Introduction
Stroke is the loss of brain function due to a
disturbance in the blood supply to the brain, also referred to as a
cerebrovascular accident, or, ‘brain attack’ (1,2).
Stroke is the second leading cause of mortality worldwide, and
another primary manifestation of stroke is disability. According to
statistics, up to 30% of stroke survivors are permanently disabled,
significantly influencing quality of life and safety (3–6).
Therefore, the search for novel ways of treating and preventing
stroke is of primary concern.
Of all types of stroke, cerebral ischemic injury is
of great importance, however its pathophysiological process is
complex. Of all types of pathophysiology in neuronal cells,
enhanced extent of neuronal apoptosis and levels of reactive oxygen
species (ROS) are considered very significant (7). In addition, inflammation is important
in the course of cerebral ischemia injury (8). Studies have demonstrated that
cerebral ischemia involves an increase in the release of
proinflammatory cytokines [interleukin (IL)-6, IL-1β and tumor
necrosis factor (TNF)-α], leading to aggravated brain injury
(9). The modulation of
anti-oxidation, anti-inflammation, and anti-apoptosis is considered
a crucial and effective therapeutic strategy in the treatment of
cerebral ischemia injury.
Myricetin (3,30,40,5,50,7-hexahydroxyflavone) is a
natural herbal flavonoid that exists abundantly in various plant
sources, including medicinal herbs, vegetables, fruits, berries and
tea leaves. Myricetin has diverse pharmacological activities,
including anti-apoptotic function, anti-inflammatory effects, and
antioxidant properties (10–12).
Studies have revealed that myricetin acts against
ischemia/reperfusion-induced acute injury through the activation of
cyclooxygenase-2, cytochrome P450 and p38 mitogen-activated protein
kinase (p38 MAPK) signaling pathways, and the inhibition of fatty
acid synthase and 6-phosphogluconate dehydrogenase signaling
pathways in the isolated heart of rats (13). Studies have additionally
demonstrated that myricetin attenuates brain injury in a rat model
of cerebral ischemia via activation of the nuclear factor,
erythroid 2 like 2 pathway and reducing oxidative stress (14). Despite these research advances, the
anti-inflammatory effect of myricetin on ischemic cerebral injury
remains unclear. Therefore, the aim of the present study was to
investigate whether myricetin could prevent cerebral ischemic
injury and to identify the potential mechanisms involved.
Materials and methods
Test compounds, chemicals and
reagents
Myricetin (purity ≥98%) was purchased from Chengdu
Technology and Market Co., Ltd., (Chengdu, China). Rat superoxide
dismutase (SOD), rat malondiadehyde (MDA), rat glutathione (GSH),
rat glutathione disulfide (GSSG), rat IL-1β, rat IL-6 and rat TNF-α
kits was purchased from Enzyme-linked Biotechnology Co., Ltd.
(Shanghai, China). In Situ Cell Death Detection kit, POD was
purchased from Roche Diagnostics GmbH (Mannheim, Germany).
Anti-protein kinase B (AKT) antibody produced in rabbit,
anti-phospho-AKT (pSer129) antibody produced in rabbit,
anti-nuclear factor (NF)-κB p65 antibody produced in rabbit,
anti-phospho-NF-κB p65 (pSer281) antibody produced in rabbit,
anti-p38 MAPK antibody produced in rabbit, and anti-p38 MAPK
phospho (pT180/pY182) antibody produced in rabbit were obtained
from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
2,3,5-triphenyltetrazolium chloride staining (TTC) and the other
chemicals and reagents were purchased from Sigma-Aldrich (Merck
KGaA).
Animals
Adult Sprague-Dawley rats (230–280 g) were purchased
from Jinan Pengyue Experimental Animal Breeding Co. Ltd. (Zhangqiu,
China: http://www.cndss.net/company-jinanpengyueshiyandongwufanyu923.html).
All experimental procedures were approved by the Institutional
Animal Care and Use Committee of the National Institute
Pharmaceutical Education and Research (Linyi, China).
Cerebral ischemia model
Transient focal cerebral ischemia was induced in the
rats via the left middle cerebral artery (MCA) occlusion technique,
as previously described (7,9). The
experiments were performed as follows: Rats were anesthetized by
intraperitoneal injection of 10% chloral hydrate, following a
ventral midline neck incision, the right common carotid artery
(CCA), the external carotid artery, and the internal carotid artery
(ICA) were carefully exposed and dissected away from adjacent
nerves. Microvascular aneurysm clips were applied to the right CCA
and the ICA. A filament rounded by gentle heating near a flame was
introduced through a small incision into the CCA and then into the
ICA. The suture was inserted 18–20 mm from the carotid bifurcation,
which was verified by mild resistance, to occlude the MCA. The
sham-operated rats underwent the same procedure, except that the
filament was not inserted. During the surgery, body temperature was
kept at 37°C with a heating pad and a warm light. Following
incision closure, the rats were returned to standard housing
conditions and allowed free access to food and water.
