Introduction
Psoriasis vulgaris is one of the most proliferative
inflammatory skin diseases in humans. Histologically, psoriasis is
characterized by epidermal thickening as a result of abnormal
proliferation, impaired maturation of keratinocytes, leucocyte
infiltration, and new blood vessel formation (angiogenesis)
(1,2). Keratinocytes of psoriatic skin can
reach the surface of the skin from the basal layer in just 6–8
days, compared to 30 days in normal skin (3). Studies have found the growth and
proliferation of psoriatic keratinocytes to be intrinsically
dysregulated by several mechanisms. First, phosphatase and tensin
homolog (PTEN) expression is downregulated in psoriatic lesions by
overactivation of the phosphoinositide-3 kinase/protein kinase-B
(PI3K/Akt) pathway. It is also correlated with the
hyperproliferation of psoriatic keratinocytes (4). Interestingly, the PI3K/Akt pathway is
tightly linked with the extracellular signaling-regulated kinase
(ERK)/mitogen-activated protein kinase (MAPK) pathway (5). Second, the final downstream target of
the ERK/MAPK pathway is a positive cell cycle regulator, cyclin D1,
which is overexpressed in psoriatic skin lesions (6–8).
Third, FOS-like antigen 1 (Fra-1; a proto-oncogene) is highly
expressed in tissues affected by psoriasis (9). Fra-1 also promotes the growth of
HaCaT keratinocyte cell lines and inhibits the apoptosis of cells
in vitro (9). Although it
seems highly probable that psoriatic skin lesions can transform
into malignancies by the accumulation of specific molecular events,
skin cancer does not usually occur in psoriatic tissues.
Psoriatic skin is usually abnormally thickened
compared to normal skin due to hyperproliferation of the epidermis.
In fact, cell proliferation is accurately regulated by cell cycle
regulatory proteins. Cyclin D1 and cyclin E are the key positive
regulatory proteins involved in the progression of the G1/S
transition phases (10–12). Therefore, the upregulation of
cyclin D1 and cyclin E makes cells rapidly transition into G1/S
phases, resulting in overall cell proliferation. Thus, the
hyperproliferation of the epidermis is correlated with the
upregulation of cyclin D1 or cyclin E in the epidermis. Under the
hyperproliferative state, p53 and pRb play important roles in
preventing cancer formation by inducing apoptosis or by the
senescence of hyperproliferated cells (13,14).
The expression of cell cycle regulatory proteins such as cyclin D
and cyclin E and tumor suppressor proteins such as p53 and pRb have
not been well investigated in psoriasis.
In this study, to elucidate why psoriasis does not
transform into malignancy under keratinocyte hyperproliferation, we
compared the expression levels of p53, pRb, and cell cycle
regulatory proteins in psoriatic skin lesions with those in
squamous cell carcinoma (SCC) tissues.s
Materials and methods
Materials
Antibodies against cell cycle regulatory proteins
[cyclin D1 (no. sc-717), cyclin E (no. sc-481), p16 (no. sc-468),
β-actin (no. sc-1616), and, Ki-67 (sc-7846)] were obtained from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-p53 (no.
48818) and anti-pRb (no. 554136) antibodies were purchased from
Cell Signaling (Beverly, MA, USA) and PharMingen (BD Biosciences,
CA, USA), respectively.
Patients
Twenty patients with psoriasis were enrolled for the
study. The criteria for entry in the study were the manifestation
of well-demarcated, erythematous, scaly psoriatic plaques on the
trunk and extremities. Study subjects did not use any systemic
anti-psoriatic treatments for 2 weeks before skin biopsy. Informed
consent was obtained from all subjects under protocols approved by
the Investigational Review Board of Dongsan Hospital of Keimyung
University (IRB-09-28).
