 (chr16) | 296.5/296.5 | 294.6/296.5 | 296.6 | Uninformative |
| STS4 (chr16) |
345.32/352.4/357.8 | 352.4/359.7 | 345.4/357.38 | Maternal |
Bioinformatic analyses
A possible cause of rearrangements, duplications and
deletions is the occurrence of recombination events. To search for
a possible breakpoint for recombination, the BLAST algorithm was
used to find sequence similarity in the breakpoint regions of the
two patients. The breakpoint regions were revealed to contain
similar sequences residing in Alu elements of Patient 1 and in L1
elements of Patient 2.
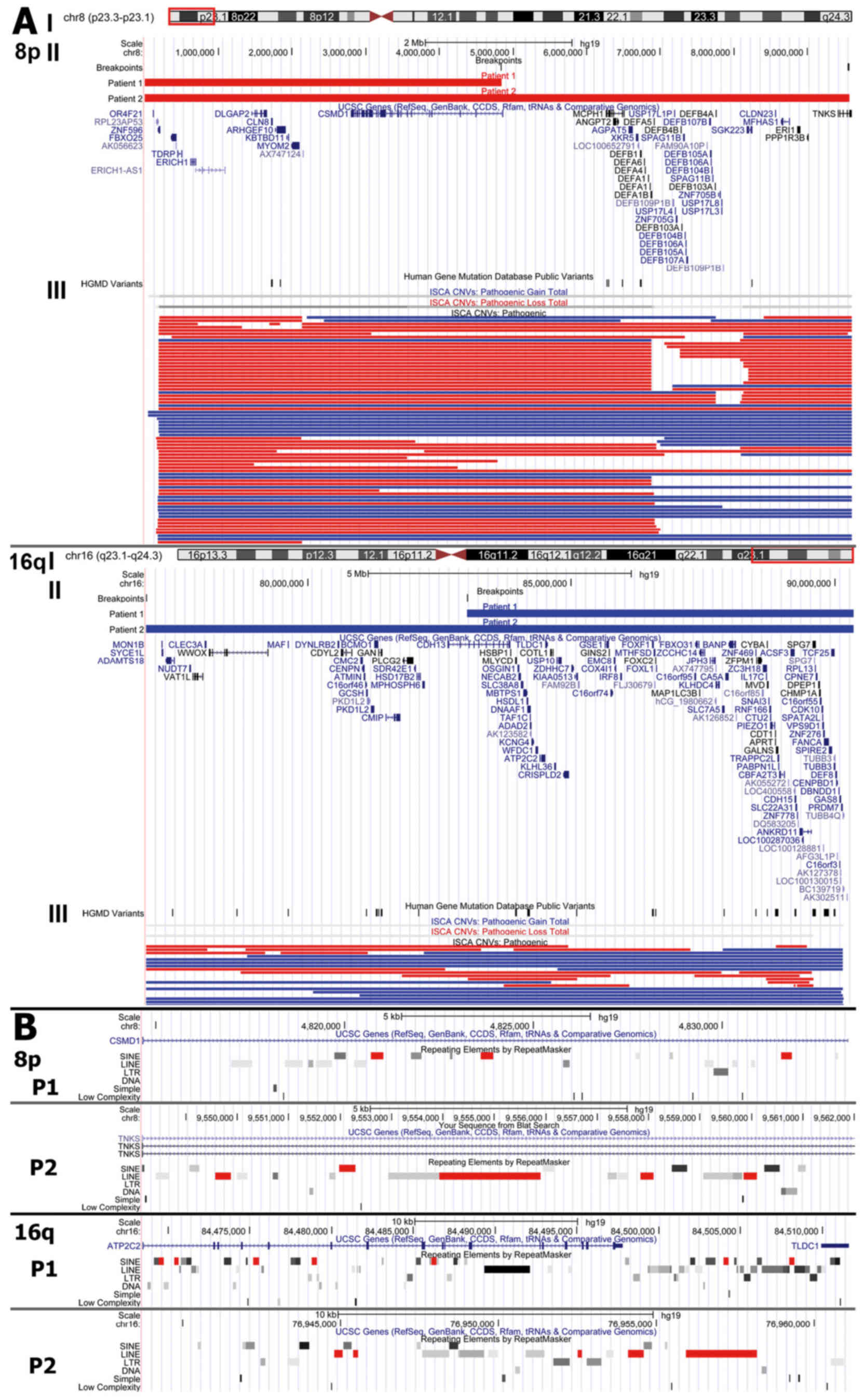
The rearranged regions were viewed in parallel with
ISCA consortium data in the UCSC Genome Browser. The 8p region
contained multiple pathogenic copy number variations (74 deletions
and 31 duplications) described in the ISCA database, while
rearrangements in 16q were less frequent (containing 19 deletions
and 16 duplications). Manual computations of the ISCA data revealed
that 66% of patients with 8p23.3-p23.1 rearrangements (deletions or
duplications), and 62% of patients with 16q23.1-q24.3
rearrangements had developmental delay in their pathogenic
phenotype (Fig. 3).