Drug treatment and surgical
operation
The 48 rats were randomly divided into five groups
for administration of myricetin by oral gavage: Sham group (n=12),
model group (n=12), low (L)-myricetin group (1 mg/kg myricetin +
model, n=6), medium (M)-myricetin group (5 mg/kg myricetin + model,
n=6), and high (H)-myricetin group (25 mg/kg myricetin + model,
n=12). The sham group received normal saline intra-gastrically
(i.g.) once per day for one week, and the sham-operation (the
sham-operated rats underwent the same procedure, except that the
filament was not inserted) was performed 1 h following the last
administration of normal saline. The model group received normal
saline i.g. once per day for one week, and the cerebral ischemia
model operation was performed 1 h following the last administration
of normal saline. The three myricetin groups received myricetin
i.g. once per day for one week, and the operation was performed 1 h
following the last administration of myricetin. Note: TTC staining
required the whole brain, so the brain tissue could not be tested
for other indicators (except for evaluation of neurological
deficit) following TTC staining. TTC staining and evaluation of
neurological deficit occurred only in the L-myricetin and
M-myricetin groups, so the number of animals in these two groups
was 6. However, in addition to the TTC staining and evaluation of
neurological deficit, there were other tests [including terminal
deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)
staining, western blot assay, and assay of oxidative stress and
inflammatory factors] in the other three groups (sham, model and
H-myricetin groups), therefore the number of animals in these three
groups was 12.
Evaluation of neurological
deficit
Following 1 day (24 h) of ischemia, evaluation of
the neurological deficit scores was performed by an examiner
blinded to the experimental groups. Neurological findings were
scored on a five-point scale: No neurological deficits=0; failure
to extend the right forepaw fully=1; circling to the right=2;
falling to the right=3; failure to walk spontaneously with
depressed levels of consciousness=4. The greater the neurological
deficit score, the more severe the impairment (15). A total of 5 groups (sham group,
model group, L-myricetin group, M-myricetin group and H-myricetin
group) were used for the analysis of neurological deficit.
Determination of cerebral infarct
areas
The rats were euthanized 24 h following permanent
middle cerebral artery occlusion (pMCAO). The brains (whole brain)
were quickly removed, sliced, fixed, stained, and photographed as
described previously (9). The
specific experimental operations were as follows: Rats were
sacrificed and brains were quickly removed and placed at −20°C for
5 min. Following 5 min of freezing, the brains were sliced into
three 2 mm-thick coronal sections. The sections were incubated in
1% TTC at 37°C for 30 min, and the sections were turned over in 15
min. Normal brain tissues were dyed bright red, and the unstained
area (white area) was defined as the ischemic lesion. Infarct areas
were measured using Photoshop CS6 image analysis software (Adobe
Systems Europe, Ltd., Maidenhead, UK). To compensate for brain
edema, the corrected volume was calculated as follows: Percentage
hemisphere lesion volume (% HLV)=[total infarct volume-(left
hemisphere volume-right hemisphere volume)]/right hemisphere volume
×100%.
Assay of oxidative stress and
inflammatory factors
The brains were harvested 24 h following the
operation and stored at −70°C for later analysis. The homogenates
extracted from the cortex of the ischemic brain hemispheres were
analyzed for biochemical parameters. According to the
manufacturers' protocol, the cortexes of the ischemic brain
hemispheres were homogenized in 10% homogenate solution, and the
supernatant was collected for biochemical marker analyses.
The levels of SOD activity, MDA, GSG/GSSH ratio,
IL-1β, IL-6 and TNF-α levels in brain tissue were determined using
a rat SOD kit (cat. no. ml540172), rat malondialdehyde (MDA) kit
(cat. no. ml016824), rat T-GSH/HSSG kit (cat. no. ml022385), rat
interleukin 1β (IL-1βa) kit (cat. no. ml037361), rat interleukin 6
(IL-6) kit (cat. no. ml102828) and rat TNF-α kit (cat. no.
ml002859) following the manufacturer's protocol (Enzyme-linked
Biotechnology Co., Ltd.). Briefly, the standards and sample
diluents were added and incubated for 30 min at 37°C, followed by
washing 5 times, and then horseradish peroxidase-conjugate reagent
was added and incubated for 30 min at 37°C. The samples were washed
5 times, and Chromogen solutions A and B were added, incubated for
another 30 min at 37°C, and then the stop solution was added and
the absorbance was measured at the corresponding wavelength ranges
within 15 min, and calculated.
TUNEL
The degree of brain cell apoptosis was determined
using TUNEL assay as previously described (9). Briefly, the brain tissue was fixed
with 10% neutral-buffered formalin for 48 h at room temperature
(about 25°C). Then the brain tissue was embedded in paraffin (cat.
no. YA0015; Beijing Solarbio Science and Technology Co., Ltd.,
Beijing, China), following deparaffinization and gradient ethanol
rehydration the sections were treated with proteinase K (10 mM) for
15 min at 37°C, and the slides were immersed in the TUNEL reaction
mixture for 60 min at 37°C in a humidified atmosphere in the dark.