Immunoblot analysis
Tissues were prepared in lysis buffer [10 mM Tris
(pH 7.4), 5 mM EDTA, 130 mM NaCl, 1% Triton X-100, 50 mM
phenylmethylsulfonyl fluoride (PMSF), 10 mM pepstatin A, 20 mM
leupeptin, 50 mM bestatin, 100 mM benzamidine, 10 mM sodium
fluoride, and 1 mM sodium orthovanadate]. The protein
concentrations of extracts were estimated with the Bradford reagent
(Bio-Rad, Hercules, CA, USA) using bovine serum albumin as the
standard. Equal amounts of protein (40 µg/lane) were resolved by
6.5–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
and transferred onto a nitrocellulose membrane. The membrane was
incubated with the respective specific antibodies (anti-pRb,
anti-cyclin D1, anti-cyclin E, and anti-p16). The membrane was
continuously incubated with appropriate secondary antibodies
coupled to horseradish peroxidase and developed in ECL western
blotting detection reagents (Amersham Pharmacia Biotech,
Piscataway, NJ, USA).
Immunohistochemistry
Formalin-fixed and paraffin-embedded tissue
specimens were cut on a microtome into 5-µm thick sections. The
sections were deparaffinized in xylenes and hydrated in alcohols of
decreasing concentration. To visualize the antigen, the sections
were heated in citrate buffer (pH 6.0). After cooling to room
temperature, 5-min incubation was performed with
H2O2 to block endogenous peroxidase activity.
After sections were rinsed in PBS (pH 7.4) for 15 min and blocked
with the antibody diluent (Golden Bridge International, Mukilteo,
WA, USA) for 5 min, they were incubated with specific primary
antibodies. Next, the anti-biotinyl antibody was used in the
reaction, and the streptavidin-peroxidase complex (LASB®
+ System-HRP; Dako, Carpintera, CA, USA) was applied. The
antigen-antibody complex was visualized by DAB chromogen.
Immunohistochemical scoring and
statistical analysis
The positively stained cells were counted (per
