Introduction
Neurodegenerative diseases, which are generally
observed in the elderly, comprise a range of chronic degenerative
disorders of the central nervous system (CNS), including
Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's
disease and amyotrophic lateral sclerosis (1). These diseases are characterized by
accumulation of toxic proteins, and gradual and progressive loss of
neural cells, which negatively affects the mental and physical
functions of patients (1). Over
the last two decades, numerous potential mechanisms for the
pathogenesis of neurodegenerative diseases have been proposed,
including certain genetic polymorphisms, advanced age, endocrine
disorders, endoplasmic reticulum (ER) stress, oxidative stress,
inflammation and chemical exposure (1,2).
Despite progress being made in the identification of initiating
factors of neurodegenerative diseases, the causative reagents
associated with neuronal degeneration remain poorly understood.
Among the proposed mechanisms, the roles of ER
stress and oxidative stress in the development of neuronal
degeneration are of particular interest. It has previously been
suggested that increased ER stress in neurons contributes to the
development of AD, which is considered one of the most common types
of neurodegenerative diseases, and related cognitive impairment
(3). Volgyi et al (4) reported that formation of toxic
β-amyloid (Aβ), which affects the ER and disturbs neurons, occurs
during the development of AD, thus suggesting that ER dysfunction
may be one of the main pathological pathways in AD. Reactive oxygen
species (ROS)-induced oxidative stress has also been implicated in
the pathogenesis of AD and PD (5,6).
Furthermore, ROS formation and ER stress are closely linked
processes, since ROS can be produced in ER due to protein
misfolding and serves an important role in ER stress-induced
apoptosis (7).
Recent studies have identified dietary factors, in
particular the relative intake of saturated fatty acids and
unsaturated fats, as being closely associated with numerous
neurodegenerative disorders (8,9).
Trans fatty acids (TFAs) are structurally unstable saturated
fatty acids with at least one double bond in the trans
configuration (10). TFAs within
the human diet are predominantly obtained from industrial partial
hydrogenation of vegetable oils, and from the natural TFAs present
in the milk and body fat of ruminants. Elaidic acid (18:1
trans-9) is the main isomer of TFAs. The pathogenic effects
of TFAs have been associated with alterations in cholesterol
metabolism and in the structure of biomembranes (11). Previous studies have reported that
TFAs are associated with chronic diseases, including coronary heart
disease, diabetes and arteriosclerosis (12,13).
However, the influence of TFAs on neurodegenerative disorders
remains poorly understood. Epidemiological studies have revealed
inconsistent results regarding the association of TFAs with the
relative risk of developing AD. Morris et al reported that
high dietary intake of TFAs was linked with an elevated risk of
developing AD (14). However,
another study failed to establish the relationship between TFA
consumption and AD (15). Grimm
et al reported that TFAs increased amyloidogenic processing
of amyloid precursor protein, resulting in an increased production
of Aβ peptides in SH-SY5Y cells (16). Therefore, the effects of dietary
intake of TFAs on neurodegenerative diseases and the mechanism
underlying its neurotoxicity require further study.
Morinaga et al reported that the predominant
isomer of TFAs, elaidic acid, plus fructose significantly increased
oxidative stress and ER stress in mice and in primarily cultured
hepatocytes (17). Cassagno et
al reported that mice fed a diet rich in TFAs developed
increased hepatic oxidative stress and ER stress (18). Given the essential roles of ER
stress and oxidative stress in the development of neurodegenerative
disorders, the present study explored the effects of TFAs on
oxidative damage and ER stress in neurons. The molecules and
signaling pathways involved in oxidative damage and ER stress were
also determined, with the aim of identifying the possible mechanism
underlying elaidic acid-induced neuronal damage in
vitro.
Materials and methods
Cell lines and cell culture
The SH-SY5Y human neuroblastoma cell line was
obtained from Peking Union Medical Center Laboratory (Beijing,
China). SH-SY5Y cells were cultured in Dulbecco's modified Eagle's
medium (DMEM) supplemented with 10% fetal bovine serum (HyClone; GE
Healthcare Life Science, Logan, UT, USA) at 37°C in an atmosphere
containing 5% CO2. Elaidic acid was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and was dissolved in
DMEM to a concentration of 1 M, finally the stock solution was
diluted with DMEM to a concentration of 1,000 µM and stored at
−80°C until further use. All experiments were conducted under
treatment with various doses (10, 20, 50, 100, 200, 400 or 800 µM)
of elaidic acid or vehicle (DMEM) for 24 h.
MTT test
The viability of SH-SY5Y cells was determined using
the MTT assay, as previously described (19). Briefly, SH-SY5Y cells were plated
at 1×104 cells/well in 96-well plates in 6 replicates,
and were incubated in DMEM containing elaidic acid (10, 20, 50,
100, 200, 400 and 800 µM) or DMEM alone (control) for 24 h at 37°C.
Subsequently, 20 µl MTT solution (5 mg/ml) was added to each well
and the plates were incubated for 4 h at 37°C. The supernatant was
discarded and 200 µl DMSO was then added to each well. The
absorbance was measured at 570 nm using a microplate reader. In
addition, SH-SY5Y cells were treated with 50 µM elaidic acid for
12, 24 and 48 h, respectively, and the viability of cells were also
tested by the same methods.
Measurement of ROS
Formation of ROS in cells was detected using the
Fluorometric Intracellular ROS kit (Sigma-Aldrich; Merck KGaA)
according to the manufacturer's protocol with a slight modification
(an increase in incubation time). Briefly, the collected cells were
incubated with 10 µM 2′,7′-dichlorofluorescin diacetate dissolved
in serum-free DMEM at 37°C for 20 min. The fluorescent signal was
then measured using flow cytometry (BD LSRFortessa with FACSDiva
software version 7.0; BD Biosciences, Franklin Lakes, NJ) with an
excitation wavelength of 488 nm and an emission wavelength of 530
nm.
Measurement of mitochondrial membrane
potential (MMP)
Alterations in MMP were measured using the
MitoScreen kit (BD Bioscience, Franklin Lakes, NJ, USA) according
to the manufacturer's protocol. Briefly, the SH-SY5Y cells
(1×105 cells/35 mm Petri dish) were collected and
incubated with 0.5 ml JC-1 working solution for 15 min at 37°C.
Cell fluorescence was measured by flow cytometry with an excitation
wavelength of 488 nm and an emission wavelength of 530 nm.
Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) staining for apoptosis
Cells in early and late apoptosis were quantified
using an Annexin V-FITC/PI double staining assay kit (KGA107;
Nanjing KeyGen Biotech Co., Ltd., Nanjing, China) as described
previously (20). Cells were
harvested by trypsinization and washed three times with PBS.
Subsequently, 1×105 cells from each 35 mm Petri dish
were resuspended in 500 µl binding buffer and stained with 5 µl
Annexin V and 5 µl PI in the dark at room temperature for 20 min.
The stained cells were immediately examined using flow cytometry
with an excitation wavelength of 488 nm for the green fluorescence
of FITC-Annexin V and 561 nm for the red fluorescence of PI.