Discussion
To the best of our knowledge, this is the first
report of a rearrangement involving an 8p deletion and 16q
duplication. The two patients presented in this report had subtle
facial feature dysmorphia, dysmorphic body features, borderline
intelligence and marginal follow up progress, low birth weight and
vertebral anomalies, and one presented with cardiovascular
abnormalities. The majority of the clinical characteristics of the
two patients were associated with those of 8p or 16q chromosome
imbalances, but it is difficult to estimate if the clinical
phenotype and developmental delay were due to the rearrangement or
whether they were the result of 8p monosomy and 16q trisomy
separately. It has previously been recognized that deletions in the
distal region of chromosome 8p are associated with growth and
mental impairment, minor facial dysmorphisms, microcephaly,
congenital heart defects and behavioral problems (23). According to all references, 16q
trisomy is a rare abnormality due to high rates of mortality and
lethality in the prenatal and neonatal period (11,24).
Partial 16q trisomy is most often the result of balanced or
unbalanced rearrangements, and therefore it is difficult to
understand if the commonly observed phenotypic characteristics
(dysmorphic facial features, developmental delay, intellectual
disability, central nervous system malformations and congenital
heart defects) are due to 16q or whether they are the result of
changes in genome architecture (24).
More than 2/3 of patients with 8p syndrome have
congenital heart defects, suggesting that 8p23.1 maybe critical for
heart development (5,25). One of the candidate genes for heart
disease is GATA4 because haploinsufficiency and mutations
have been documented in patients and families with atrial septal
defects and other cardiac defects associated with 8p23.1 deletion
(4,26–29).
Chen et al (30) studied a
four-generation Chinese atrial septal defect family and suggested
that a mutation in the GATA4 gene (c.A899C, p.K300T) may
contribute to this congenital heart disease. However, the
GATA4 gene was not deleted in either patient in the present
study. The fact that Patient 1 has heart problems suggested either
that other genes were responsible for these problems, or that the
rearrangement resulted in a structural alteration affecting the
function of genes associated with the heart. The CSMD1 gene
(8p23.2) was deleted in Patient 2 but only partially deleted in
Patient 1. According to the literature, CSMD1 loss of
function is correlated with head and neck squamous cell carcinoma
(31,32), and liver (33,34),
lung, breast and skin cancers (31). Deletion of this gene has been
reported in a case of craniofacial and body dysmorphisms and mental
retardation (35). In Patient 2,
two of the deleted genes were TNKS and MCPH1. These
genes are involved in meiosis and mitosis mechanisms. The
TNKS gene, located at 8p23.1, is involved in sister
chromatid cohesion and deletions result in anaphase arrest
(36). Páez et al (4) identified deletions of TNKS
gene in patients with mental retardation and behavioral problems.
TNKS protein positively regulates the Wnt/β-catenin signaling
pathway (37). This pathway is
critical for healthy embryonic development and cellular
differentiation (38).