Next, the slides were incubated in a converter-POD (50 µl) for 30
min at 37°C, resulting in the characteristic blue nuclear staining,
and were then analyzed using an Eclipse Ti2 inverted microscope
(Nikon Corporation, Tokyo, Japan). To evaluate the apoptosis index
of TUNEL-stained brain tissues, the TUNEL index (%) was calculated
as the ratio of the number of TUNEL-positive cells divided by the
total number of cells. For each sample, the average was calculated
from the counting of eight randomly selected areas of TUNEL-stained
slices at ×400 magnification.
Determination of protein levels via
western blotting assay
The frozen brain tissues (cortex) were ground with a
mortar and pestle, and then were homogenized with total protein
extraction lysis buffer (Beijing Solarbio Science and Technology
Co., Ltd). The protein concentration was determined using a Bio-Rad
protein assay (Bio-Rad Laboratories Inc., Hercules, CA, USA). The
lysates (30 µg) were separated by electrophoresis on 10% sodium
dodecyl sulfate-polyacrylamide gels (SDS-PAGE), then transferred to
nitrocellulose membranes (GE Healthcare, Chicago, IL, USA), and
blocked at room temperature for 2 h with 5% non-fat milk in
Tris-buffered saline with Tween (TBST). Membranes were incubated
with an antibody against rabbit anti-AKT (cat. no. SAB4300574),
rabbit anti-phospho-AKT (pSer129; cat. no. SAB4300259),
rabbit anti-NF-κB p65 (cat. no. SAB4502610), rabbit
anti-phospho-NF-κB p65 (pSer281; Cat. no. SAB4301496), rabbit
anti-p38 MAPK (cat. no. SAB4500491) and rabbit anti-p38 MAPK
phospho (pT180/pY182; cat. no. SAB5600057) all at dilution:
1:10,000 in 5% milk/TBST at 4°C overnight (15 h). The membrane was
then washed with TBST and incubated with horseradish
peroxidase-conjugated antibody (mouse anti-rabbit IgG cat. no.
93702; 1:5,000; Cell Signaling Technology Inc., Danvers, MA, USA)
at room temperature (25°C) for 1 h. The blots were developed with
an enhanced chemiluminescence detection kit (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and were exposed on Kodak
radiographic film. The immunoblots were visualized with an EC3
Imaging System, and analyzed with VisionWorks®LS
Analysis Software (UVP, LLC, Phoenix, AZ, USA).
Ethics statement
All experimental protocols were approved by the