Measurement of oxidative
stress-related biomarkers
Cells (1×105 cells/35 mm Petri dish) were
washed with PBS, harvested by trypsin and resuspended in 0.5 ml
PBS. An ultrasonic cell disruption system was used and the cells
ruptured with ultrasonic waves for 5 sec twice, with a 5 sec
interval, at 37°C. Protein concentrations of the cell lysates were
measured using the bicinchoninic acid (BCA) protein assay kit
(Pierce; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Subsequently, the levels of lipid peroxide (LPO; A106) and
malondialdehyde (MDA; A003-3), the activities of superoxide
dismutase (SOD; A001-3) and glutathione peroxidase (GSH-Px; A005),
and reduced glutathione (GSH)/oxidized glutathione (GSSG) ratio
(A061-1) were measured using corresponding assay kits (Nanjing
Jiancheng Bioengineering Institute, Nanjing, China) according to
the manufacturer's protocols.
Western blot analysis
Cells were harvested using trypsin and cell pellets
were lysed in radioimmunoprecipitation assay buffer (Beijing
Solarbio Science & Technology Co., Ltd., Beijing, China) and
phenylmethylsulfonyl fluoride with vigorous agitation for 15 min at
4°C. The lysates were then centrifuged at 16,000 × g for 15 min at
4°C and the protein-containing supernatant was collected. Total
protein concentration was determined using the BCA assay. Western
blotting was performed as previously described (21). Briefly, 40 µg cell lysate was
separated by 12% SDS-PAGE and the proteins were transferred onto an
activated polyvinylidene fluoride membrane at 60 V for 2 h. The
membrane was blocked with 5% non-fat dry milk in TBS containing 20%
Tween (TBST) at room temperature for 1 h, and was then incubated
with anti-β-actin, anti-nuclear factor erythroid 2-related factor 2
(Nrf2; ab31163, Abcam, Cambridge, MA, USA), anti-heme oxygenase
(HO)-1 (ab13248; Abcam), anti-inositol requiring enzyme (IRE)1α
(9956; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA),
anti-glucose-regulated protein (GRP)78 (9956; 1:1,000; Cell
Signaling Technology, Inc.), anti-CCAAT/enhancer-binding protein
homologous protein (CHOP; 9956; 1:1,000; Cell Signaling Technology,
Inc.), anti-activating transcription factor (ATF)4 (11815; 1:1,000;
Cell Signaling Technology, Inc.), anti-protein disulfide isomerase
(PDI; 9956; 1:1,000; Cell Signaling Technology, Inc.) and
anti-endoplasmic oxidoreductin-1-like protein (Ero1-Lα; 9956;
1:1,000; Cell Signaling Technology, Inc.) primary antibodies
overnight at 4°C. Subsequently, the membrane was washed three times
with TBST and was incubated with the corresponding secondary
antibodies (Anti-mouse IgG, HRP-linked Antibody 7076 and
Anti-rabbit IgG, HRP-linked Antibody 7074; 1:2,000; Cell Signaling
Technology, Inc.) for 1 h at room temperature. Electrochemical
luminescence (W101B; Promega Corporation, Madison, WI, USA) was
used to visualize the target proteins. Images were obtained using
the FluoChem®FC2 imaging system (ProteinSimple;
Bio-Techne, Minneapolis, MN, USA) and semi-quantification was
performed by analyzing the gray values of protein bands.
Statistical analysis
All data are presented as the mean ± standard error
of at least three independent experiments. All data analyses were
performed using SPSS software version 16.0 (SPSS, Inc., Chicago,
IL, USA). One-way analysis of variance followed by least
significant difference or Dunnett's T3 post-hoc tests were used to
compare the means of different groups according to the homogeneity
of variance. P<0.05 was considered to indicate a statistically
significant difference.
Results
Treatment with elaidic acid decreases
cell viability
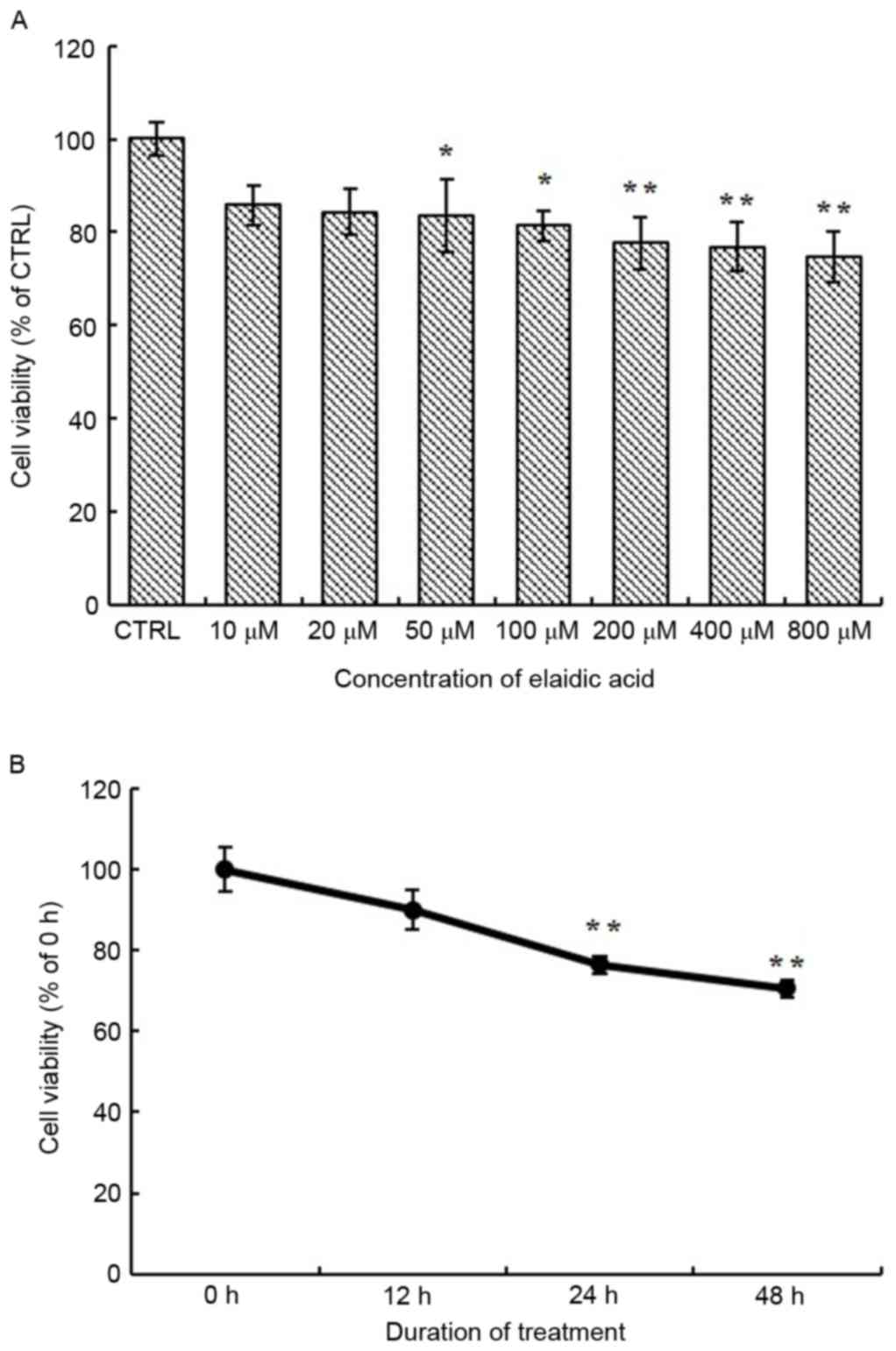
To investigate the effects of elaidic acid on cell
viability, SH-SY5Y cells were treated with various concentrations
of elaidic acid or vehicle for 24 h and cell viability was measured
using the MTT assay. As presented in Fig. 1A, the viability of SH-SY5Y cells
was significantly reduced following treatment with 50–800 µM
elaidic acid compared with the control group (P=0.044, 0.022,
0.007, 0.005 and 0.002, respectively). In addition, SH-SY5Y cells
were treated with 50 µM elaidic acid for 12, 24 and 48 h. Cell
viability was gradually decreased as the duration of treatment was
prolonged (Fig. 1B), thus
suggesting that elaidic acid induced the inhibition of cell
viability in a time-dependent manner.