mm2) for total epidermal immunostaining in the same ×100
magnification field area. Data from the microscopic analysis were
expressed as the mean ± standard error. The Kruskal-Wallis one-way
analysis of variance (ANOVA) and Mann-Whitney U test were
used to compare the cell count/mm2 as determined by
immunostaining in the epidermal layers of the psoriasis group and
the normal epidermis group. P-values of less than 0.05 were
considered statistically significant. The statistical analysis was
performed by using SPSS statistical software version 21.0 (SPSS,
Chicago, IL, USA).
Results
Expression of cyclin D1, cyclin E,
pRb, p53, and p16 in psoriasis
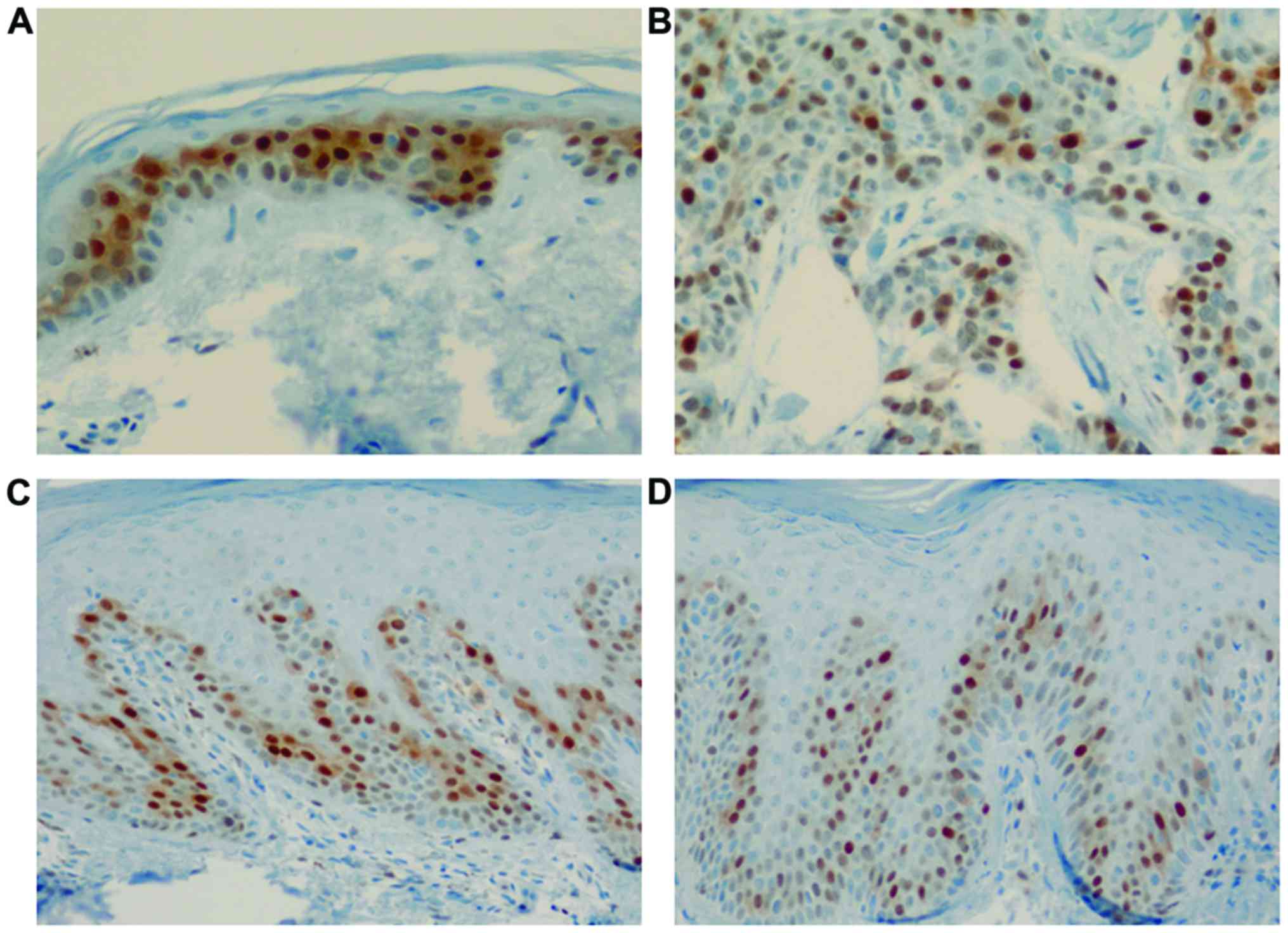
Cyclin D1 expression was clearly observed in the
basal and suprabasal layers of the normal epidermis, showing
positively stained nuclei. Cyclin D1 immunoreactivity in the normal
epidermis was observed at regular intervals in the basal layer.
Interestingly, psoriasis lesions showed a strong intensity of
positive nuclear staining for cyclin D1 among several normally
stained nuclei in the basal layer (Fig. 1). Furthermore, the number of cells
stained with cyclin D1 differed psoriasis group (111.2±65.2
cells/mm2; acute, 53±30.1 cells/mm2 and
chronic, 157.8±41.6 cells/mm2) compared with that of the
normal skin (81.5±10.6 cells/mm2) and SCC groups
(399±14.1 cells/mm2) (χ2=10.35, P=0.016,
Kruskal-Wallis test). A post hoc Mann-Whitney U test showed
the chronic psoriasis group to have stronger cyclin D1 expression
than that in than the acute psoriasis group (Table I).
 | Table I.Expressions differences of cyclin D1,
pRb, p53 and p16 between groups by Kruskal-Wallis test and post hoc
test (Mann-Whitney U test). |
Table I.
Expressions differences of cyclin D1,
pRb, p53 and p16 between groups by Kruskal-Wallis test and post hoc
test (Mann-Whitney U test).