Institutional Animal Care and Use Committee of Lin Yi Central
Hospital (Linyi, China). The methods were carried out in accordance
with the approved guidelines.
Statistical analysis
Data are presented as the mean ± standard deviation
from at least six independent experiments. Statistical differences
were determined using analysis of variance followed by Bonferroni
correction. The analyses were performed using SPSS version 22.0
software (IBM SPSS, Armonk, NY, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Myricetin decreases brain infarct area
and neurological deficit scores
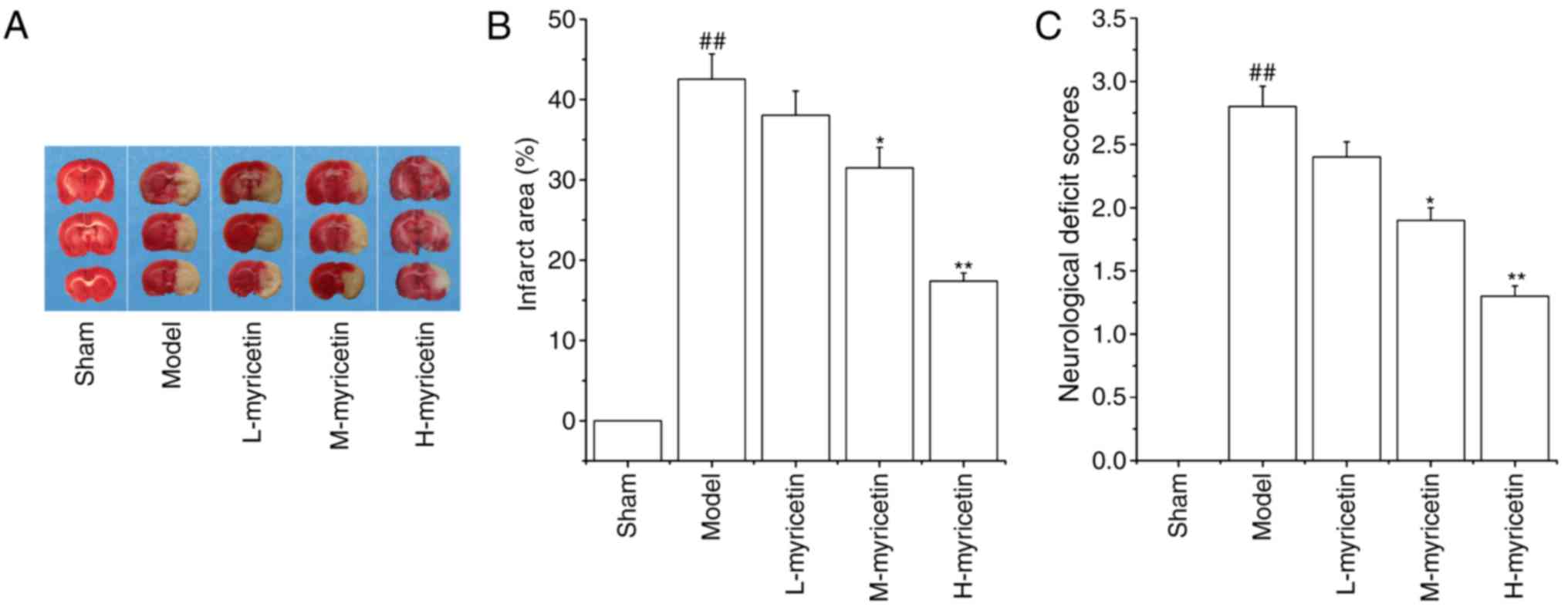
The area of brain infarction was measured with TTC
staining. As presented in Fig. 1A and
B, no infarct area was observed in the sham group. When
compared with the sham group, the model group demonstrated a marked
increase in brain infarct area (P<0.01). Pretreatment with
M-myricetin and H-myricetin significantly decreased the brain
infarct size induced by ischemia (P<0.05 and P<0.01). As
presented in Fig. 1C, pMCAO
resulted in a significant increase in the neurological deficit
score of the model group (P<0.01), and a significant decrease
was then observed in the M-myricetin and H-myricetin groups
(P<0.05 and P<0.01). The results indicated that myricetin
protected against ischemic brain injury, and that the
neuroprotective effect of H-myricetin was increased compared with
M-myricetin. Considering the significant neuroprotective effects
observed with myricetin at 25 mg/kg body weight (H-myricetin), this
concentration was used for the subsequent studies.
Myricetin treatment alleviates
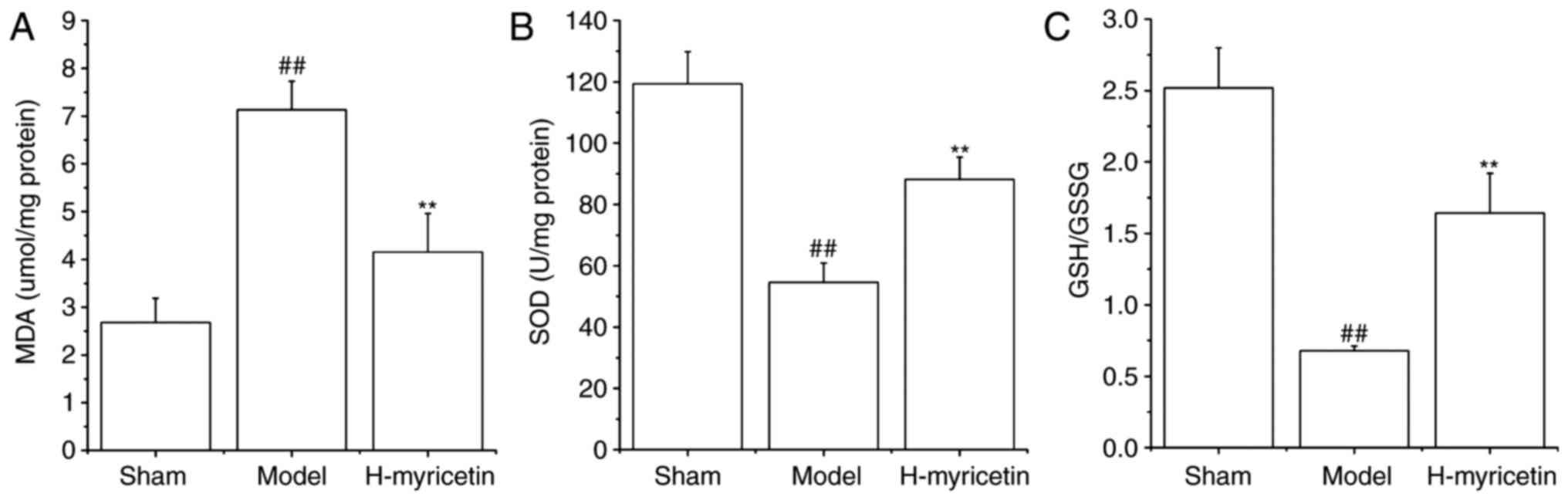
oxidative stress in ischemic brain tissue
Previous studies have indicated that oxidative
stress results in ischemic cerebral injury. Therefore, the GSH/GSSG
ratio, MDA contents, and the SOD activity in the brain tissue were
detected. As presented in Fig. 2,
compared with sham group, the MDA content increased significantly,
whereas the SOD activity and GSH/GSSG ratio decreased significantly
in the model group (P<0.01). However, treatment with H-myricetin
significantly reversed these alterations (P<0.01). Decreased MDA
content and increased GSH/GSSG ratio and SOD activity were observed
following treatment with H-myricetin.