Elaidic acid reduces MMP in SH-SY5Y
cells
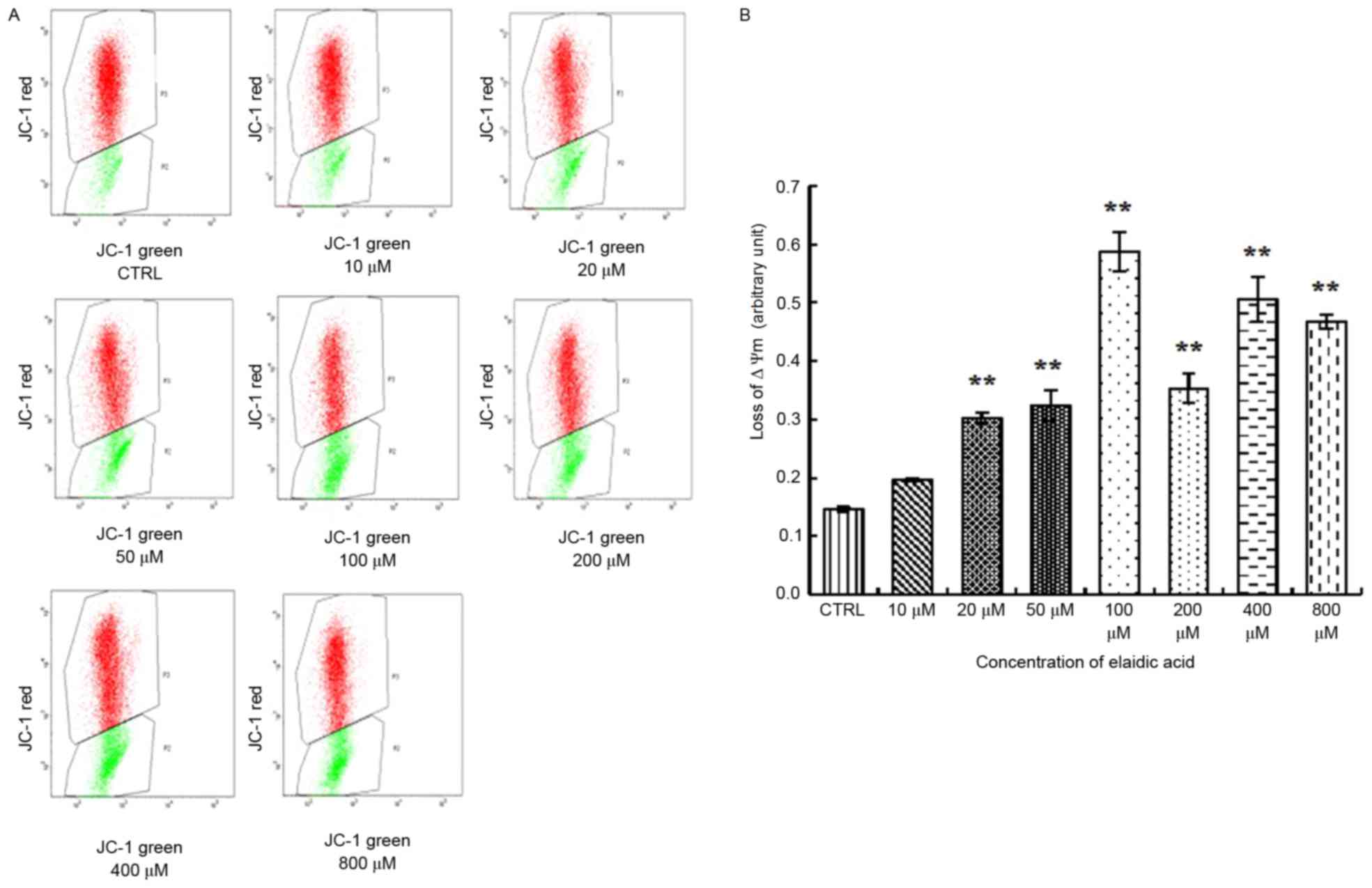
A distinctive feature of the early stages of
apoptosis is the disruption of active mitochondria, including
alterations in the MMP and the oxidation-reduction potential of
mitochondria. Therefore, the present study investigated the effects
of elaidic acid on MMP. The results indicated that the ratio of
green fluorescence to red fluorescence was significantly elevated
in cells treated with 20–800 µM elaidic acid (P<0.01),
indicating the loss of MMP and the occurrence of early apoptotic
events. In addition, the extent of the MMP was increased as the
concentration of elaidic acid increased (Fig. 2A and B). These findings indicated
that elaidic acid induced early apoptosis of SH-SY5Y cells in a
dose-dependent manner.
