Introduction
Pancreatic ductal adenocarcinoma (PDAC), is a type
of cancer with one of the highest rates of mortality (1). Pancreatitis, which is a significant
risk factor of PDAC (2), has been
identified to effectively induce initiation of PDAC in mice with
K-ras oncogene (3). Acinar-ductal
metaplasia (ADM) is a phenomenon that appears during pancreatitis
and is characterized by the replacement of normal acinus with a
ductal-like structure, which contains metaplastic acinar cells that
express ductal markers, such as cytokeratin 19 (CK19) (4). Pancreatitis-induced ADM is a
reversible process in normal adult mice where metaplastic acinar
cells are replaced by normal acinar cells following diminishing of
inflammation. However, with mutant K-ras oncogenes, acinar
regeneration is blocked and metaplastic acini are driven to form
pancreatic intraepithelial neoplasia (PanIN) (4), which is the most common precursor of
PDAC (5). Therefore, ADM has been
demonstrated to be an essential step in pancreatitis-induced,
K-ras-dependent carcinogenesis of pancreatic cancer (4,6).
SRY-box 9 (SOX9) is a human motility group-box
transcription factor that has been observed to regulate pancreatic
exocrine and endocrine development (7). In the exocrine part of normal adult
pancreas, SOX9 is expressed only in centroacinar cells and ductal
cells (4). However, SOX9 is also
expressed in acinar cells during pancreatitis (8). Previous studies identified that SOX9
serves a critical role in pancreatitis-induced ADM and pancreatic
carcinogenesis (8–10). However, the regulation of SOX9
expression remains unclear, and further research is required in
order for SOX9 to become a therapeutic target of PDAC.
In addition to the SOX9 gene, numerous other
proteins and pathways were activated or upregulated during
pancreatitis, and certain pathways have been demonstrated to be
essential for ADM formation and pancreatic carcinogenesis. The
ductal transcription factor hepatocyte nuclear factor 6 has been
demonstrated to serve an important role in ADM (9). Human and mouse pancreatic lesions
express abundant epidermal growth factor receptor (EGFR) and EGFR
is required for ADM formation, Kras-induced PanIN formation and
pancreatic carcinogenesis (11,12).
Also, the downward pathways of EGFR, such as mitogen-activated
protein kinase signaling and phosphatidyl inositol 3-kinase
signaling, are required for PanIN formation and pancreatic
carcinogenesis (13,14). Signal transducer and activator of
transcription 3 (STAT3) signaling is also significantly upregulated
during pancreatitis and the phosphorylation of STAT3 has been
indicated to be essential for the proliferation of metaplastic
acinar cells and initiation of PDAC (15–17).
Other pathways including Notch signaling, canonical Wnt signaling
and the Hedgehog pathway have been observed to promote ADM and
pancreatic carcinogenesis (6,18,19).
Epithelial-mesenchymal transition (EMT) is a
biological process in which epithelial cells transform into special
cells with mesenchymal phenotypes. EMT has a close association with
the metastasis of pancreatic cancer. Certain mesenchymal markers,
such as zinc finger E-box binding homeobox 1 (Zeb1) and
fibroblast-specific protein 1, serve as independent predictors of
mortality in patients with pancreatic cancer (20,21).
It has been confirmed that Kras-dependent PanIN formation and
pancreatic carcinogenesis were accompanied with EMT of acinar
cells, and EMT promotes dissemination of acinar cells under
malignant transformation (22).
Flavonoids are the effective constituents of many
herbs used in traditional Chinese medicine and have produced
preventive and therapeutic effects against certain diseases
(23). As a flavone widely present
in various foodstuffs, plants and herbs, luteolin has been
identified as one of the most potent flavonoids (24). Luteolin has been identified to
exhibit numerous biological activities and therapeutic effects,
including anti-inflammatory and pro-apoptotic effects, and the
ability to reverse anti-neoplastic drug resistance (25–27).
In a previous study, it was demonstrated that luteolin inhibits
auto-phosphorylation of STAT3 and EMT of pancreatic cancer cell
in vitro (28). The current
study determined if luteolin inhibits certain
carcinogenesis-associated pathways and proteins that were
upregulated during pancreatitis. In addition, the effects of
luteolin on ADM and certain other pathological changes that were
essential for pancreatic carcinogenesis or metastasis were
examined.
Materials and methods
Induction of acute pancreatitis and
mouse treatment
Experiments were approved by the Animal Experimental
Ethical Inspection of Laboratory Animal Centre, Wenzhou Medical
University, Wenzhou, China (permit no. wydw2015-0128). A total of
45 male C57/BL6 mice were supplied by Laboratory Animal Centre of
Wenzhou Medical University, housed in specific pathogen-free
condition with constant temperature of 24°C and 12-h light/dark
cycle. The mice had free access to food and water until 24 h before
experiments. When mice were 6 weeks old and about 25 g, acute
pancreatitis was induced by 2 sets of 6 hourly intra-peritoneal
injections of cearulein (50 µg/kg; Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany) on alternating days. Luteolin (Chengdu Must
Bio-Technology, Chengdu, China) was dissolved by DMSO at a
concentration of 40 mg/ml, and then diluted by normal saline to
0.05 mg/ml. Mice were intra-peritoneally injected with luteolin
daily at a dosage of 2 mg/kg. The first injection of luteolin was
performed immediately subsequent to the completion of cearulein
injections on the first day. The last injection of luteolin was
performed on the day prior to sacrifice. The next day after
finishing induction of pancreatitis was defined as day 1. Mice were
sacrificed on days 1, 3 and 5 in the 9 different groups (day
1-CTRL, day 1-AP and day 1-AP+Luteolin, and so on for days 3 and
5). Each group comprised 5 mice.