|
| Mean rank |
|
|
|
|---|
|
|
|
|
|
|
|---|
| Immunostaining | Control | Acute psoriasis | Chronic
psoriasis | Squamous cell
carcinoma | χ2 | P-value | Post hoc test |
|---|
| Cyclin D1 | 4.5 | 3.0 | 9.0 | 12.5 | 10.352 | 0.016 | 3>2 |
| pRb | 1.5 | 7.0 | 12.17 | 4.5 | 10.098 | 0.012 | 3>2 |
| p53 | 1.5 | 5.2 | 9.8 | 13.5 | 11.301 | 0.010 | 3>2 |
| Ki-67 | 1.5 | 5.6 | 11.50 | 10.00 | 9.740 | 0.021 | 3>2 |
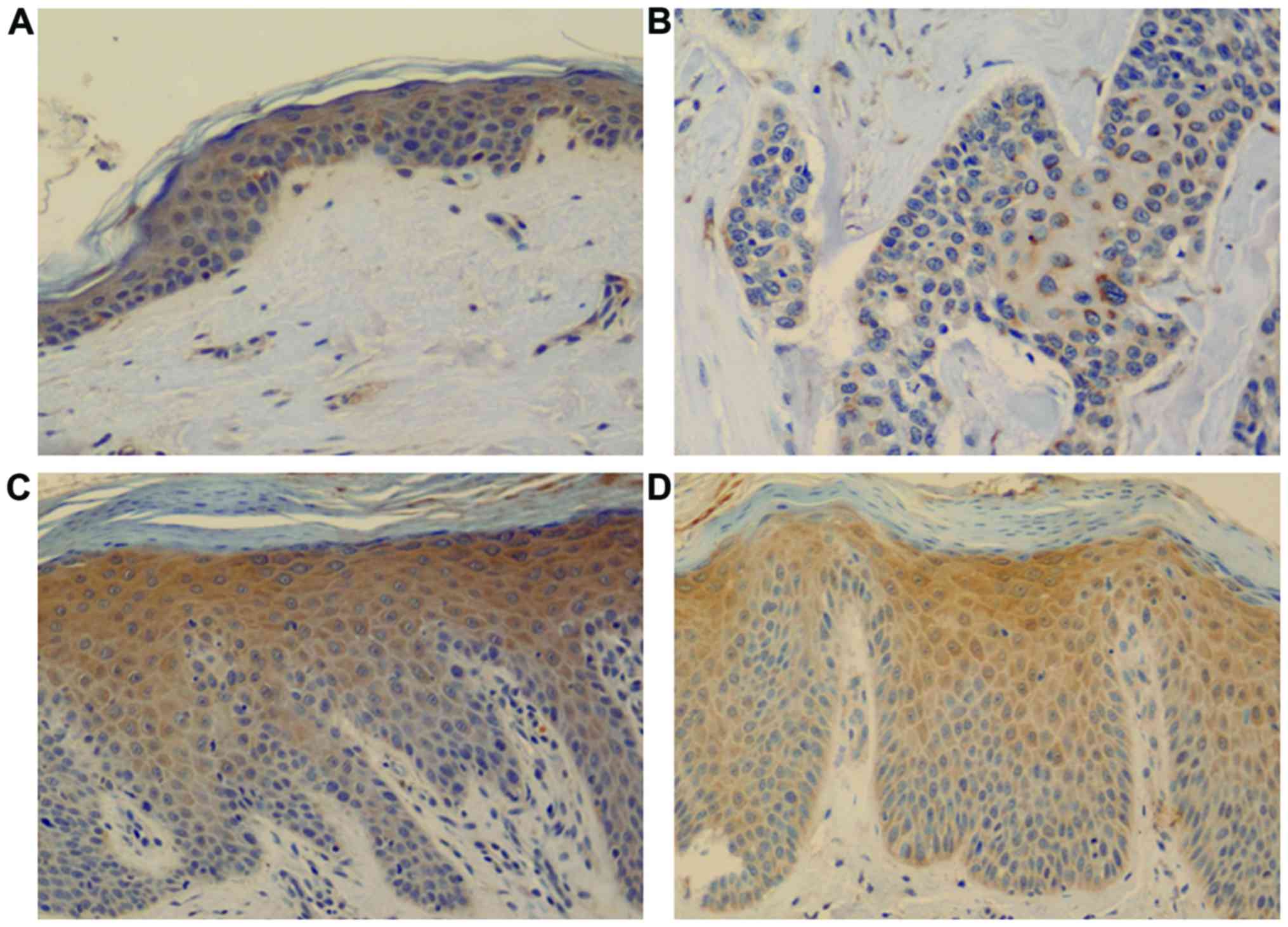
Cyclin E expression was observed throughout the
normal epidermis, showing positively stained nuclei and cytoplasm.