Elaidic acid induces SH-SY5Y cell
apoptosis
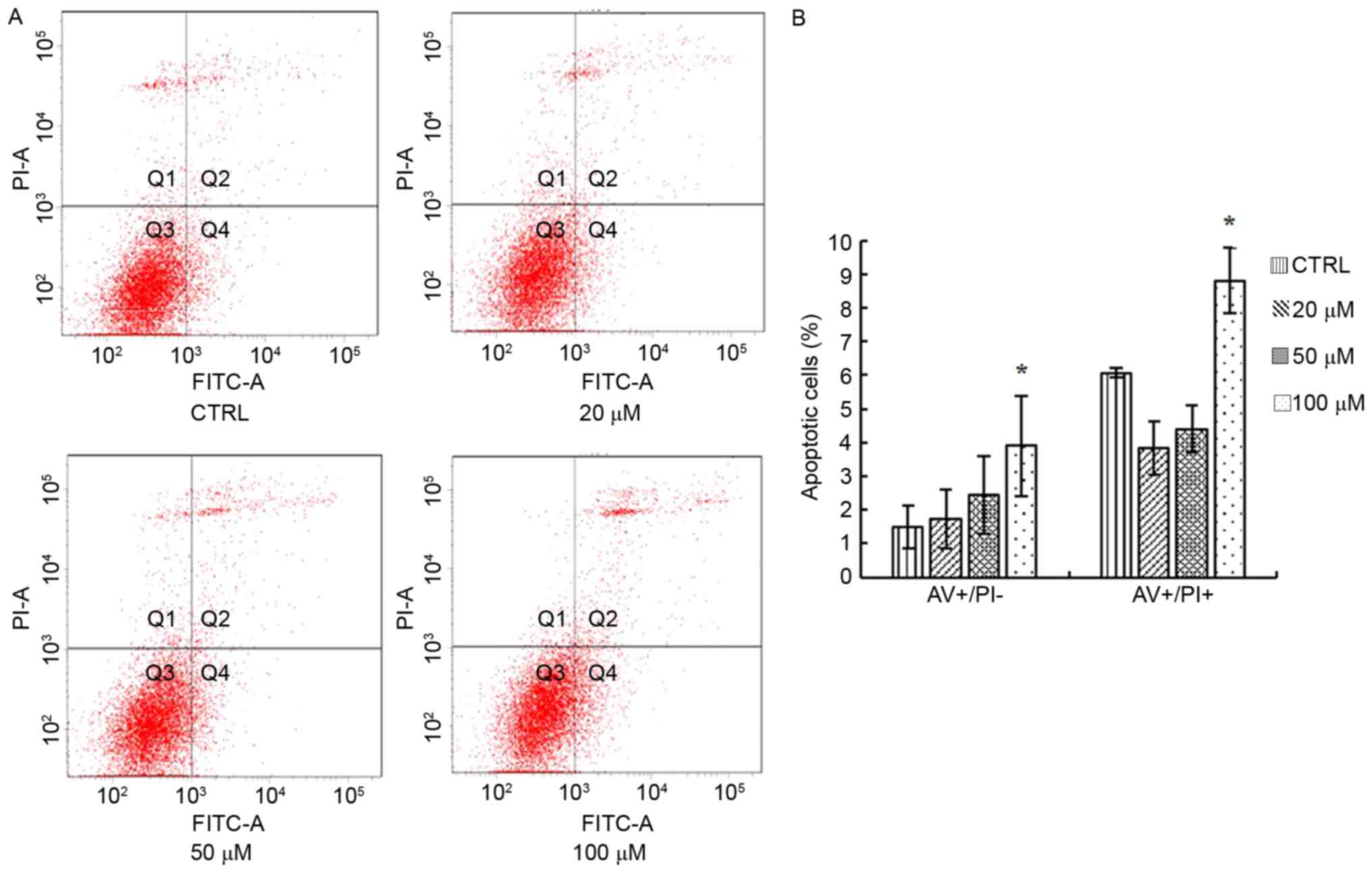
The present study detected the apoptosis of SH-SY5Y
cells following treatment with 20, 50 and 100 µM elaidic acid using
Annexin V-FITC/PI double staining and flow cytometry. As presented
in Fig. 3A and B, compared with
the control group, cells treated with 100 µM elaidic acid exhibited
a significantly higher percentage of apoptotic cells.
Elaidic acid induces elevated ROS
release from SH-SY5Y cells
Previous studies have indicated that elaidic acid
may induce oxidative stress, which is an important trigger of cell
apoptosis, in liver cells (17,18).
Therefore, the present study examined whether elaidic acid was able
to promote the generation of ROS, which is an inducer of oxidative
stress in SH-SY5Y cells. As presented in Fig. 4, increased ROS release was observed
in cells treated with 200, 400 or 800 µM elaidic acid (P=0.044,
0.002 and 0.019, respectively).
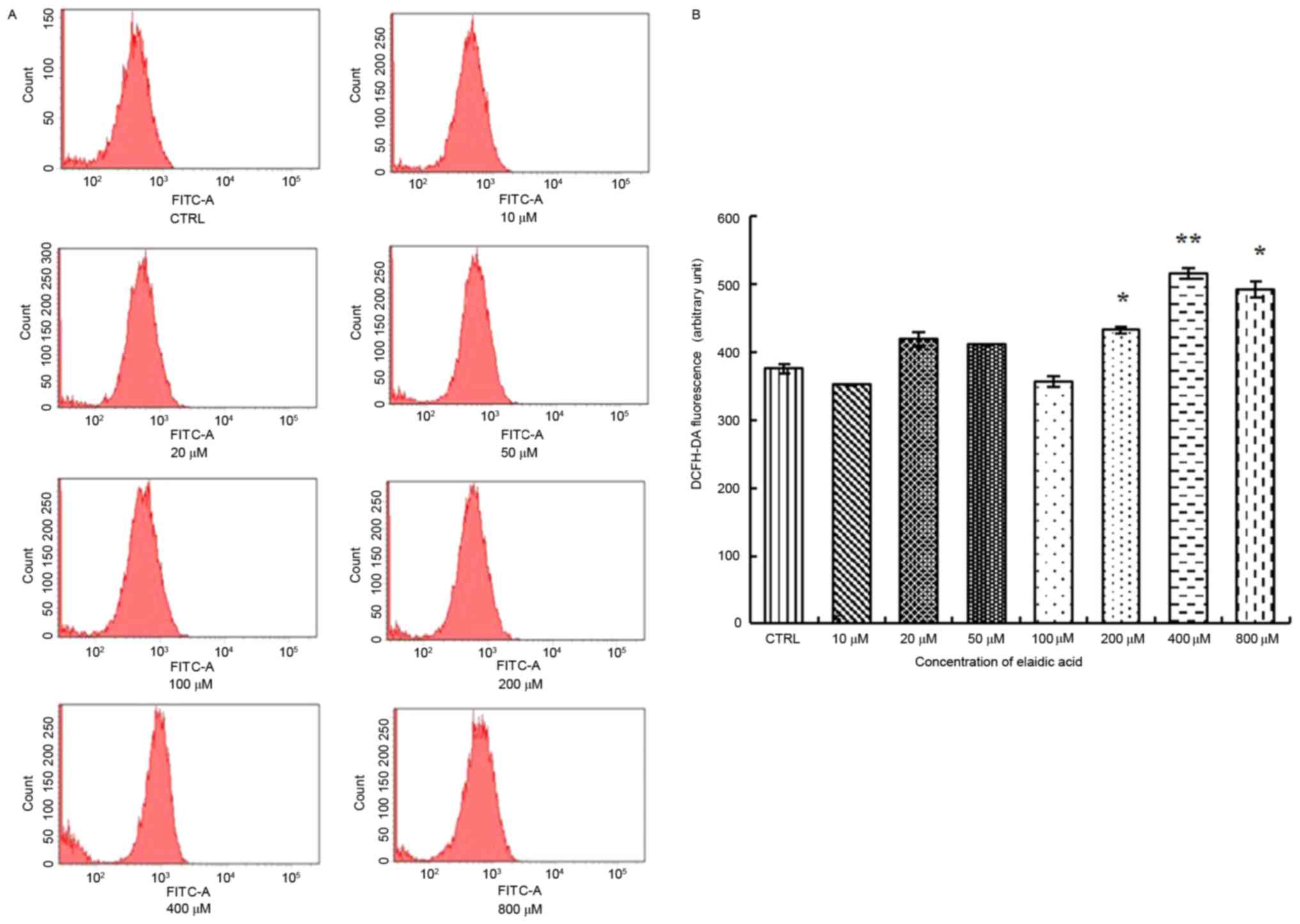 | Figure 4.Elaidic acid induces elevated ROS
release from SH-SY5Y cells. Cells were treated with Dulbecco's
modified Eagle's medium or elaidic acid (10, 20, 50, 100, 200, 400
and 800 µM) for 24 h. (A) Production of intracellular ROS was
examined using a fluorometric ROS detection kit. The fluorescent
signal was then measured using flow cytometry with an excitation
wavelength of 488 nm and an emission wavelength of 530 nm. (B)
Quantification of flow cytometry results. Data are presented as the
mean ± standard error of three replicates. *P<0.05, **P<0.01
vs. control group. DCFH-DA, 2′,7′-dichlorofluorescin diacetate;
FITC, fluorescein isothiocyanate; ROS, reactive oxygen species. |
Effects of elaidic acid on cellular
redox indicators
The MDA levels in cells treated with 100–800 µM
elaidic acid were significantly upregulated compared with in the
control group (Table I). LPO
levels were also increased following treatment with 50–800 µM
elaidic acid (Table I). Elaidic
acid only affected SOD activity in the 800 µM treatment group, in
which SOD activity was significantly reduced (P<0.05; Table II). In addition, 400 and 800 µM
elaidic acid decreased GSH-Px activity in SH-SY5Y cells (P<0.05;
Table II). In addition, decreased
GSH levels were detected in the 800 µM elaidic acid group, whereas
the levels of GSSG were increased in the 200 µM elaidic acid group
(P<0.05). However, the ratio of GSH and GSSG showed no
significant difference among the groups (P>0.05; Table III).
 | Table I.Effects of EA on MDA and LPO levels
in SH-SY5Y cells. |
Table I.