Myricetin treatment alleviates
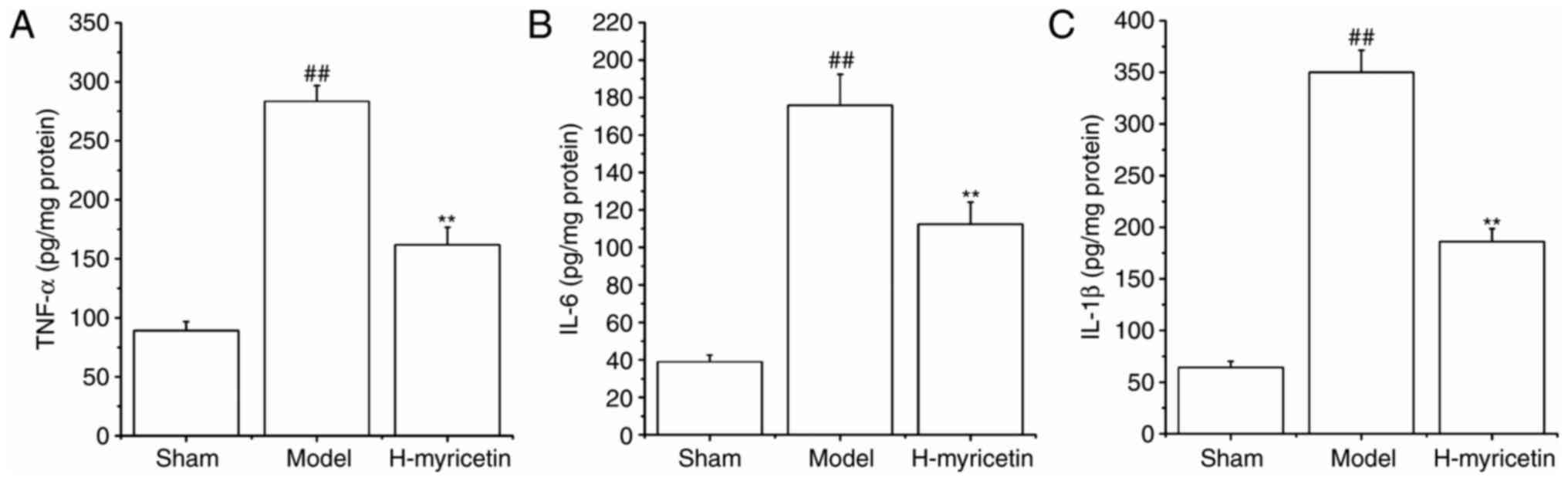
inflammation in ischemic brain tissue
It has previously been demonstrated that the
inflammatory response is critical in ischemic cerebral injury, and
that myricetin has an anti-inflammatory effect (13), therefore the present study detected
the effects of myricetin on levels of inflammatory cytokines
(IL-1β, TNF-α and IL-6) in brain tissue. As presented in Fig. 3, compared with the sham group, the
levels of IL-1β, IL-6 and TNF-α increased significantly in the
model group (P<0.01). Treatment with H-myricetin significantly
reversed these alterations in levels of inflammatory factors.
Levels of inflammatory cytokines were significantly decreased in
the H-myricetin group (P<0.01) compared with the model
group.
Myricetin suppresses apoptosis in
ischemic brain tissue
As presented in Fig.
4, there was almost no apoptosis in the sham group. pMCAO
induced an increase in apoptosis in the model group (P<0.01,
Apoptosis index: 65.94±4.89). Compared with the model group,
treatment with H-myricetin substantially reduced the
ischemia-induced neuronal apoptosis (P<0.01, apoptosis index:
38.79±3.12).
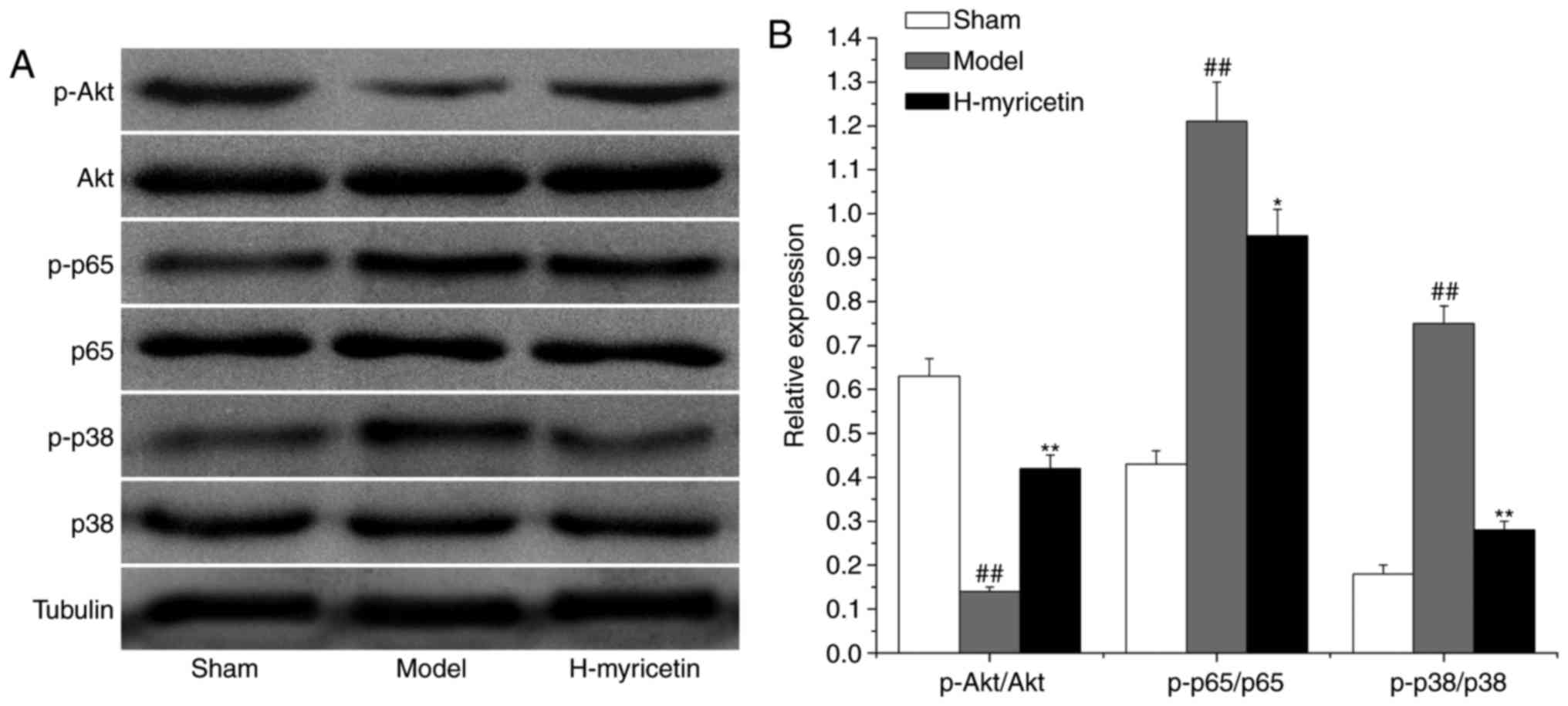
Effect of myricetin treatment on
proteins associated with oxidative stress, inflammation and
apoptosis in the ischemic brain tissue
To study the possible mechanisms of myricetin's
neuroprotective effects, the expression of proteins associated with
apoptosis, inflammation, and oxidative stress (AKT, NF-κB/p65 and
p38 MAPK, respectively) were measured via western blot analysis. As
presented in Fig. 5, compared with
the sham group, a decreased level of phosphorylated AKT and
increased levels of phosphorylated p38 MAPK and NF-κB/p65 were
observed in the model group (P<0.01). Compared with the model
group, treatment with H-myricetin significantly increased the
relative level of phosphorylated AKT and decreased the levels of
phosphorylated p38 MAPK and NF-κB/p65.
Discussion
The results of the present study demonstrated that
myricetin treatment markedly attenuated ischemia-induced cerebral
injury, considerably abated the ischemia-induced brain injury, and
reduced inflammatory response, apoptosis, and oxidative stress in
the brain tissue. The neuroprotective effect of myricetin may be
associated with the activation of the AKT pathway and the
inactivation of the NF-κB/p65 and p38 MAPK pathways.