Furthermore, TNKS is a candidate gene for Cornelia de Lange
Syndrome (CdLS) (3,36), a syndrome characterized by
distinctive facial features including well-defined curved and
confluent eyebrows, long eyelashes, anteverted nares, micrognathia
and downturned corners of the mouth with a thin upper lip. Patient
2 resembled the CdLS facial phenotype, and she is expected to have
psychomotor retardation, language acquisition difficulties and
behavioral disorders in the autistic spectrum, typical aspects of
CdLS. The MCPH1 gene, additionally located in 8p23.1, is
involved in preventing cells from prematurely entering mitosis, and
truncated mutations have been associated with premature chromosome
condensation and were observed in patients with microcephaly,
growth impairment and mental retardation (36,39).
Another gene located in 8p23.1 is RP1 like 1 (RP1L1). Its
expression is restricted to the postnatal retina, potentially being
involved in retinal development (40). The RP1L1 gene may not be
haploinsufficient in Patient 2, who was diagnosed with diffuse
depigmentation of the retina. This gene maybe under the control of
translocated regulatory elements, being in the proximity of the
breakpoint, and may have resulted in this retinal disorder.
In total, >30 cases with distal 8p deletion have
been described in the literature, and 9 with 16q24 duplication, but
only a few have been characterized with high resolution molecular
techniques (1,11). The 8p region is more often reported
to be involved in rearrangements than 16q. Giglio et al
(41) demonstrated that the
olfactory receptor (OR) gene clusters are the substrate for the
formation of intrachromosomal rearrangements involving chromosome
8p. Different rearrangements, most of them recurring, are
associated with the distal 8p region. Among them there are inv
dup(8p), del(8p22) and small marker chromosomes der(8)(p23-pter)
(41). Furthermore, seven
individuals with balanced and unbalanced translocations between
4p16 and 8p23 demonstrated that the breakpoints fell within the 4p
and 8p OR-gene clusters (42).
BLAST alignment in the 8p and 16q regions revealed
high similarity regions with several Alu elements in Patient 1 and
two similar long interspersed nuclear element 1 elements in Patient
2. Retroelements are known to facilitate recombination events
(43). Consequently, a potential
mechanism of their appearance maybe unequal cross over between
repetitive DNA regions with high sequence similarity (44). The deleted and duplicated regions
are regions often correlated with developmental delay, according to
the ISCA Consortium (20).
Novel diagnostic methods with great potential have
facilitated the study and interpretation of the consequences of
chromosome aberrations and revealed that the pathogenicity may be
due to complex molecular mechanisms (45,46).
A number of the genes identified as deleted or duplicated in these
cases may have resulted in developmental delay, but developmental
delay may also be the result of rearrangements and changes of
important parts of gene structure functional elements, truncated or
fusion genes. A multidisciplinary effort aiming to study all cases
with 8p;16q rearrangements with combined and accurate methods and
tools (cytogenetic, molecular cytogenetic, NGS mapping) along with
their clinical phenotypes may elucidate the involvement of the
rearrangement, genes involved, participation of control elements
and/or interactions with polymorphic regions, and potentially a
clear phenotype-genotype correlation.
References
|
1
|
Reddy KS: A paternally inherited terminal
deletion, del(8)(p23.1)pat, detected prenatally in an amniotic
fluid sample: A review of deletion 8p23.1 cases. Prenat Diagn.
19:868–872. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fagan KA and Morris RB: Del(4)(q33-qter):
Another case report of a child with mild dysmorphism. J Med Genet.
26:776–778. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ballarati L, Cereda A, Caselli R,
Selicorni A, Recalcati MP, Maitz S, Finelli P, Larizza L and
Giardino D: Genotype-phenotype correlations in a new case of 8p23.1
deletion and review of the literature. Eur J Med Genet. 54:55–59.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Páez MT, Yamamoto T, Hayashi K, Yasuda T,
Harada N, Matsumoto N, Kurosawa K, Furutani Y, Asakawa S, Shimizu N
and Matsuoka R: Two patients with atypical interstitial deletions
of 8p23.1: Mapping of phenotypical traits. Am J Med Genet A.