Effects of EA on MDA and LPO levels
in SH-SY5Y cells.
| EA concentration
(µM) | MDA levels (nmol/mg
prot) |
P-valuea | LPO levels (nmol/mg
prot) |
P-valuea |
|---|
| Control | 0.38±0.02 | – | 4.33±0.36 | – |
| 10 | 1.06±0.20 | 0.230 | 5.65±0.15 | 0.072 |
| 20 | 1.03±0.27 | 0.513 | 5.74±0.34 | 0.056 |
| 50 | 3.39±0.73 | 0.143 | 5.87±0.22 | 0.038 |
| 100 | 3.50±0.26 | 0.003 | 6.25±0.45 | 0.011 |
| 200 | 5.91±0.63 | 0.010 | 6.12±0.84 | 0.017 |
| 400 | 2.46±0.14 | 0.001 | 5.89±0.53 | 0.035 |
| 800 | 4.57±0.21 | 0.013 | 8.04±0.67 | <0.001 |
| Table II.Effects of EA on SOD activity and
GSH-Px levels. |
Table II.
Effects of EA on SOD activity and
GSH-Px levels.
| EA concentration
(µM) | SOD activity
(%) |
P-valuea | GSH-Px levels (U/mg
prot) |
P-valuea |
|---|
| Control | 50.0±1.62 | – | 39.74±7.87 | – |
| 10 | 41.1±2.35 | 0.203 | 24.43±4.39 | 0.066 |
| 20 | 40.3±1.93 | 0.073 | 45.33±6.31 | 0.491 |
| 50 | 37.0±3.01 | 0.093 | 40.42±3.98 | 0.934 |
| 100 | 49.1±5.23 | 1.000 | 32.65±4.07 | 0.356 |
| 200 | 48.5±5.13 | 1.000 | 47.48±4.52 | 0.314 |
| 400 | 37.6±5.45 | 0.607 | 23.03±4.80 | 0.028 |
| 800 | 26.8±1.43 | <0.001 | 23.08±5.99 | 0.035 |
| Table III.Effects of EA on GSH/GSSG ratio. |
Table III.
Effects of EA on GSH/GSSG ratio.
| EA concentration
(µM) | GSSG (µmol/l) |
P-valuea | GSH (µmol/l) |
P-valuea | GSH/GSSG ratio |
P-valuea |
|---|
| Control | 9.84±0.94 | – | 34.24±2.24 | – | 3.68±0.58 | – |
| 10 | 10.40±0.56 | 0.502 | 36.78±2.68 | 0.520 | 3.62±0.43 | 1.000 |
| 20 | 9.29±0.38 | 0.502 | 50.59±3.29 | <0.001 | 5.50±0.47 | 0.461 |
| 50 | 9.79±0.20 | 0.949 | 40.89±3.05 | 0.098 | 4.21±0.39 | 1.000 |
| 100 | 10.46±0.54 | 0.453 | 37.21±3.24 | 0.452 | 3.55±0.20 | 1.000 |
| 200 | 11.78±0.38 | 0.024 | 29.33±1.77 | 0.217 | 2.49±0.11 | 0.688 |
| 400 | 11.37±0.66 | 0.070 | 33.14±2.12 | 0.779 | 2.99±0.34 | 0.997 |
| 800 | 10.87±0.65 | 0.216 | 23.96±3.21 | 0.013 | 2.28±0.42 | 0.730 |
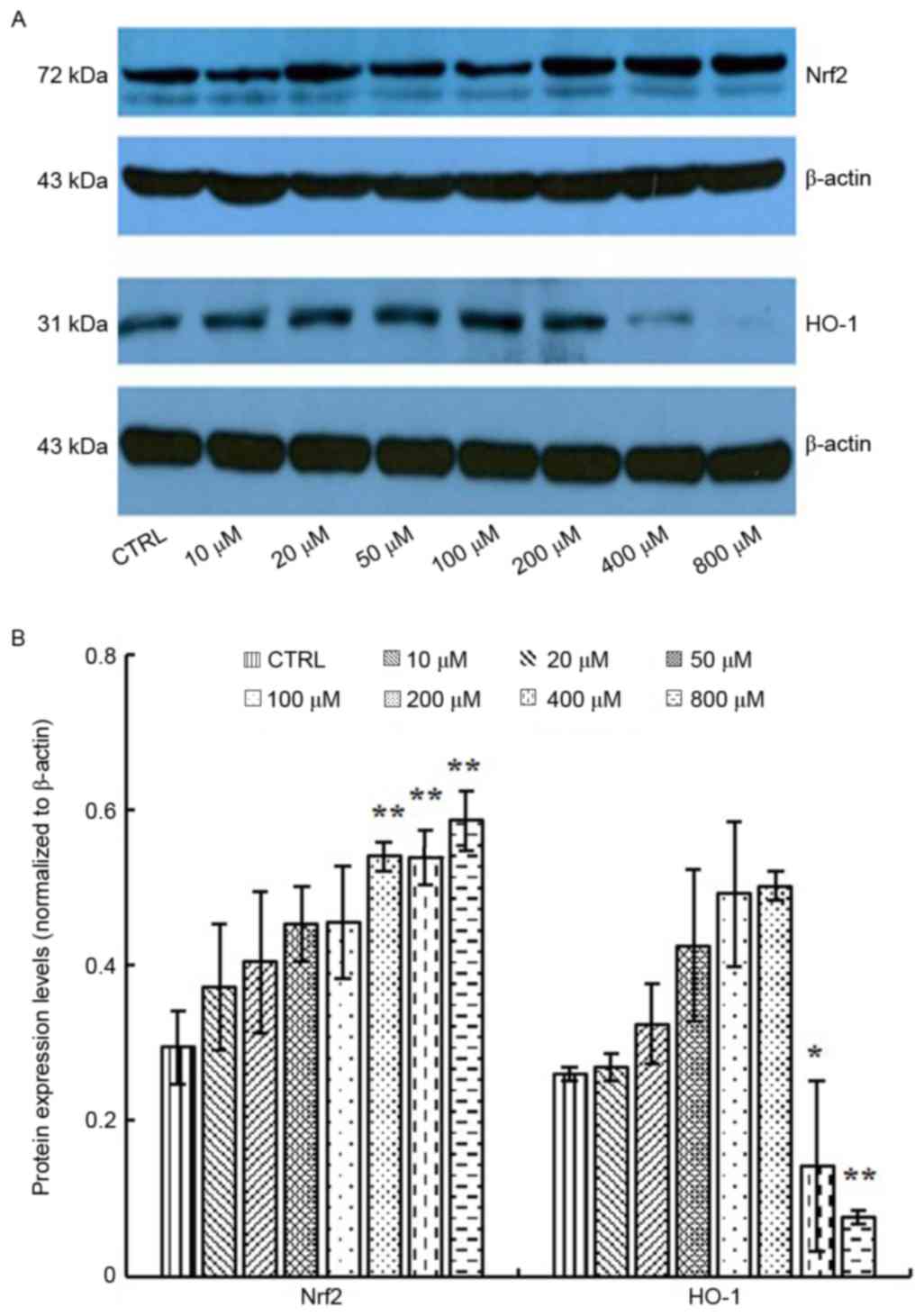
Effects of elaidic acid on expression
of antioxidative factors
Nrf2 regulates the expression of a series of
antioxidant proteins, including HO-1, which protect cells against
oxidative damage (22). In
response to oxidative stress, Nrf2 and HO-1 expression may be
upregulated (23). Therefore, the
present study measured the expression levels of Nrf2 and HO-1 in
response to elaidic acid-induced ROS accumulation. As presented in
Fig. 5A and B, the protein
expression levels of Nrf2 were upregulated following treatment with
200, 400 and 800 µM elaidic acid at (P=0.008, 0.008 and 0.002,
respectively), whereas the expression of HO-1 was significantly
inhibited by elaidic acid at 400 and 800 µM (P=0.013 and 0.001,
respectively). Although HO-1 protein expression appeared increased
following treatment with elaidic acid between 10 and 200 µM, no
statistical significance was detected. These results suggested that
other mechanisms may participate in the regulation of HO-1
expression, and the loss of HO-1-induced protection following
treatment with high concentrations of elaidic acid may contribute
to elaidic acid-induced apoptosis.