Ischemia-induced cerebral injury leads to cerebral
infarction and, ultimately, brain dysfunction (16,17).
In the present study, it was observed that ischemia exacerbated
neurological deficits and increased cerebral infarction. A previous
study demonstrated that myricetin protects against ischemic
cerebral injury via antioxidant properties (14). It was observed that treatment with
myricetin attenuated cerebral infarction and the neurological
deficits induced by ischemia. Additionally, treatment with
myricetin significantly reduced the content of MDA (a critical
component of reduced nicotinamide adenine dinucleotide phosphate
oxidase), and reversed the ischemia-induced reduction of total
antioxidant capacity (SOD activity), in addition to the GSH/GSSG
ratio. Therefore, these findings suggest that myricetin decreased
cerebral ischemia injury via the regulation of oxidative
stress.
The inflammatory reaction is additionally involved
in ischemia-induced cerebral injury (18). Inflammatory cell infiltration is an
early step in the process of cerebral ischemic injury, and the
activation of NF-κB/p65 is a primary pathway that modulates
cerebral inflammation (19,20).
Downregulation of the NF-κB pathway reduces the release of
pro-inflammatory mediators (intercellular adhesion molecule-1,
IL-1β, IL-6, and IL-8), therefore reducing the cerebral ischemic
injury in rats (21). A previous
study revealed that myricetin exhibits a potent anti-inflammatory
effect (22). The present study
demonstrated that treatment with myricetin markedly inhibited the
ischemia-induced increased levels of proinflammatory factors and
activities of cytokines, including IL-1β, TNF-α, IL-6 and
NF-κB/p-p65. The findings suggested that myricetin is a promising
natural product that may be administered to counteract the
ischemia-induced cerebral inflammatory response.
Cell apoptosis exhibits a key role in the
development of cerebral infarction and brain dysfunction following
ischemia (23). Consequently,
inhibiting oxidative stress is considered a viable way of treating
ischemia-induced cerebral injury. The results of the present study
demonstrated that ischemia significantly increased neuronal
apoptosis. It was observed that treatment with myricetin
significantly reduced neuronal apoptosis. This finding suggested
that myricetin exhibits a neuroprotective role, which may be via
anti-apoptosis.