Hematoxylin and eosin (H&E)
staining and immunohistochemistry
Pancreas tissues were fixed overnight in 4%
paraformaldehyde (Sigma-Aldrich; Merck Millipore), embedded in
paraffin, cut into 4-µm-thick sections, and placed on slides.
Sections were subjected to H&E and immunohistochemical staining
as previously described (19,29).
For H&E staining, sections were stained with hematoxylin for 4
min and with eosin for 1.5 min. For immunohistochemistry,
3–3′-diaminobenzidine tetrahydrochloride was used as a chromogen.
Primary antibodies were diluted according to the instructions and
were incubated overnight at 4°C (Table
I). Bright-field images were acquired using a DM4000B
microscope (Leica Microsystems GmbH, Wetzlar, Germany).
 | Table I.Antibodies used. |
Table I.
Antibodies used.
| Antibody | Supplier | Catalog number | Dilution |
|---|
| SOX9 | Cell Signaling
Technology, Inc. (Danvers, MA, USA) | 82630 |
1:400 |
| p-STAT3 | Abcam (Cambridge,
UK) | Ab76315 |
1:200 |
| p-EGFR
(Tyr1068) | Cell Signaling
Technology, Inc. | 3777 |
1:100 |
| CK19 | Abcam | Ab52625 |
1:500 |
| Ki67 | Abcam | Ab16667 |
1:100 |
| N-cadherin | Abcam | Ab98952 | 1:1,000 |
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Pancreas tissue was preserved in liquid nitrogen.
Total RNA was extracted by tissue dissociation in TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
according to the manufacturer's protocol. cDNA was synthesized
using 1 µg of total RNA in a 20 µl final volume by reverse
transcription with RevertAid RT Reverse Transcription kit (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). qPCR was performed with
0.3 µl of cDNA, 1.5 µl forward and reverse primers (2 µM), 1.7 µl
double distilled water, and 5 µl SYBR Green Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Expression levels were
normalized using a custom primer set for GAPDH. cDNA was amplified
using ABI Prism 7500 Sequence Detection System (Applied Biosystems;
Thermo Fisher Scientific, Inc.). Primer sequences are listed in
Table II. The thermocycling
conditions were: Stage 1: 95°C, 5 min; Stage 2: 95°C, 30 sec, 60°C,
1 min, 40 cycles; melt curve: 95°C, 5 min; and, cooling at
1.6°C/sec to 60°C.
| Table II.Primers used. |
Table II.
Primers used.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| Snail |
GAGCTGCAGGACGCGTGTGT |
TTGAGGACCTCGGGCGGAGG |
| Slug |
TCGTCGGCAGCTCCACTCCA |
CGGGGGACTTACACGCCCCA |
| Twist |
AGACCCAGCGGGTCATGGCT |
CTTGTCCGAGGGCAGCGTGG |
| Zeb1 |
CGGTGCCAAGAACTGCTGGCA |
CGGCGGTGTCTTGTTGCTGC |
| Vimentin |
AATCCAAGTTTGCTGACCTCTCTGA |
ACTGCACCTGTCTCCGGTACTC |
| CDH1 |
AGCCCCTGCTGCCACCAGAT |
CATCCAGGCCCCTGTGCAGC |
| EpCAM |
GCAGGTCCAGTGTGGTACTC |
GGAAGCGCTAACCCTCCTAC |
| CDH2 |
GCGGGATAAAGAGCG |
GGAGTCATACGGTGGC |
| ZO1 |
CCACAAGCGCAGCCACAAGCTA |
GCTGGGGTTGTTTCAGGCGAA |
| GAPDH |
AATGGGGTGAGGCCGGTGCT |
CACCCTTCAAGTGGGCCCCG |
Statistical analysis
Comparisons among multiple groups were made with a
one-way analysis of variance followed by least significant
difference or Dunnett's t-test. P<0.05 was considered to
indicate a statistically significant difference. Results are
represented as the mean ± standard error. All data presented
represent at least 3 mice and represent at least three 4-µM
sections unless otherwise noted.
Results
Luteolin inhibits pancreatitis-induced
ADM formation
H&E staining was performed and an
immunohistochemistry assay was conducted to determine whether
luteolin has effects on ADM formation induced by acute
pancreatitis. By H&E staining, it was identified that ADM was
most evident on day 3, compared with days and day 5. A prominent
change of ADM induced by acute pancreatitis was the formation of
tubular complexes, which was characterized by circular arrangement
of acinar cells that resembled ducts, and the tubular complexes of
each field were counted (magnification, ×200) (30,31).
The H&E staining slides exhibited no tubular complex in the
control group (Fig. 1A), and 3
days subsequent to acute pancreatitis, this number increased to
32.27±5.58 (Fig. 1B). However,
with the treatment with luteolin, 22±3.79 tubular complexes were
observed per field (P<0.05) (Fig.
1C and D). This indicates that acute pancreatitis-induced the
formation of tubular complexes, which was then significantly
inhibited by luteolin.