Elaidic acid induces ER stress in
SH-SY5Y cells
Production of ROS is known to be associated with ER
stress and activation of the UPR. Overactivation of the UPR has
been reported to induce apoptotic cell death and contribute to
various degenerative diseases. In the present study, the protein
expression levels of GRP78 were significantly upregulated following
treatment with 400 and 800 µM elaidic acid (P=0.019 and 0.007,
respectively; Fig. 6A and B). ATF4
protein expression was upregulated following treatment with 20–400
µM (P=0.019, 0.001, <0.001, 0.002 and 0.043, respectively;
Fig. 6A and B). However, elaidic
acid downregulated CHOP expression at the concentrations of 200,
400 and 800 µM (P=0.028, 0.016 and 0.011, respectively; Fig. 6A and B). In addition, no
significant alterations were detected in the protein expression
levels of IRE1α following treatment with elaidic acid (P>0.05;
Fig. 6A and B). During ER stress,
disulfide bond formation is dysregulated, since disulfide bond
formation in the ER lumen is highly sensitive to altered redox
balance, which in turn exacerbates ROS accumulation and oxidative
stress. However, the present study failed to detect significant
alterations in the protein expression levels of PDI and Ero1-Lα,
which are two major proteins associated with disulfide bond
formation, in cells treated with elaidic acid (P>0.05; Fig. 6C and D).
 | Figure 6.Elaidic acid induces endoplasmic
reticulum stress in SH-SY5Y cells. Cell were treated with
Dulbecco's modified Eagle's medium or elaidic acid (10, 20, 50,
100, 200, 400 and 800 µM) for 24 h. (A and B) Protein expression
levels of GRP78, IRE1α, CHOP and ATF4 were detected by western
blotting. (A) Representative blotting image. (B)
Semi-quantification of the western blotting data. (C and D) Protein
expression levels of PDI and Ero1-Lα were detected by western
blotting. (C) Representative blotting image. (D)
Semi-quantification of the western blotting data. Data are
presented as the mean ± standard error of five independent
experiments. *P<0.05, **P<0.01 vs. control group. ATF4,
activating transcription factor 4; CHOP, CCAAT/enhancer-binding
protein homologous protein; Ero1-Lα, endoplasmic
oxidoreductin-1-like protein; GRP78, glucose-regulated protein 78;
IRE1α, inositol requiring enzyme 1α; PDI, protein disulfide
isomerase. |
Discussion
In the present study, elaidic acid between 10 and
800 µM was selected to investigate its neurotoxic effects in
SH-SY5Y cells in vitro, according to the physiological dose
of elaidic acid, which is between 10 and 80 µM (24–29).
The results demonstrated that the cell viability was decreased
following treatment with elaidic acid in a dose- and time-dependent
manner, indicating the neurotoxic effects of elaidic acid.
Destruction of MMP is considered an early event in cell apoptosis.
The results of the present study demonstrated that various doses of
elaidic acid were able to decrease MMP of cells in a dose-dependent
manner, indicating that elaidic acid-induced loss of SH-SY5Y cell
viability was at least partially attributed to apoptosis. Annexin
V/PI double staining also confirmed that 100 µM elaidic acid
induced cell apoptosis. Taken together, these data indicated that
elaidic acid could induce growth inhibition and apoptosis of
SH-SY5Y neuroblastoma cells, thus suggesting a potential role for
elaidic acid in the development of neuronal loss in
neurodegenerative diseases.
Morinaga et al (17) and Cassagno et al (18) reported that elaidic acid caused
oxidative stress and ER stress in mouse hepatocytes and liver
tissues. Neurons are particularly vulnerable to oxidative stress
due to their high oxygen consumption. Oxidative stress is known to
serve an essential role in the pathogenesis of neurodegenerative
disorders. ROS can be generated in the mitochondria, ER, plasma
membrane and cytoplasm, and induces oxidative stress and ER stress.
The present study demonstrated that, at high concentrations,
elaidic acid enhanced ROS release, which may lead to cell oxidative
damage and ultimately cell apoptosis.
To limit overaccumulation of ROS in the body,
enzymatic and non-enzymatic systems exist to maintain ROS balance.
Enzymatic antioxidant defenses include SOD and GSH-Px. GSH in the
nucleus maintains the redox state of sulfhydryls of critical
proteins for DNA repair and gene expression. Under oxidative
stress, GSH-Px is a peroxide decomposition enzyme and has a
specific catalytic role in the oxide reduction reaction of GSH,
whose functions are eliminating peroxide metabolites and protecting
cell membrane structure and function. SOD converts ROS to
H2O2; therefore, SOD possesses the ability to
act as a free radical scavenging enzyme. Lipid peroxidation of
unsaturated fatty acids in the cell membrane, which is triggered by
free radicals, results in the formation of LPO; therefore, LPO
content may reflect free radical content and lipid peroxidation in
cells. A reduction in SOD activity, which in turn may trigger the
breakdown of LPO to MDA, indicates cell toxicity. The results of
the present study demonstrated that, compared with the control
group, cells treated with 800 µM elaidic acid exhibited decreased
GSH content and SOD and GSH-Px activities, whereas LPO and MDA
levels were increased. These results indicated that elaidic acid
impaired the ability of SH-SY5Y cells to scavenge ROS and in turn
resulted in the formation of LPO and its metabolic product, MDA,
thus suggesting the existence of oxidative damage in cells. In
addition, elaidic acid caused a decrease in GSH-Px, which
contributed to GSSG formation by GSH; therefore, reduced GSH but
increased GSSG levels were detected.