146A:1–1165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Devriendt K, Matthijs G, Van Dael R,
Gewillig M, Eyskens B, Hjalgrim H, Dolmer B, McGaughran J,
Bröndum-Nielsen K, Marynen P, et al: Delineation of the critical
deletion region for congenital heart defects, on chromosome 8p23.1.
Am J Hum Genet. 64:1119–1126. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shin WJ, Kim SD and Kim KH: The general
anesthesia experience of deletion 8p syndrome patient-A case
report. Korean J Anesthesiol. 61:332–335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brisset S, Joly G, Ozilou C, Lapierre JM,
Gosset P, LeLorc'h M, Raoul O, Turleau C, Vekemans M and Romana SP:
Molecular characterization of partial trisomy 16q24.1-qter:
Clinical report and review of the literature. Am J Med Genet.
113:339–345. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ferrero GB, Belligni E, Sorasio L,
Delmonaco AG, Oggero R, Faravelli F, Pierluigi M and Silengo M:
Phenotype resembling donnai-barrow syndrome in a patient with
9qter;16qter unbalanced translocation. Am J Med Genet A.
140:892–894. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hutchinson R, Wilson M and Voullaire L:
Distal 8p deletion (8p23. 1-8pter): A common deletion? J Med Genet.
29:407–411. 1992.
|
|
10
|
Zahn S, Ehrbrecht A, Bosse K, Kalscheuer
V, Propping P, Schwanitz G, Albrecht B and Engels H: Further
delineation of the phenotype maps for partial trisomy 16q24 and
jacobsen syndrome by a subtle familial translocation
t(11;16)(q24.2;q24.1). Am J Med Genet A. 139:19–24. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou Y, Yao Q, Cui YX, Yao B, Fan K, Xia
XY, Hu YA and Li XJ: Clinical and cytogenetic characterization of a
boy with a de novo pure partial trisomy 16q24.1q24.3 and complex
chromosome rearrangement. Am J Med Genet A. 161A:1–900.
2013.PubMed/NCBI
|
|
12
|
Burnside RD, Pappas JG, Sacharow S,
Applegate C, Hamosh A, Gadi IK, Jaswaney V, Keitges E, Phillips KK,
Potluri VR, et al: Three cases of isolated terminal deletion of
chromosome 8p without heart defects presenting with a mild
phenotype. Am J Med Genet A. 161A:1–828. 2013.PubMed/NCBI
|
|
13
|
Baker E, Hinton L, Callen DF, Altree M,
Dobbie A, Eyre HJ, Sutherland GR, Thompson E, Thompson P, Woollatt
E and Haan E: Study of 250 children with idiopathic mental
retardation reveals nine cryptic and diverse subtelomeric
chromosome anomalies. Am J Med Genet. 107:285–293. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Giardino D, Finelli P, Gottardi G, Clerici
D, Mosca F, Briscioli V and Larizza L: Cryptic subtelomeric
translocation t(2;16)(q37;q24) segregating in a family with
unexplained stillbirths and a dysmorphic, slightly retarded child.
Eur J Hum Genet. 9:881–886. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maher ER, Willatt L, Cuthbert G, Chapman C
and Hodgson SV: Three cases of 16q duplication. J Med Genet.
28:801–802. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bayley N: Scales of Infant Development.