In psoriasis, positively stained nuclei and cytoplasm were observed
in the epidermis, especially above the suprabasal layer till the
granular layer; this was not observed in normal epidermis or SCC
(Fig. 2).
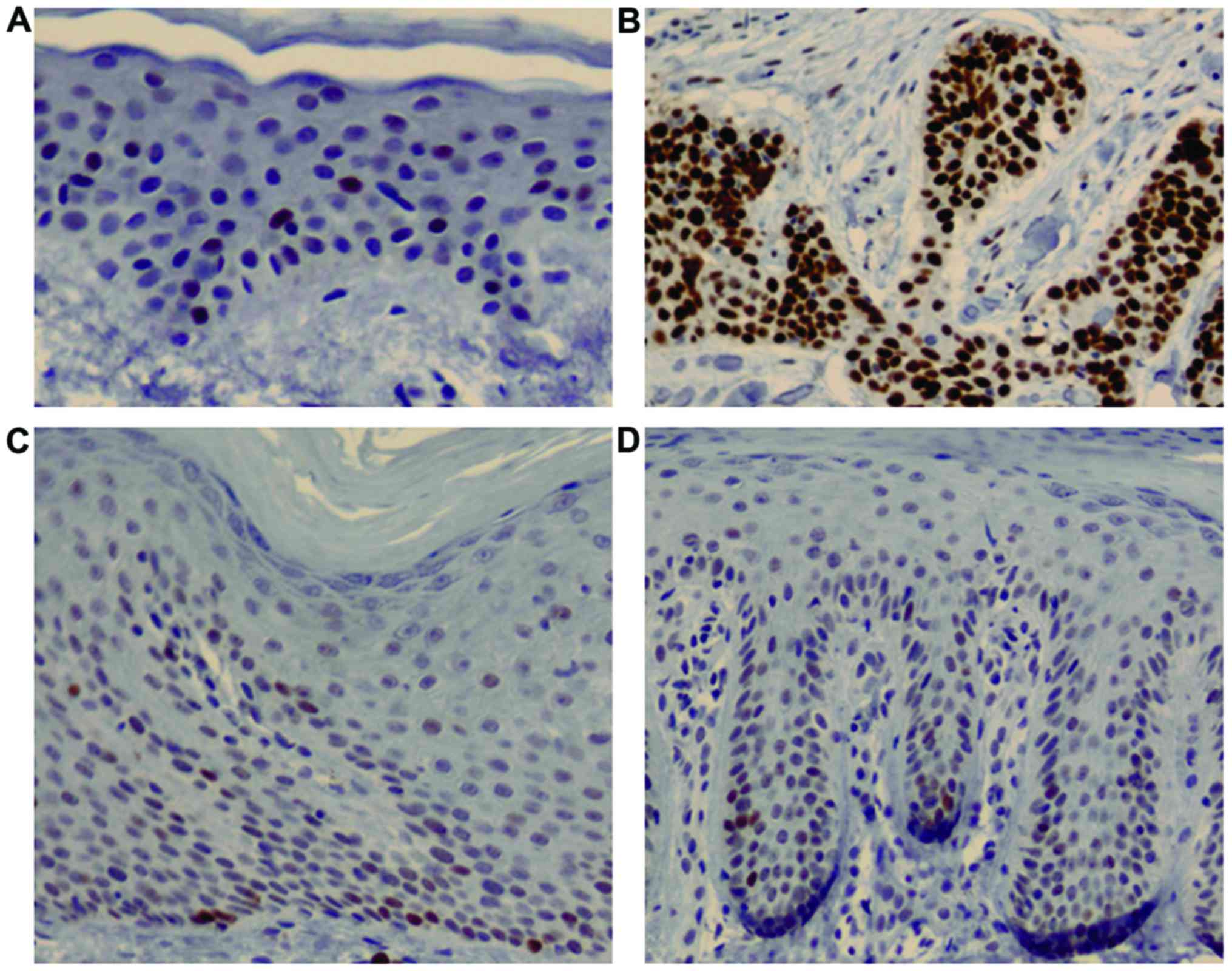
Total basal layer cell counts for pRb expression
were found to be higher in the psoriasis group (316.8±212.2
cells/mm2; acute, 141.6+113.4 cells/mm2 and
chronic, 462.8+153.2 cells/mm2) compared with the normal
(2.5±0.7 cells/mm2) and SCC groups (58.5±14.8
cells/mm2) (χ2=10.91, P=0.012, Kruskal-Wallis
test) (Fig. 3). However, in the
post hoc test, significant differences were found only between the
acute and chronic psoriasis groups (P=0.16) (Table I).