Various signaling pathways are associated with
neuroprotective effects, including NF-κB, p38 MAPK and AKT
(24,25). AKT exhibits a key role in
strengthening cell survival through the inhibition of
caspase-activated apoptosis (26).
Studies have demonstrated that increasing phosphorylation of AKT
inhibits the activation of glycogen synthase kinase (GSK)-3β,
resulting in inhibition of caspase-activated apoptosis. A previous
study additionally demonstrated that the AKT inhibitor (MK2206)
suppresses the phosphorylation of AKT, followed by the activation
of GSK-3β, which increases apoptosis (27). p38 MAPK is also important in
response to ischemic cerebral injury, and is involved in various
physiological processes, specifically, oxidative stress injury,
inflammatory response injury and neuronal apoptosis (28,29).
Various studies have demonstrated that inhibition of p38 MAPK
reduces ischemia-induced cerebral injury (30,31).
The results of the present study indicated that treatment with
myricetin following cerebral ischemic injury led to inhibition of
p38 MAPK and activation of AKT signaling pathways. Therefore,
myricetin may protect against ischemia-induced cerebral injury,
possibly through the modulation of multiple signaling pathways.
In accordance with the results of a previous study
(14), the present study
demonstrated that myricetin reduced the oxidative stress in brain
ischemia injury. Additionally, the present study revealed that
myricetin reduced the inflammatory response in ischemic brain
injury tissue. Furthermore, it was indicated that the effects of
myricetin in decreasing cerebral ischemia injury may be associated
with inhibition of p38 MAPK and NF-κB pathway, and activation of
AKT signaling pathways.
In conclusion, the results from the present study
suggested that treatment with myricetin decreased the size of
cerebral infarction, inhibited apoptosis, reduced inflammation and
decreased oxidative stress. Additionally, it was observed that
these effects may be associated with inhibition of p38 MAPK and
activation of AKT pathways in the ischemia-injured brain.
Acknowledgements
The present study (grant no. 20150102516) was
supported by Lin Yi Central Hospital.
References
|
1
|
Brill AK, Rösti R, Hefti JP, Bassetti C,
Gugger M and Ott SR: Adaptive servo-ventilation as treatment of
persistent central sleep apnea in post-acute ischemic stroke
patients. Sleep Med. 15:1309–1313. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jovanovic A, Stolic RV, Rasic DV,
Markovic-Jovanovic SR and Peric VM: Stroke and diabetic
ketoacidosis-some diagnostic and therapeutic considerations. Vasc
Health Risk Manag. 10:201–204. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lozano R, Naghavi M, Foreman K, Lim S,
Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et
al: Global and regional mortality from 235 causes of death for 20
age groups in 1990 and 2010: A systematic analysis for the Global
Burden of Disease Study 2010. Lancet. 380:2095–2128. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Murray CJ, Vos T, Lozano R, Naghavi M,
Flaxman AD, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S,
et al: Disability-adjusted life years (DALYs) for 291 diseases and
injuries in 21 regions, 1990–2010: A systematic analysis for the
Global Burden of Disease Study 2010. Lancet. 380:2197–2223. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Writing Group Members, ; Mozaffarian D,
Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de
Ferranti S, Després JP, et al: Executive summary: Heart disease and
stroke statistics-2016 update: A report from the American Heart
Association. Circulation. 133:447–454. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Strong K, Mathers C and Bonita R:
Preventing stroke: Saving lives around the world. Lancet Neurol.
6:182–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tewari A, Mahendru V, Sinha A and Bilotta
F: Antioxidants: The new frontier for translational research in
cerebroprotection. J Anaesthesiol Clin Pharmacol. 30:160–171. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liang LJ, Yang JM and Jin XC: Cocktail
treatment, a promising strategy to treat acute cerebral ischemic
stroke. Med Gas Res. 6:33–38. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang W, Ma X, Han J, Zhou M, Ren H, Pan Q,
Zheng C and Zheng Q: Neuroprotective effect of scutellarin on
ischemic cerebral injury by down-regulating the expression of
angiotensin-converting enzyme and AT1 receptor. PLoS One.
11:e01461972016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kang KA, Wang ZH, Zhang R, Piao MJ, Kim
KC, Kang SS, Kim YW, Lee J, Park D and Hyun JW: Myricetin protects
cells against oxidative stress-induced apoptosis via regulation of
PI3K/Akt and MAPK signaling pathways. Int J Mol Sci. 11:4348–4360.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Majid M, Khan MR, Shah NA, UI Haq I,
Farooq MA, Ullah S, Sharif A, Zahra Z, Younis T and Sajid M:
Studies on phytochemical, antioxidant, anti-inflammatory and
analgesic activities of Euphorbia dracunculoides. BMC Complement
Altern Med. 15:3492015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mendes V, Vilaça R, de Freitas V, Ferreira
PM, Mateus N and Costa V: Effect of myricetin, pyrogallol, and
phloroglucinol on yeast resistance to oxidative stress. Oxid Med
Cell Longev. 2015:7825042015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qiu Y, Cong N, Liang M, Wang Y and Wang J:
Systems pharmacology dissection of the protective effect of
myricetin against acute ischemia/reperfusion-induced myocardial
injury in isolated rat heart. Cardiovasc Toxicol. 17:277–286. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu S, Yue Y, Peng A, Zhang L, Xiang J, Cao
X, Ding H and Yin S: Myricetin ameliorates brain injury and
neurological deficits via Nrf2 activation after experimental stroke
in middle-aged rats. Food Funct. 7:2624–2634. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livnat A, Barbiro-Michaely E and Mayevsky
A: Mitochondrial function and cerebral blood flow variable
responses to middle cerebral artery occlusion. J Neurosci Methods.