Under oxidative stress, Nrf2 translocates into the
nucleus of cells, where it binds with the antioxidant response
element (30). HO-1 protein
expression, which is regulated by Nrf2 (22), may be enhanced in response to
oxidative stress (23). In the
present study, upregulation of Nrf2 and HO-1 were detected in
response to low doses of elaidic acid, which indicated that Nrf2
and HO-1 exerted protective effects against ROS accumulation
induced by low concentrations of elaidic acid. However, when
SH-SY5Y cells were treated with high concentrations of elaidic
acid, HO-1 expression was downregulated, whereas Nrf2 expression
was further upregulated, indicating that other mechanisms
superseded the regulation of HO-1 by Nrf2 and that the toxic
effects of high doses of elaidic acid exhausted the protective
capacity of HO-1.
ER serves a pivotal role in the synthesis, folding,
post-translational modifications and trafficking of secretory and
membrane proteins, calcium storage and release, lipid biogenesis
and apoptosis (31,32). Physiological and pathological
stimuli may lead to disruption of ER homeostasis and result in an
accumulation of misfolded and unfolded proteins; this condition is
known as ER stress. ER stress activates three main signaling
pathways (ATF6, IRE1 and PERK) to reduce ER stress and restore
homeostasis, which is referred to as the UPR. However, if ER stress
is excessive and prolonged, these adaptive responses fail to
compensate and overactivation of the UPR leads to cell death
(33). It has previously been
reported that oxidative stress-induced ER stress may be crucial in
the regulation of cell apoptosis and may contribute to various
degenerative diseases (34). ROS
production has been associated with ER stress and activation of the
UPR. In addition, some UPR components, such as CHOP, can contribute
to oxidative stress (35).
Meanwhile, ER stress can cause mitochondrial dysfunction and
increase mitochondrial ROS production. In numerous ER stress
models, ER stress and oxidative stress accentuate each other in a
positive feedback loop, which interferes with cell function and
activates proapoptotic signaling (36).
IRE1α is a highly conserved protein in neurons and
the major neuronal UPR sensor (37,38).
Under normal physiological conditions, IRE1α is activated by
dimerization and trans-autophosphorylation upon its release from
GRP78 (39,40). The released IRE1α migrates to the
nucleus and induces the transcription of ER chaperone proteins,
including GRP78 and CHOP (41).
ATF4 is a master regulator that has a crucial role in stress
adaptation via regulation of the transcription of numerous genes,
including CHOP. In the present study, following elaidic acid
treatment, the expression levels of GRP78 were upregulated,
indicating that ER stress was induced. However, IRE1α expression
was not significantly altered, whereas the expression levels of
CHOP and ATF4 were upregulated, and then downregulated, following
treatment with various doses of elaidic acid, indicating that as
the dose of elaidic was increased, the damage to cells was
aggravated. These results suggested that the effects of elaidic
acid on ER stress were mainly mediated via activation of the
GRP78/ATF4/CHOP signaling pathway.
The ER redox state is closely associated with ER
protein-folding homeostasis. Disulfide bond formation in the ER
lumen is highly sensitive to altered redox balance, where both
reducing and oxidizing reagents disrupt protein folding and cause
ER stress. During ER stress, dysregulated disulfide bond formation
and breakage may result in ROS accumulation and the induction of
oxidative stress. Two of the major contributors to disulfide bond
formation in the ER are PDI and Ero1 (42). However, the present study did not
observe alterations in the expression of PDI and Ero1 in SH-SY5Y
cells treated with elaidic acid, indicating that elaidic acid has
no effect on the formation of disulfide bonds and protein
conformation.
In conclusion, elaidic acid may induce apoptosis of
SH-SY5Y cells via the induction of oxidative stress and ER stress,
and through activation of the GRP78/ATF4/CHOP signaling pathway.
These findings suggested a potential role for dietary TFAs in the
development of neurodegenerative disorders, providing the basis for
the development of novel strategies for prevention of
neurodegenerative disorders. Animal-based in vivo
experiments are required to confirm the conclusion of this research
in further studies.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81472982),
the Support Project of High-level Teachers in Beijing Municipal
Universities in the Period of 13th Five-year Plan (grant no.
CIT&TCD201704096) and the Scientific Research Common Program of
Beijing Municipal Commission of Education (grant no.
KM201710025007).
References
|
1
|
Forman MS, Trojanowski JQ and Lee VM:
Neurodegenerative diseases: A decade of discoveries paves the way
for therapeutic breakthroughs. Nat Med. 10:1055–1063. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brown RC, Lockwood AH and Sonawane BR:
Neurodegenerative diseases: An overview of environmental risk
factors. Environ Health Perspect. 113:1250–1256. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rozpedek W, Markiewicz L, Diehl JA, Pytel
D and Majsterek I: Unfolded protein response and PERK kinase as a
new therapeutic target in the pathogenesis of alzheimer's disease.
Curr Med Chem. 22:3169–3184. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Volgyi K, Juhász G, Kovacs Z and Penke B:
Dysfunction of endoplasmic reticulum (ER) and mitochondria (MT) in
Alzheimer's disease: The role of the ER-MT cross-talk. Curr
Alzheimer Res. 12:655–672. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Castellani R, Hirai K, Aliev G, Drew KL,
Nunomura A, Takeda A, Cash AD, Obrenovich ME, Perry G and Smith MA:
Role of mitochondrial dysfunction in Alzheimer's disease. J
Neurosci Res. 70:357–360. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Blesa J, Trigo-Damas I, Quiroga-Varela A
and Jackson-Lewis VR: Oxidative stress and Parkinson's disease.
Front Neuroanat. 9:912015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: A vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barnard ND, Bunner AE and Agarwal U:
Saturated and trans fats and dementia: A systematic review.
Neurobiol Aging. 35 Suppl 2:S65–S73. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morris MC and Tangney CC: Dietary fat
composition and dementia risk. Neurobiol Aging. 35 Suppl 2:S59–S64.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lichtenstein AH: Dietary trans fatty acids
and cardiovascular disease risk: Past and present. Curr Atheroscler
Rep. 16:4332014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nielsen L Vendel, Krogager TP, Young C,
Ferreri C, Chatgilialoglu C, Nørregaard Jensen O and Enghild JJ:
Effects of elaidic acid on lipid metabolism in HepG2 cells,
investigated by an integrated approach of lipidomics,
transcriptomics and proteomics. PLoS One. 8:e742832013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Perova NV, Metel'skaia VA and Boĭtsov SA:
Trans isomers of unsaturated fatty acids increase the risk of
atherosclerosis-related circulatory system diseases. Ter Arkh.
85:113–117. 2013.(In Russian). PubMed/NCBI
|
|
13
|
de Souza RJ, Mente A, Maroleanu A, Cozma
AI, Ha V, Kishibe T, Uleryk E, Budylowski P, Schünemann H, Beyene J
and Anand SS: Intake of saturated and trans unsaturated fatty acids
and risk of all cause mortality, cardiovascular disease, and type 2
diabetes: Systematic review and meta-analysis of observational
studies. BMJ. 351:h39782015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morris MC, Evans DA, Bienias JL, Tangney
CC, Bennett DA, Aggarwal N, Schneider J and Wilson RS: Dietary fats
and the risk of incident Alzheimer disease. Arch Neurol.
60:194–200. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Engelhart MJ, Geerlings MI, Ruitenberg A,
Van Swieten JC, Hofman A, Witteman JC and Breteler MM: Diet and
risk of dementia: Does fat matter? The rotterdam study. Neurology.