In addition, total basal layer cell counts for p53WT
expression were found to be higher in the psoriasis group
(25.1+15.8 cells/mm2; acute, 13.61+4.9
cells/mm2 and chronic, 36.6+14.5 cells/mm2)
compared with the normal epidermis (5±1.4 cells/mm2).
Strong p53 WT/MUT expression was observed in SCC (1063.5±328.8
cells/mm2) (Fig.
4).
The expression of p16 tumor suppressor protein in
the epidermis was very weak in the normal and psoriasis groups
compared with that in the SCC group (Fig. 5).
Ki-67 immunoreactive cells were found to be higher
in the psoriasis group (452.9±242.5 cells/mm2; acute,
267.2+109.6 cells/mm2 and chronic, 607.6+211.6
cells/mm2) compared with the normal epidermis group
(38.5±4.9 cells/mm2). Ki-67 expression levels in
psoriasis were similar to those in SCC (498±134.3
cells/mm2) (Fig.
6).
However, the Kruskal-Wallis one-way ANOVA and
Mann-Whitney U test for the cell count/mm2 in all
immunostaining results in the epidermal layer among psoriasis,
control, and SCC groups showed no statistically significant
differences. The only significant difference was between the acute
and chronic psoriasis groups (Table
I).
Expression of cyclin D1, cyclin E,
pRb, and Ki-67 in treated psoriatic skin lesions
We investigated whether the expression patterns of
cell cycle regulatory proteins in psoriasis were normalized in
treated psoriatic skin lesions. Immunohistochemical analysis
revealed that the expression of cyclin D1, cyclin E, pRb, and Ki-67
decreased in treated psoriatic skin, and was similar with that of
normal epidermis (Fig. 7).
Consistent with immunohistochemical data, western blot analysis
also showed that the expression levels of cyclin D1, cyclin E, pRb,
and Ki-67 were markedly decreased in treated psoriatic lesions
(Fig. 8).
Discussion
Psoriasis is a complex epidermal disorder
characterized by keratinocyte hyperproliferation and abnormal
differentiation owing to intricate interactions with the immune
system (15,16). Even though hyperproliferation and
abnormal differentiation are the hallmarks of psoriasis, epidermal
carcinogenesis is not observed in psoriatic skin lesions. To
elucidate why psoriasis does not transform into malignancy under
hyperproliferation of keratinocytes, we compared the expression
levels of p53, pRb, and cell cycle regulatory proteins in psoriatic
skin lesions to those of SCC tissues.
The upregulation of cyclin D1 is well-established in
hyperproliferative tissues (10,11).
In this study, we found that cyclin D1 was overexpressed in thick
plaques of chronic psoriasis and SCC tissues. When compared to the
normal epidermis and acute psoriatic epidermis, cyclin D1 in
chronic psoriatic epidermis had a different pattern in terms of
localization and expression level. We found that cyclin D1
immunoreactivity in chronic psoriasis was demonstrated as strong
positive staining among several normal stained nuclei and at
regular intervals in the basal layer. This finding is in agreement
with previous studies, which also found increased cyclin D1
expression in psoriasis (8,17,18).
However, further studies are warranted to elucidate whether the
cells in basal layers that were strongly positively stained for
cyclin D1 are psoriatic stem cells or transient amplifying cells,
which are important in the hyperproliferation of the epidermis.
Furthermore, p16 tumor suppressor protein showed
weak activity in the psoriasis group compared to that in the SCC
group. The increased expression level of p16 inhibits cell cycle
progression and induces cell senescence and the prevention of
aberrant cell proliferation (19,20).