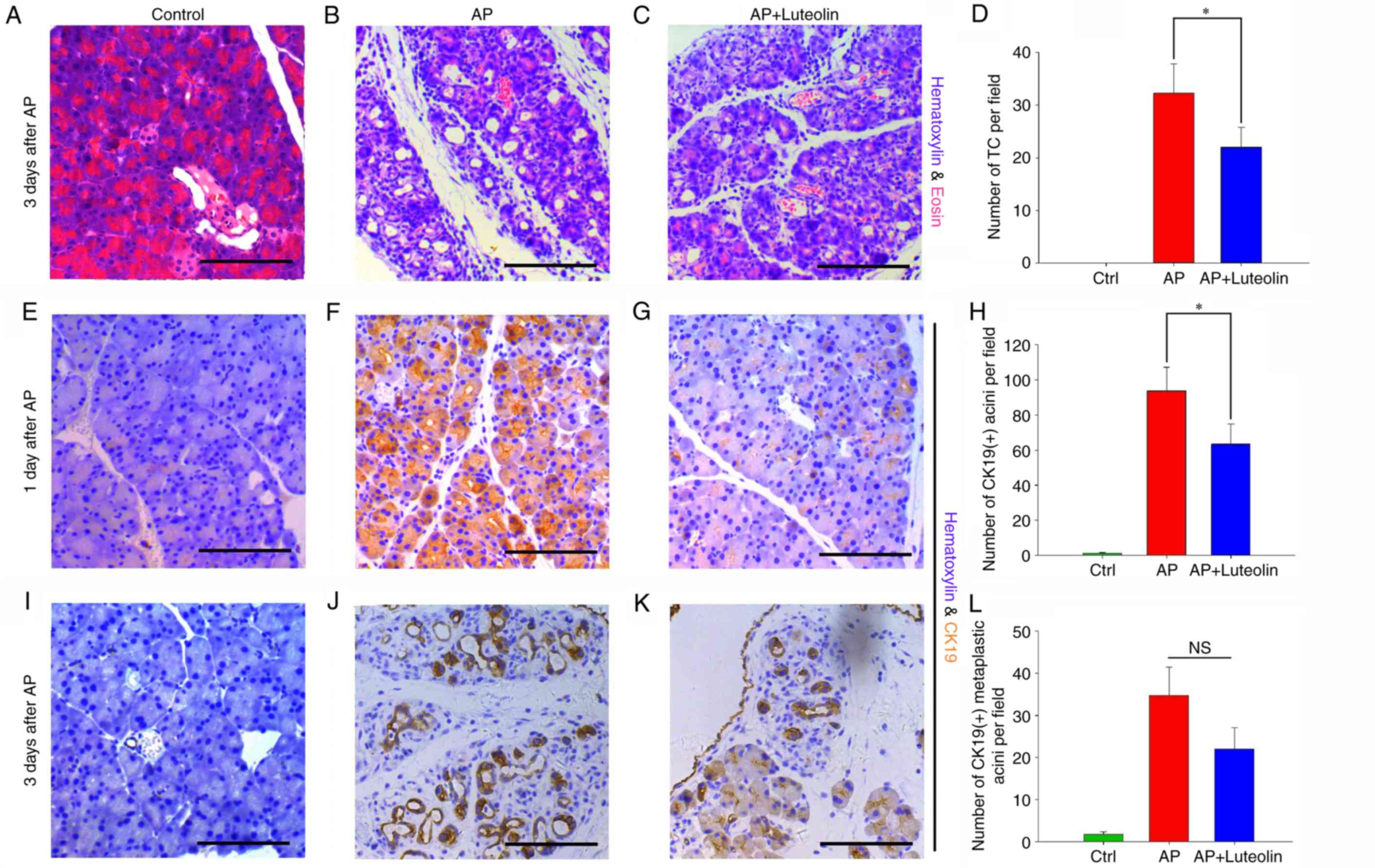 | Figure 1.Luteolin inhibits
pancreatitis-induced ADM formation. Hematoxylin and eosin staining
of the pancreas in the (A) Ctrl, (B) AP and (C) luteolin groups on
day 3. (D) Number of TC per field (magnification, ×200) in the
Ctrl, AP and luteolin groups. Immunohistochemistry of CK19 in the
(E) Ctrl, (F) AP and (G) luteolin groups on day 1. (H) Number of
CK19(+) acini per field (×200) in the Ctrl, AP and luteolin groups
on day 1. Immunohistochemistry of CK19 in the (I) Ctrl, (J) AP and
(K) luteolin groups on day 3. (L) Number of CK19(+) acini per field
(×200) in the Ctrl, AP and luteolin groups on day 3. *P<0.05.
Scale bars, 100 µm. ADM, acinar-ductal metaplasia; Ctrl, control;
AP, acute pancreatitis group; AP + luteolin, luteolin group; TC,
tubular complex; NS, not significant. |
To confirm luteolin's effects on ADM further, an
immunohistochemistry assay was performed in order to detect ectopic
expression of CK19 in acinar cells. CK19-positive acini were then
counted to determine the extent of ADM. As the immunohistochemistry
assay indicated, unlike tubular complexes which were clearer on day
3, CK19-positive acini rapidly appeared on day 1 and could last up
to day 5. The number of CK19-positive acini were counted on days 1
and 3 to assess whether luteolin has effects on ADM. It was
identified that on day 1, the number of CK19-positive acini
increased from 1.13±0.45 to 93.67±13.54 per field with the
injection of cearulein (P<0.05) (Fig. 1E and F), and this number decreased
to 63.47±11.48 with the treatment of luteolin (P<0.05) (Fig. 1G and H). A total of 3 days
subsequent to the induction of acute pancreatitis, mice which were
not treated with luteolin had 34.73±6.69 CK19-positive acini per
field (Fig. 1J). In mice treated
with luteolin, this number decreased to 22.07±5.04 however this was
not significant (P=0.072) (Fig. 1K and
L). The results of H&E staining and immunohistochemistry
assay both indicated that luteolin inhibits ADM induced by acute
pancreatitis.