2nd edition. The Psychological Corporation; San Antonio, TX:
1993
|
|
17
|
Griffiths R: A comprehensive system of
measurement for the first eight years of lifeThe Abilities of Young
Children. Bucks: Association for Research in Infant and Child
Development. The test Agency; Oxford: pp. 101–172. 1984
|
|
18
|
Manolakos E, Peitsidis P, Eleftheriades M,
Dedoulis E, Ziegler M, Orru S, Liehr T and Petersen MB: Prenatal
detection of full monosomy 21 in a fetus with increased nuchal
translucency: Molecular cytogenetic analysis and review of the
literature. J Obstet Gynaecol Res. 36:435–440. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kent WJ, Sugnet CW, Furey TS, Roskin KM,
Pringle TH, Zahler AM and Haussler D: The human genome browser at
UCSC. Genome Res. 12:996–1006. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kaminsky EB, Kaul V, Paschall J, Church
DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG,
Warren ST, et al: An evidence-based approach to establish the
functional and clinical significance of copy number variants in
intellectual and developmental disabilities. Genet Med. 13:777–784.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miller DT, Adam MP, Aradhya S, Biesecker
LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE,
Epstein CJ, et al: Consensus statement: Chromosomal microarray is a
first-tier clinical diagnostic test for individuals with
developmental disabilities or congenital anomalies. Am J Hum Genet.
86:749–764. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Altschul SF, Gish W, Miller W, Myers EW
and Lipman DJ: Basic local alignment search tool. J Mol Biol.
215:403–410. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Digilio MC, Marino B, Guccione P,
Giannotti A, Mingarelli R and Dallapiccola B: Deletion 8p syndrome.
Am J Med Genet. 75:534–536. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lonardo F, Perone L, Maioli M, Ciavarella
M, Ciccone R, Monica MD, Lombardi C, Forino L, Cantalupo G, Masella
L and Scarano F: Clinical, cytogenetic and molecular-cytogenetic
characterization of a patient with a de novo tandem
proximal-intermediate duplication of 16q and review of the
literature. Am J Med Genet A. 155A:1–777. 2011.PubMed/NCBI
|
|
25
|
Johnson MC, Hing A, Wood MK and Watson MS:
Chromosome abnormalities in congenital heart disease. Am J Med
Genet. 70:292–298. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mei M, Yang L, Zhan G, Wang H, Ma D, Zhou
W and Huang G: Analysis of genomic copy number variations in two
unrelated neonates with 8p deletion and duplication associated with
congenital heart disease. Zhonghua Er Ke Za Zhi. 52:460–463.
2014.(In Chinese). PubMed/NCBI
|
|
27
|
Pehlivan T, Pober BR, Brueckner M, Garrett
S, Slaugh R, Van Rheeden R, Wilson DB, Watson MS and Hing AV: Gata4
haploinsufficiency in patients with interstitial deletion of
chromosome region 8p23.1 and congenital heart disease. Am J Med
Genet. 83:201–206. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wat MJ, Shchelochkov OA, Holder AM, Breman
AM, Dagli A, Bacino C, Scaglia F, Zori RT, Cheung SW, Scott DA and
Kang SH: Chromosome 8p23.1 deletions as a cause of complex
congenital heart defects and diaphragmatic hernia. Am J Med Genet
A. 149A:1–1677. 2009. View Article : Google Scholar
|
|
29
|
Yang YQ, Wang J, Liu XY, Chen XZ, Zhang W,
Wang XZ, Liu X and Fang WY: Novel GATA4 mutations in patients with
congenital ventricular septal defects. Med Sci Monit.
18:CR344–CR350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen J, Qi B, Zhao J, Liu W, Duan R and
Zhang M: A novel mutation of GATA4 (K300T) associated with familial
atrial septal defect. Gene. 575:473–477. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma C, Quesnelle KM, Sparano A, Rao S, Park
MS, Cohen MA, Wang Y, Samanta M, Kumar MS, Aziz MU, et al:
Characterization CSMD1 in a large set of primary lung, head and
neck, breast and skin cancer tissues. Cancer Biol Ther. 8:907–916.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Toomes C, Jackson A, Maguire K, Wood J,
Gollin S, Ishwad C, Paterson I, Prime S, Parkinson K, Bell S, et
al: The presence of multiple regions of homozygous deletion at the
CSMD1 locus in oral squamous cell carcinoma question the role of
CSMD1 in head and neck carcinogenesis. Genes Chromosomes Cancer.