Our data suggested that p16 may not be involved in the process of
hyperproliferation to inhibit cell cycle progression, such as
processes underlying SCC. These findings are in agreement with
those of previous studies, indicating a low basal level of p16 in
psoriasis (17). The levels of
cyclin D1, cyclin E, and Ki-67 decreased significantly after
psoriasis treatment in this study, which is also in accordance with
previous studies (18). From these
findings, we conclude that altered expression of cyclin D1 and
cyclin E may contribute to the proliferation of psoriatic lesions.
The phosphorylation status of pRb is specifically regulated by the
activities of cyclin D1, cyclin E, and p16 (21,22).
In this study, chronic psoriatic skin lesions showed increased
expression of cyclin D1 and cyclin E and low expression of p16
compared to those of the normal epidermis. According to these data,
pRb phosphorylation in psoriasis may be increased by the activity
of cyclin D1 and cyclin E. In this study, psoriatic epidermis
showed a higher expression of pRb than that of normal
epidermis.
In terms of expression patterns of cyclin D1, cyclin
E, and p16, psoriatic skin lesions may easily transform into
malignancies under long inflammatory processes, such as actinic
keratosis (23). Our data
suggested that increased expression of p53 in chronic psoriatic
skin lesions could inhibit malignant transformation and create
homeostasis between normal and hyperproliferative epidermal
conditions. The involvement of the hyperproliferative state in
psoriatic skin lesions is observed in the basal layer of the
epidermis, which is supported by a high labeling index with Ki-67.
Our data showed a similar distribution of p53- and Ki67-positive
cells, which supports the hypothesis that increased proliferation
enhances wild-type p53 protein synthesis for the prevention of
malignant transformation (24).
Indeed, p53 mutation gives a chance for the epidermis to undergo
malignant transformation under constant inflammatory stress, e.g.,
in actinic keratosis and SCC (25). Fortunately, p53 mutation is not
observed in psoriatic skin lesions. Normal p53 plays an important
role in preventing psoriatic carcinogenesis.
It has been still largely unknown which mechanisms
contribute to transit acute form psoriatic papules into chronic
plaques form psoriasis. Our data indicate that up-regulation of
cyclin D1 and cyclin E plays important roles to drive maturation
and hyperproliferation of epidermis, eventually forming of thick
plaques in psoriasis.
In conclusion, we demonstrated that altered
expression of cyclin D1 and cyclin E may be involved in cell cycle
progression in psoriatic epidermis, whereas p53 may be playing an
important role for the prevention of malignant transformation under
a hyperproliferative state in psoriasis.
Acknowledgements
The present study was supported by the Bisa Research
Grant of Keimyung University in 2012.
References
|
1
|
Deng Y, Chang C and Lu Q: The inflammatory
response in psoriasis: A comprehensive review. Clin Rev Allergy
Immunol. 50:377–389. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nestle FO, Kaplan DH and Barker J:
Psoriasis. N Engl J Med. 361:496–509. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weinstein GD, McCullough JL and Ross P:
Cell proliferation in normal epidermis. J Invest Dermatol.