Pancreatitis-induced expression of
SOX9 in acinar cells is inhibited by luteolin
Numerous kinds of transcriptional factors, such as
SOX9 and HNF6, have been confirmed to be essential for ADM
formation (9). In order to
investigate how luteolin inhibits pancreatitis-induced ADM
formation, immunohistochemistry was conducted to determine the
effects of luteolin on SOX9 expression (Fig. 2). SOX9 was expressed in ductal
cells and centroacinar cells alone in the normal pancreas tissues,
while it was induced in acinar cells following acute pancreatitis.
SOX9-positive cells increased significantly one day subsequent to
pancreatitis (Fig. 2A and B). On
day 1, for the control group, 93.67±11.83 of pancreatic cells per
field were detected to exhibit expression of SOX9. However, for
mice with acute pancreatitis, this number increased to 213.9 ±17.98
per field (P<0.05). Furthermore, when treated with luteolin,
this number decreased to 151.4±16.99 (P<0.05) (Fig. 2C and J). Acute pancreatitis-induced
ectopic SOX9 expression lasted up to day 3. On day 3, for mice
injected with cearulein and treated with luteolin, SOX9-positive
cells decreased significantly from 232.45±16.02 to 184.07±21.22 per
field (P<0.05) (Fig. 2D-F and
J). Luteolin also caused the decrease of SOX9-positive cells on
day 5, however not significantly (P=0.112; Fig. 2G-I and J), and this is suggested to
be due to the fact that ectopic SOX9 expression was almost
decreased to normal levels. With the results of
immunohistochemistry assay, it was concluded that luteolin inhibits
acute pancreatitis-induced SOX9 expression in acinar cells, and as
SOX9 is essential for ADM formation, luteolin's effects of
inhibiting ADM may partly depend on the inhibition of SOX9
expression.
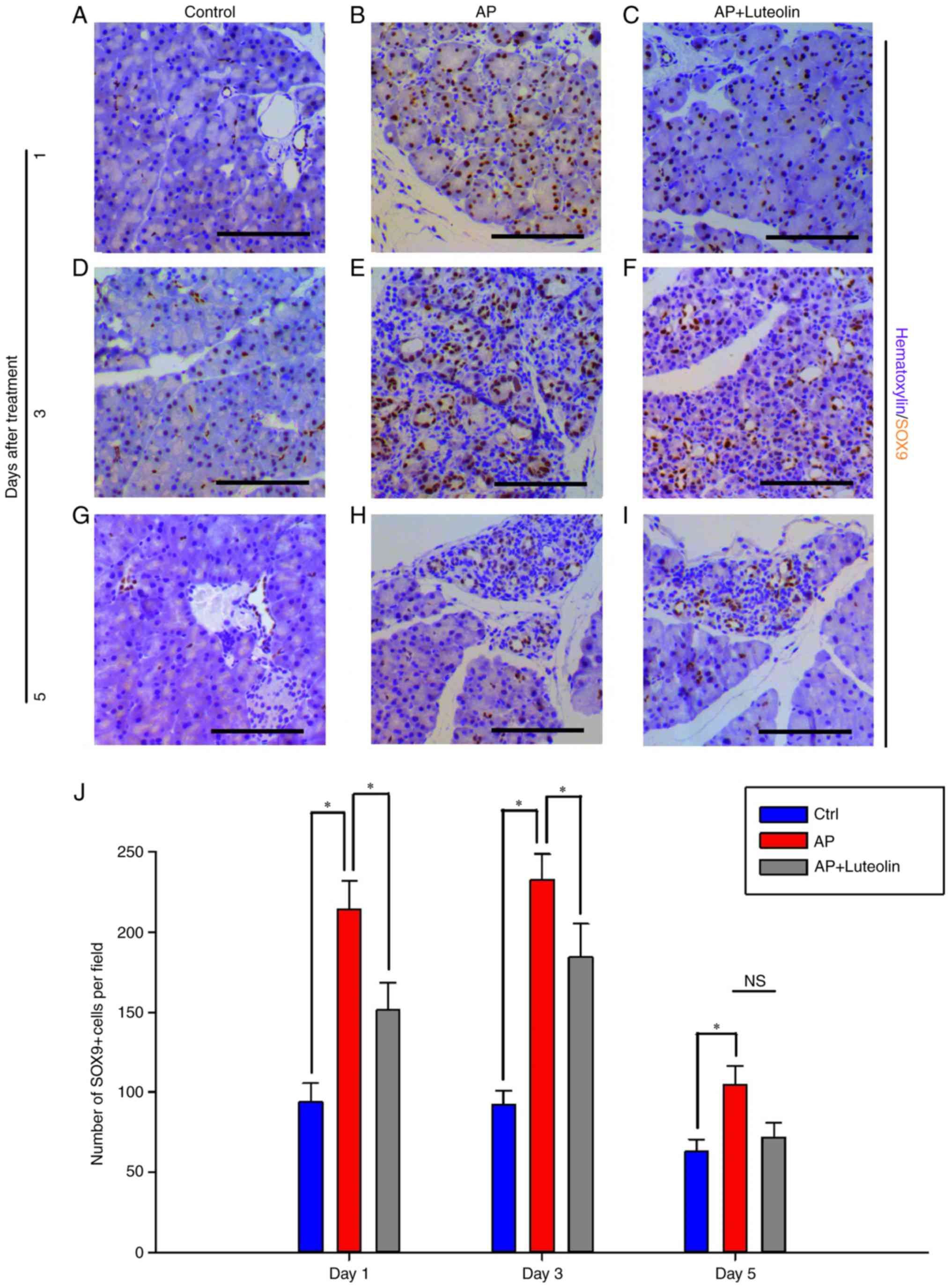 | Figure 2.Pancreatitis-induced expression of
SOX9 in acinar cells is inhibited by luteolin. Immunohistochemistry
for SOX9 in the (A) Ctrl, (B) AP and (C) luteolin groups 1 day
after AP. Immunohistochemistry for SOX9 in the (D) Ctrl, (E) AP and
(F) luteolin groups 3 days after AP. Immunohistochemistry for SOX9
in the (G) Ctrl, (H) AP and (I) luteolin groups 5 days after AP.