188:76–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ahn HC, Yoo KY, Hwang IK, Cho JH, Lee CH,
Choi JH, Li H, Cho BR, Kim YM and Won MH: Ischemia-related changes
in naive and mutant forms of ubiquitin and neuroprotective effects
of ubiquitin in the hippocampus following experimental transient
ischemic damage. Exp Neurol. 220:120–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiang Y, Zhao H, Wang J, Zhang L, Liu A
and Chen Y: Inflammatory mechanisms involved in brain injury
following cardiac arrest and cardiopulmonary resuscitation. Biomed
Rep. 5:11–17. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen YF, Wang YW, Huang WS, Lee MM, Wood
WG, Leung YM and Tsai HY: Trans-Cinnamaldehyde, An essential oil in
cinnamon powder, ameliorates cerebral ischemia-induced brain injury
via inhibition of neuroinflammation through attenuation of iNOS,
COX-2 expression and NFκ-B signaling pathway. Neuromolecular Med.
18:322–333. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang YS, Li YX, Zhao P, Wang HB, Zhou R,
Hao YJ, Wang J, Wang SJ, Du J, Ma L, et al: Anti-inflammation
effects of oxysophoridine on cerebral ischemia-reperfusion injury
in mice. Inflammation. 38:2259–2268. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu S, Zhong A, Ma H, Li D, Hu Y, Xu Y and
Zhang J: Neuroprotective effect of salvianolic acid B against
cerebral ischemic injury in rats via the CD40/NF-κB pathway
associated with suppression of platelets activation and
neuroinflammation. Brain Res. 1661:37–48. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chanput W, Krueyos N and Ritthiruangdej P:
Anti-oxidative assays as markers for anti-inflammatory activity of
flavonoids. Int Immunopharmacol. 40:170–175. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen YF, Wu KJ, Huang WS, Hsieh YW, Wang
YW, Tsai HY and Lee MM: Neuroprotection of Gueichih-Fuling-Wan on
cerebral ischemia/reperfusion injury in streptozotocin-induced
hyperglycemic rats via the inhibition of the cellular apoptosis
pathway and neuroinflammation. Biomedicine (Taipei). 6:212016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang S, Shao SY, Song XY, Xia CY, Yang
YN, Zhang PC and Chen NH: Protective effects of Forsythia suspense
extract with antioxidant and anti-inflammatory properties in a
model of rotenone induced neurotoxicity. Neurotoxicology. 52:72–83.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu L, Bi W and Lu D, Zhang C, Shu X and
Lu D: Luteolin inhibits SH-SY5Y cell apoptosis through suppression
of the nuclear transcription factor-κB, mitogen-activated protein
kinase and protein kinase B pathways in
lipopolysaccharide-stimulated cocultured BV2 cells. Exp Ther Med.
7:1065–1070. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fu P, Wu Q, Hu J, Li T and Gao F: Baclofen
protects primary rat retinal ganglion cells from chemical
hypoxia-induced apoptosis through the Akt and PERK pathways. Front
Cell Neurosci. 10:2552016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ying Y, Zhu H, Liang Z, Ma X and Li S:
GLP1 protects cardiomyocytes from palmitate-induced apoptosis via
Akt/GSK3b/b-catenin pathway. J Mol Endocrinol. 55:245–262. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qi Z, Qi S, Gui L, Shen L and Feng Z:
Daphnetin protects oxidative stress-induced neuronal apoptosis via
regulation of MAPK signaling and HSP70 expression. Oncol Lett.
12:1959–1964. 2016.PubMed/NCBI
|
|
29
|
Suchal K, Malik S, Gamad N, Malhotra RK,
Goyal SN, Chaudhary U, Bhatia J, Ojha S and Arya DS: Kaempferol
attenuates myocardial ischemic injury via inhibition of MAPK
signaling pathway in experimental model of myocardial
ischemia-reperfusion injury. Oxid Med Cell Longev.
2016:75807312016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Apostolatos A, Song S, Acosta S, Peart M,
Watson JE, Bickford P, Cooper DR and Patel NA: Insulin promotes
neuronal survival via the alternatively spliced protein kinase CδII
isoform. J Biol Chem. 287:9299–9310. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cheng CY, Lin JG, Tang NY, Kao ST and
Hsieh CL: Electroacupuncture at different frequencies (5Hz and
25Hz) ameliorates cerebral ischemia-reperfusion injury in rats:
Possible involvement of p38 MAPK-mediated anti-apoptotic signaling
pathways. BMC Complement Altern Med. 15:2412015. View Article : Google Scholar : PubMed/NCBI
|