37:132–140. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Midorikawa Y, Yamamoto S, Tsuji S,
Kamimura N, Ishikawa S, Igarashi H, Makuuchi M, Kokudo N, Sugimura
H and Aburatani H: Allelic imbalances and homozygous deletion on
8p23.2 for stepwise progression of hepatocarcinogenesis.
Hepatology. 49:513–522. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhu Q, Gong L, Liu X, Wang J, Ren P, Zhang
W, Yao L, Han X, Zhu S, Lan M, et al: Loss of heterozygosity at
D8S262: An early genetic event of hepatocarcinogenesis. Diagn
Pathol. 10:702015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Naseer MI, Chaudhary AG, Rasool M,
Kalamegam G, Ashgan FT, Assidi M, Ahmed F, Ansari SA, Zaidi SK, Jan
MM and Al-Qahtani MH: Copy number variations in Saudi family with
intellectual disability and epilepsy. BMC Genomics. 17 Suppl
9:S7572016. View Article : Google Scholar
|
|
36
|
Baynam G, Goldblatt J and Walpole I:
Deletion of 8p23.1 with features of Cornelia de Lange syndrome and
congenital diaphragmatic hernia and a review of deletions of 8p23.1
to 8pter? A further locus for Cornelia de Lange syndrome. Am J Med
Genet A. 146A:1–1570. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Roy S, Liu F and Arav-Boger R: Human
Cytomegalovirus Inhibits the PARsylation Activity of Tankyrase-A
potential strategy for suppression of the Wnt pathway. Viruses.
8:pii: E82015. View Article : Google Scholar
|
|
38
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Trimborn M, Bell SM, Felix C, Rashid Y,
Jafri H, Griffiths PD, Neumann LM, Krebs A, Reis A, Sperling K, et
al: Mutations in microcephalin cause aberrant regulation of
chromosome condensation. Am J Hum Genet. 75:261–266. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Conte I, Lestingi M, den Hollander A,
Alfano G, Ziviello C, Pugliese M, Circolo D, Caccioppoli C,
Ciccodicola A and Banfi S: Identification and characterisation of
the retinitis pigmentosa 1-like1 gene (RP1L1): A novel candidate
for retinal degenerations. Eur J Hum Genet. 11:155–162. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Giglio S, Calvari V, Gregato G, Gimelli G,
Camanini S, Giorda R, Ragusa A, Guerneri S, Selicorni A, Stumm M,
et al: Heterozygous submicroscopic inversions involving olfactory
receptor-gene clusters mediate the recurrent t(4;8)(p16;p23)
translocation. Am J Hum Genet. 71:276–285. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Giglio S, Broman KW, Matsumoto N, Calvari
V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda
R, et al: Olfactory receptor-gene clusters, genomic-inversion
polymorphisms, and common chromosome rearrangements. Am J Hum
Genet. 68:874–883. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Konkel MK and Batzer MA: A mobile threat
to genome stability: The impact of non-LTR retrotransposons upon
the human genome. Semin Cancer Biol. 20:211–221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Robberecht C, Voet T, Esteki M Zamani,
Nowakowska BA and Vermeesch JR: Nonallelic homologous recombination
between retrotransposable elements is a driver of de novo
unbalanced translocations. Genome Res. 23:411–418. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Newman S, Hermetz KE, Weckselblatt B and
Rudd MK: Next-generation sequencing of duplication CNVS reveals
that most are tandem and some create fusion genes at breakpoints.
Am J Hum Genet. 96:208–220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Utami KH, Hillmer AM, Aksoy I, Chew EG,
Teo AS, Zhang Z, Lee CW, Chen PJ, Seng CC, Ariyaratne PN, et al:
Detection of chromosomal breakpoints in patients with developmental
delay and speech disorders. PLoS One. 9:e908522014. View Article : Google Scholar : PubMed/NCBI
|