(J) Number of SOX9(+) cells per field (×200) in the Ctrl, AP and
luteolin groups; *P<0.05; scale bars, 100 µm. SOX9, SRY-box 9;
Ctrl, control; AP, acute pancreatitis group; AP + luteolin,
luteolin group; NS, not significant. |
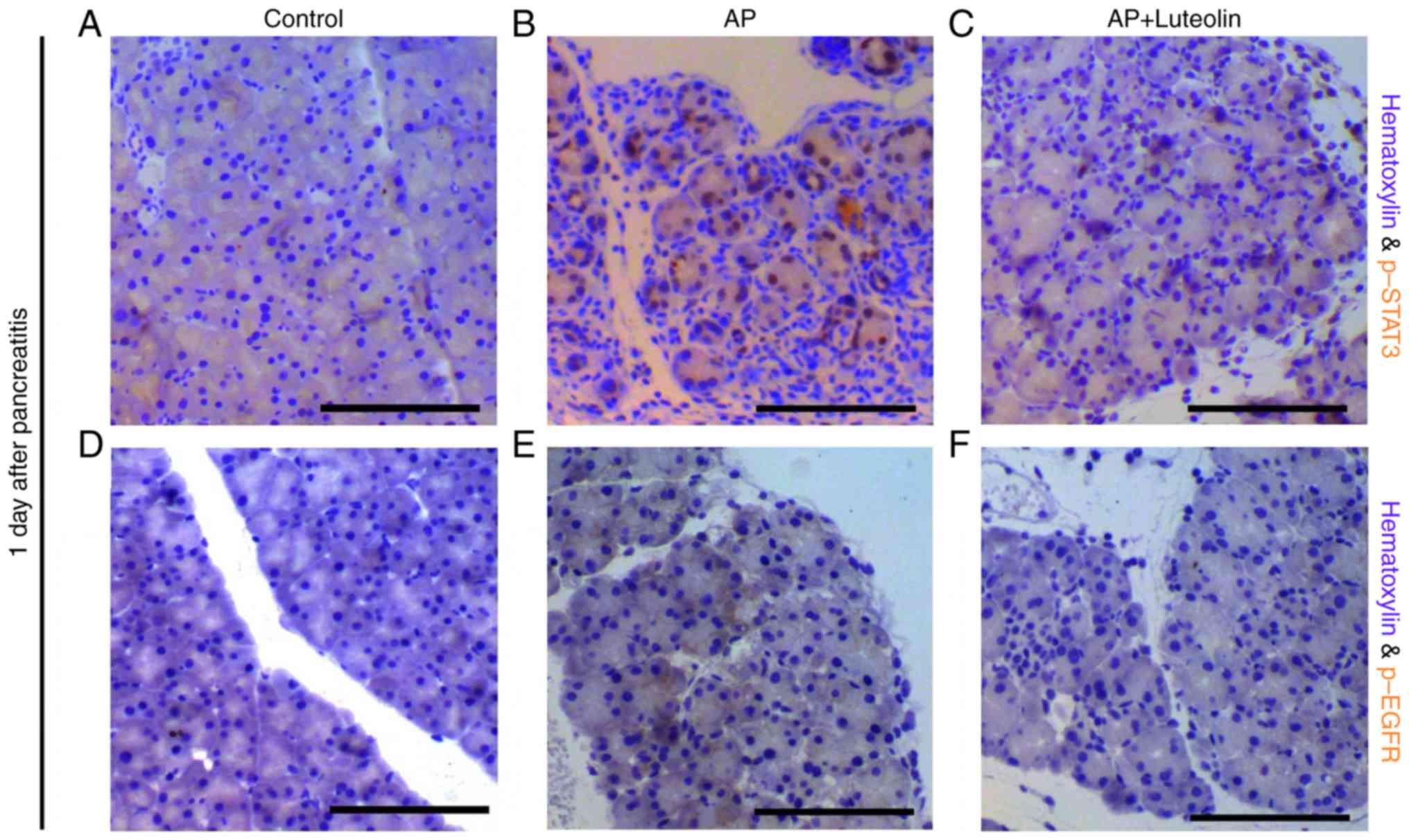
Luteolin inhibits STAT3 and EGFR
signaling pathways
A number of signaling pathways have been confirmed
to be essential for ADM and pancreatic carcinogenesis, including
the interleukin (IL)-STAT3 pathway, EGFR-ras pathway and canonical
Wnt pathway (11–13,15–18).