59:1915–1921. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Grimm MO, Rothhaar TL, Grösgen S, Burg VK,
Hundsdörfer B, Haupenthal VJ, Friess P, Kins S, Grimm HS and
Hartmann T: Trans fatty acids enhance amyloidogenic processing of
the Alzheimer amyloid precursor protein (APP). J Nutr Biochem.
23:1214–1223. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Morinaga M, Kon K, Saito H, Arai K, Kusama
H, Uchiyama A, Yamashina S, Ikejima K and Watanabe S: Sodium
4-phenylbutyrate prevents murine dietary steatohepatitis caused by
trans-fatty acid plus fructose. J Clin Biochem Nutr. 57:183–191.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cassagno N, Palos-Pinto A, Costet P,
Breilh D, Darmon M and Bérard AM: Low amounts of trans 18:1 fatty
acids elevate plasma triacylglycerols but not cholesterol and alter
the cellular defence to oxidative stress in mice. Br J Nutr.
94:346–352. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Starr TK, Scott PM, Marsh BM, Zhao L, Than
BL, O'Sullivan MG, Sarver AL, Dupuy AJ, Largaespada DA and Cormier
RT: A sleeping beauty transposon-mediated screen identifies murine
susceptibility genes for adenomatous polyposis coli (Apc)-dependent
intestinal tumorigenesis. Proc Natl Acad Sci USA. 108:pp.
5765–5770. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sakitani K, Hirata Y, Hikiba Y, Hayakawa
Y, Ihara S, Suzuki H, Suzuki N, Serizawa T, Kinoshita H, Sakamoto
K, et al: Inhibition of autophagy exerts anti-colon cancer effects
via apoptosis induced by p53 activation and ER stress. BMC Cancer.
15:7952015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sui C, Ma Q, Nan K, Xiao J, Suo A, Sha H
and Zhao L: hSSTR2 expression and octreotide treatment reverses
multidrug resistance of BxPC-3 human pancreatic cancer cells. Oncol
Rep. 22:1391–1396. 2009.PubMed/NCBI
|
|
22
|
Gong X, Zhang L, Jiang R, Ye M, Yin X and
Wan J: Anti-inflammatory effects of mangiferin on sepsis-induced
lung injury in mice via up-regulation of heme oxygenase-1. J Nutr
Biochem. 24:1173–1181. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takahashi T, Shimizu H, Morimatsu H,
Maeshima K, Inoue K, Akagi R, Matsumi M, Katayama H and Morita K:
Heme Oxygenase-1 is an essential cytoprotective component in
oxidative tissue injury induced by hemorrhagic shock. J Clin
Biochem Nutr. 44:28–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
van Poppel G: Intake of trans fatty acids
in western Europe: The TRANSFAIR study. Lancet. 351:10991998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jokela H, Kalela A, Lilja M, Salmi M,
Lehtimäki T, Kunnas T, Teisala K, Punnonen R and Nikkari ST:
Sequentially combined estradiol valerate plus levonorgestrel
therapy decreases 18:1 trans-fatty acid content of plasma lipids in
healthy postmenopausal women. Gynecol Endocrinol. 21:360–365. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Svahn JC, Feldl F, Räihä NC, Koletzko B
and Axelsson IE: Different quantities and quality of fat in milk
products given to young children: Effects on long chain
polyunsaturated fatty acids and trans fatty acids in plasma. Acta
Paediatr. 91:20–29. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Abraham RA, Bahl VK, Parshad R, Seenu V,
Roy A, Golandaz S, Dorairaj P and Ramakrishnan L: Content of trans
fatty acids in human cheek epithelium: Comparison with serum and
adipose tissue. Biomed Res Int. 2013:2761742013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun Q, Ma J, Campos H, Hankinson SE,
Manson JE, Stampfer MJ, Rexrode KM, Willett WC and Hu FB: A
prospective study of trans fatty acids in erythrocytes and risk of
coronary heart disease. Circulation. 115:1858–1865. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Burdge GC, Tricon S, Morgan R, Kliem KE,
Childs C, Jones E, Russell JJ, Grimble RF, Williams CM, Yaqoob P
and Calder PC: Incorporation of cis-9, trans-11 conjugated linoleic
acid and vaccenic acid (trans-11 18:1) into plasma and leucocyte
lipids in healthy men consuming dairy products naturally enriched
in these fatty acids. Br J Nutr. 94:237–243. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Itoh K, Tong KI and Yamamoto M: Molecular
mechanism activating Nrf2-Keap1 pathway in regulation of adaptive
response to electrophiles. Free Radic Biol Med. 36:1208–1213. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Henderson MJ, Baldwin HA, Werley CA,
Boccardo S, Whitaker LR, Yan X, Holt GT, Schreiter ER, Looger LL,
Cohen AE, et al: A low affinity GCaMP3 variant (GCaMPer) for
imaging the endoplasmic reticulum calcium store. PLoS One.
10:e01392732015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schwarz DS and Blower MD: The endoplasmic
reticulum: Structure, function and response to cellular signaling.
Cell Mol Life Sci. 73:79–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bakhshi J, Weinstein L, Poksay KS,
Nishinaga B, Bredesen DE and Rao RV: Coupling endoplasmic reticulum
stress to the cell death program in mouse melanoma cells: Effect of
curcumin. Apoptosis. 13:904–914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu D, Perez RE, Rezaiekhaligh MH, Bourdi M
and Truog WE: Knockdown of ERp57 increases BiP/GRP78 induction and
protects against hyperoxia and tunicamycin-induced apoptosis. Am J
Physiol Lung Cell Mol Physiol. 297:L44–L51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feng J, Chen X and Sun X, Wang F and Sun
X: Expression of endoplasmic reticulum stress markers GRP78 and
CHOP induced by oxidative stress in blue light-mediated damage of
A2E-containing retinal pigment epithelium cells. Ophthalmic Res.
52:224–233. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ferreiro E, Baldeiras I, Ferreira IL,
Costa RO, Rego AC, Pereira CF and Oliveira CR: Mitochondrial- and
endoplasmic reticulum-associated oxidative stress in Alzheimer's
disease: From pathogenesis to biomarkers. Int J Cell Biol.
2012:7352062012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen Y and Brandizzi F: AtIRE1A/AtIRE1B
and AGB1 independently control two essential unfolded protein
response pathways in Arabidopsis. Plant J. 69:266–277. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nagashima Y, Mishiba K, Suzuki E, Shimada
Y, Iwata Y and Koizumi N: Arabidopsis IRE1 catalyses unconventional
splicing of bZIP60 mRNA to produce the active transcription factor.
Sci Rep. 1:292011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim I, Xu W and Reed JC: Cell death and
endoplasmic reticulum stress: Disease relevance and therapeutic
opportunities. Nat Rev Drug Discov. 7:1013–1030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bertolotti A, Zhang Y, Hendershot LM,
Harding HP and Ron D: Dynamic interaction of BiP and ER stress
transducers in the unfolded-protein response. Nat Cell Biol.
2:326–332. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Benham AM, van Lith M, Sitia R and
Braakman I: Ero1-PDI interactions, the response to redox flux and
the implications for disulfide bond formation in the mammalian
endoplasmic reticulum. Philos Trans R Soc Lond B Biol Sci.
368:201104032013. View Article : Google Scholar : PubMed/NCBI
|