82:623–628. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang T, Lin X, Meng X and Lin M:
Phosphoinositide-3 kinase/protein kinase-B/mammalian target of
rapamycin pathway in psoriasis pathogenesis. A potential
therapeutic target? Acta Derm Venereol. 94:371–379. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Faes S and Dormond O: PI3K and AKT:
Unfaithful partners in cancer. Int J Mol Sci. 16:21138–21152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sticozzi C, Belmonte G, Cervellati F, Di
Capua A, Maioli E, Cappelli A, Giordani A, Biava M, Anzini M and
Valacchi G: Antiproliferative effect of two novel COX-2 inhibitors
on human keratinocytes. Eur J Pharm Sci. 49:133–141. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bhatt KV, Spofford LS, Aram G, McMullen M,
Pumiglia K and Aplin AE: Adhesion control of cyclin D1 and p27Kip1
levels is deregulated in melanoma cells through BRAF-MEK-ERK
signaling. Oncogene. 24:3459–3471. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sezer E, Böer-Auer A, Cetin E, Tokat F,
Durmaz E, Sahin S and Ince U: Diagnostic utility of Ki-67 and
Cyclin D1 immunostaining in differentiation of psoriasis vs. other
psoriasiform dermatitis. Dermatol Pract Concept. 5:7–13. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu W, Li J, Su J, Li J, Li J, Deng B, Shi
Q, Zhou Y and Chen X: FOS-like antigen1 is highly expressed in
human psoriasis tissues and promotes the growth of HaCaT cells in
vitro. Mol Med Rep. 10:2489–2494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lavoie JN, Rivard N, L'Allemain G and
Pouysségur J: A temporal and biochemical link between growth
factor-activated MAP kinases, cyclin D1 induction and cell cycle
entry. Prog Cell Cycle Res. 2:49–58. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Palmero I and Peters G: Perturbation of
cell cycle regulators in human cancer. Cancer Surv. 27:351–367.
1996.PubMed/NCBI
|
|
12
|
Sheaff RJ, Groudine M, Gordon M, Roberts
JM and Clurman BE: Cyclin E-CDK2 is a regulator of p27Kip1. Genes
Dev. 11:1464–1478. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Flatt PM, Tang LJ, Scatena CD, Szak ST and
Pietenpol JA: p53 regulation of G(2) checkpoint is retinoblastoma
protein dependent. Mol Cell Biol. 12:4210–4213. 2000. View Article : Google Scholar
|
|
14
|
Gire V and Dulic V: Senescence from G2
arrest, revisited. Cell Cycle. 14:297–304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rashmi R, Rak KS and Basavaraj KH: A
comprehensive review of biomarkers in psoriasis. Clin Exp Dermatol.
34:658–663. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schaller G and Meyer-Hermann M: A modeling
approach towards epidermal homoeostasis control. J Theor Biol.
247:554–573. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Abou EL-Ela M, Nagui N, Mahgoub D,
El-Eishi N, Fawzy M, El-Tawdy A, Abdel Hay R and Rashed L:
Expression of cyclin D1 and p16 in psoriasis before and after
phototherapy. Clin Exp Dermatol. 35:781–785. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miracco C, Pellegrino M, Flori ML, Vatti
R, Materno M and Andreassi L: Cyclin D1, B and A expression and
cell turnover in psoriatic skin lesions before and after
cyclosporin treatment. Br J Dermatol. 143:950–956. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
LaPak KM and Burd CE: The molecular
balancing act of p16(INK4a) in cancer and aging. Mol Cancer Res.
12:167–183. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim WY and Sharpless NE: The regulation of
INK4/ARF in cancer and aging. Cell. 127:265–275. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lukas J, Parry D, Aagaard L, Mann DJ,
Bartkova J, Strauss M, Peters G and Bartek J:
Retinoblastoma-protein-dependent cell-cycle inhibition by the
tumour suppressor p16. Nature. 375:503–506. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sherr CJ: The Pezcoller lecture: Cancer
cell cycles revisited. Cancer Res. 60:3689–3695. 2000.PubMed/NCBI
|
|
23
|
Brasanac D, Stojkovic-Filipovic J, Bosic
M, Tomanovic N and Manojlovic-Gacic E: Expression of G1/S-cyclins
and cyclin-dependent kinase inhibitors in actinic keratosis and
squamous cell carcinoma. J Cutan Pathol. 43:200–210. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yazici AC, Karabulut AA, Ozen O, Ekşioğlu
M and Ustün H: Expression of p53 in lesions and unaffected skin of
patients with plaque-type and guttate psoriasis: A quantitative
comparative study. J Dermatol. 34:367–374. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Florence ME, Massuda JY, Soares TC,
Stelini RF, Poppe LM, Bröcker EB, Metze K, Cintra ML and de Souza
EM: p53 immunoexpression in stepwise progression of cutaneous
squamous cell carcinoma and correlation with angiogenesis and
cellular proliferation. Pathol Res Pract. 211:782–788. 2015.
View Article : Google Scholar : PubMed/NCBI
|