An immunohistochemistry assay was conducted to determine the
effects of luteolin on these pathways. Consistent with previous
studies (15,16), p-STAT3 positive acinar cells were
not observed in the exocrine part of the pancreas in the control
group, however is widely observed in mice with pancreatitis on day
1 (Fig. 3A and B). However, with
the treatment of luteolin, p-STAT3-positive cells decreased
significantly during pancreatitis (Fig. 3C). It was also identified that
pancreatitis significantly induced phosphorylation of EGFR in
acinar cells (Fig. 3D and E) and
with the treatment of luteolin, phosphorylation of EGFR was
decreased significantly on day 1 (Fig.
3F). Therefore, not only the STAT3 pathway, however
additionally the EGFR pathway were inhibited by luteolin during
pancreatitis. The expression of β-catenin was determined following
the treatment of luteolin. However, it seems that luteolin has no
significant effect on the expression and membrane-translocation of
β-catenin (data not shown). As STAT3 and EGFR signaling pathways
serve a role in ADM, and the inhibition of them may contribute to
luteolin's effects of inhibiting ADM formation.
Luteolin inhibits acute
pancreatitis-induced proliferation of acinar cells
It has been reported that pancreatitis-induced ADM
and pancreatic carcinogenesis were accompanied with persistent
proliferation of pancreatic cells (17). Cell cycle activation marker Ki67
was used to determine proliferative level of acinar cells. Few
acinar cells were identified to be Ki67-positive on the control
group (Fig. 4A). And frequency of
Ki67-positive cells increased to 46.62±7.45 per field one day after
the induction of pancreatitis (Fig.
4B). However, with the treatment of luteolin, Ki67-positive
cells decreased to 19.4±6.45 per field (P<0.05; Fig. 4C). On day 3, mice on the acute
pancreatitis group had 68±9.54 Ki67-positive cells per field
(Fig. 4D and E). Whilst with the
treatment of luteolin, this number decreased to 47.67±6.32
(P<0.05; Fig. 4F). On day 5,
the number of Ki67-positive cells per field decreased from 30.8±3
to 27.43±4.52 with the treatment of luteolin (Fig. 4G-I), however it was not significant
(Fig. 4J). Therefore, it was
concluded that pancreatitis-induced proliferation of exocrine cells
was inhibited by luteolin.
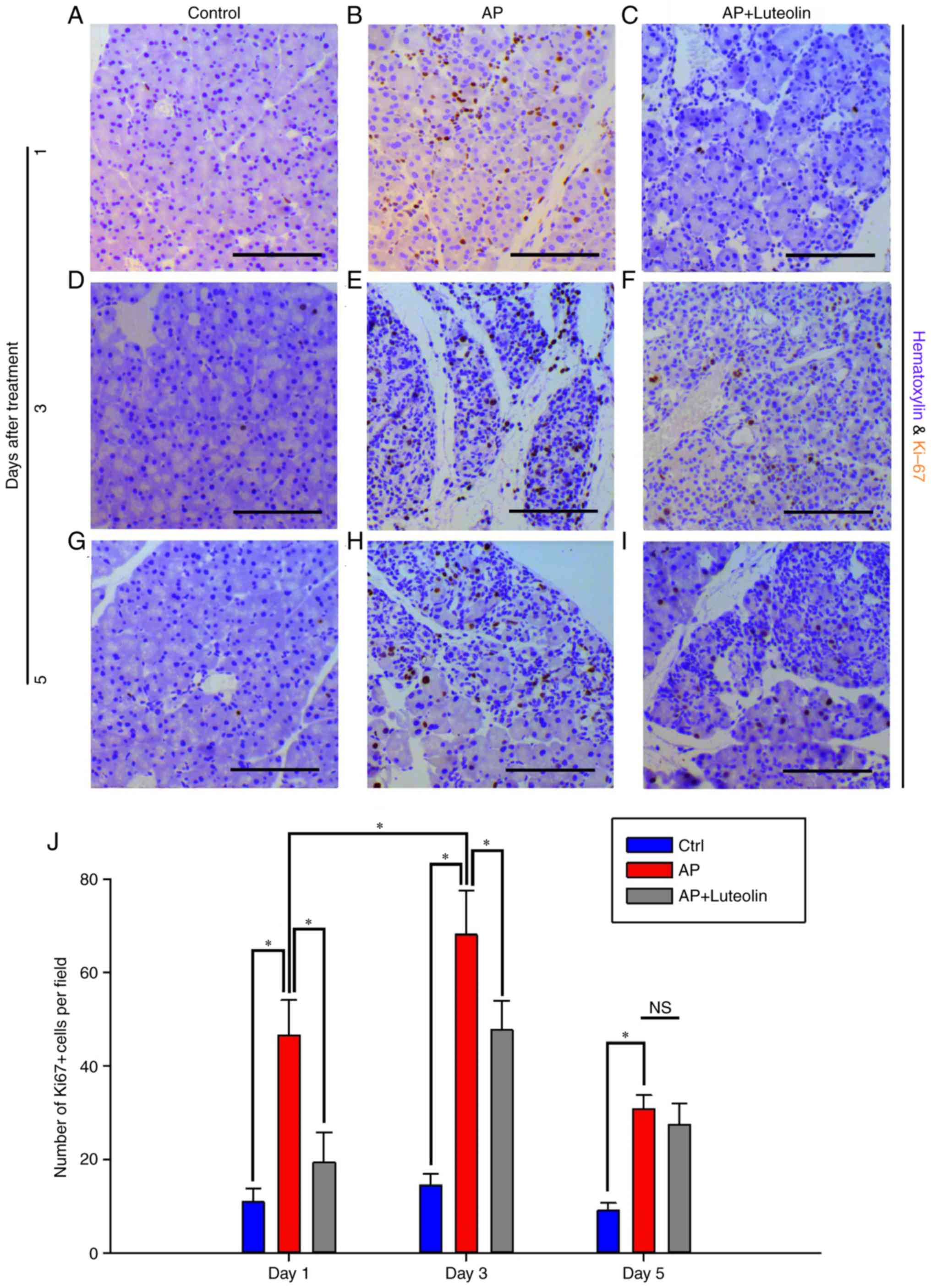 | Figure 4.Luteolin inhibits acute
pancreatitis-induced proliferation of acinar cells.
Immunohistochemistry for Ki67 in the (A) Ctrl, (B) AP and (C)
luteolin groups 1 day after AP. Immunohistochemistry for Ki67 in
the (D) Ctrl, (E) AP and (F) luteolin groups 3 days after AP.
Immunohistochemistry for Ki67 in the (G) Ctrl, (H) AP and (I)
luteolin groups 5 days after AP. (J) Number of Ki67(+) cells per
field (×200) in the Ctrl, AP group and luteolin groups. *P<0.05,
scale bars, 100 µm. Ctrl, control; AP, acute pancreatitis group; AP
+ luteolin, luteolin group; NS, not significant. |
Transient EMT occurs on acinar cells
during pancreatitis and luteolin inhibits EMT of acinar cells
Pancreatic carcinogenesis was accompanied by EMT of
acinar cells, which significantly promoted dissemination of acinar
cells under malignant transition (22). Thus, it was investigated whether
acute pancreatitis promotes EMT of acinar cells and if luteolin has
effects on it. CDH2, which was an adhesion molecule in the membrane
and a marker of mesenchyma, was chosen to indicate EMT change of
acinar cells during pancreatitis. By immunohistochemistry, it was
observed that CDH2 was highly expressed in islets and marginally
expressed in acinar cells of normal pancreas (Fig. 5A). However, during pancreatitis,
expression of CDH2 increased significantly in acinar cells on day 1
(Fig. 5B). Subsequently, the
expression of CDH2 on mice treated with luteolin was established.
The results indicated that pancreatitis-induced expression of CDH2
was effectively inhibited by luteolin (Fig. 5C).
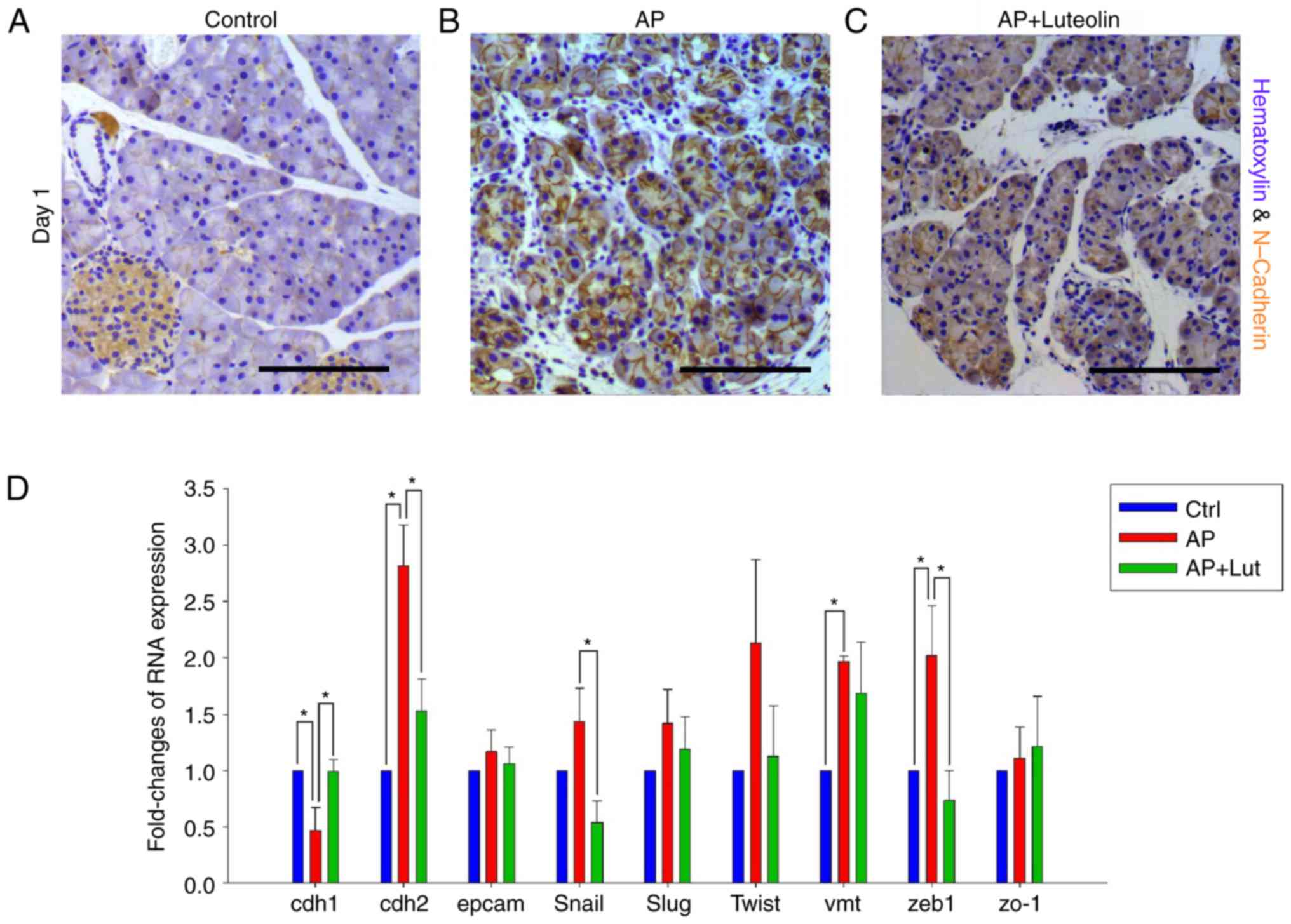 | Figure 5.Transient EMT occurs on acinar cells
during pancreatitis and luteolin inhibits EMT of acinar cells.
(A-C) Immunohistochemistry for N-Cadherin in control group (A),
acute pancreatitis group (B) and luteolin group (C) 1 day after
pancreatitis. (D) Fold change in mRNA expression of CDH1, CDH2,
Slug, Zeb1, EpCAM, ZO1, VMT, Snail, and Twist of pancreatic tissue.
GAPDH was used as an internal control. *P<0.05; scale bars, 100
µm. EMT, epithelial-mesenchymal transition; CDH1, E-cadherin; CDH2,
N-cadherin; Slug, snail family transcriptional repressor 2; Zeb1,
zinc finger E-box binding homeobox 1; EpCAM, epithelial cell
adhesion molecule; ZO1, zonula occludens 1; VMT, vimentin; Snail,
snail family transcriptional repressor 1; Twist, twist family BHLH
transcription factor 1; AP, acute pancreatitis group; AP +
luteolin, luteolin group; NS, not significant. |
RT-qPCR was conducted to further confirm the
results. Certain EMT markers, including CDH1, CDH2, EpCAM, Snail,
Slug, Twist, Vimentin, Zeb1 and ZO-1, were selected to indicate EMT
change of acinar cells. As compared with the control group, the
results of PCR indicated that CDH1, CDH2, Vimentin and Zeb1 changed
significantly 1 day after acute pancreatitis (Fig. 5D). Changes of the four markers all
indicated EMT changes to acinar cells. No marker was identified to
change significantly against EMT (Fig.
5D) and compared with the acute pancreatitis group, CDH1, CDH2,
Snail and Zeb1 changed significantly with the treatment of luteolin
(Fig. 5D). Changes of the four
markers all indicated that the EMT changes of acinar cells were
reversed. No marker suggested that luteolin would promote EMT
(Fig. 5D). Therefore, with the
results of the immunohistochemistry assay and RT-qPCR, it was
confirmed that acute pancreatitis promoted EMT of acinar cells, and
this was reversed by luteolin.
Discussion
A number of pathways and proteins have been
demonstrated to serve different roles in pancreatic carcinogenesis.
Certain pathways and proteins are also essential for ADM formation,
for example EGFR-ras pathway, IL6-STAT3 pathway, and SOX9 protein
(8,11,17).
These pathways and proteins are upregulated during pancreatitis,
and serve as a bridge connecting pancreatitis and pancreatic
cancer. Thus, pancreatitis-induced ADM formation and pancreatic
carcinogenesis were blocked by inhibiting those pathways and
proteins. Luteolin has been demonstrated to exhibit various
anti-neoplastic properties, for example pro-apoptosis, inhibition
of EMT and reversing chemotherapeutic-resistance (26–28).
It has been identified to affect a number of pathways (28,29,32),
and some of those pathways additionally serve roles on
pancreatitis-induced ADM formation and pancreatic carcinogenesis.
Thus, it was suggested that luteolin may inhibit
pancreatitis-induced ADM formation. At present, the majority of
studies about pancreatic carcinogenesis focused on the pathogenetic
mechanisms and regulatory pathways of ADM and PDAC formation, few
studies focused on interference of them with exogenous agents. The
current study used luteolin, a herbal extract, to interfere with
ADM by inhibiting pro-ADM pathways and proteins.
The current study identified that
inflammation-associated pathways such as IL6-STAT3 and EGFR
pathways were all activated on day 1 and then decreased to normal
levels by day 3. It was observed that cearulein-induced
inflammation of the pancreas was most prominent on day 1, compared
with days 3 and 5. However, it was identified that morphological
changes of acini were most prominent on day 3, not day 1. On day 3,
acinar cells under ADM transformed into a more duct-like structure,
accompanied by high levels of non-acinar cell infiltration.
Notably, on day 3, metaplastic acini always clustered together in a
lobule, single metaplastic acinus were not identified between
non-ADM acini. ADM is considered as a pathological change caused by
inflammation during pancreatitis (9). However, considering the difference
between the time of inflammation and ADM and the characteristics of
ADM on day 3, it was hypothesized that ADM may be not a
pathological process directly caused by inflammation, however
rather a physiological change actively mediated via acinar cells
subsequent to exposure to inflammation. ADM has been regarded as a
necessary step for Kras-dependent pancreatic carcinogenesis, and
acinar cells under ADM exhibit increased tendency for malignant
transition (8). However, it was
suggested that ADM may have positive or protective effects for
acinar cells during pancreatitis. Nevertheless, further study is
required in order to confirm these hypotheses.
Early metastasis serves an important role in the
poor prognosis of pancreatic cancer. It was reported that during
the process of pancreatic carcinogenesis on mice with Kras
mutation, EMT and dissemination of acinar cells precede tumor
formation, and the EMT of acinar cells was promoted by pancreatitis
(22). In addition,
pancreatitis-induced pancreatic carcinogenesis has been observed to
be accompanied by increased expression of, matrix
metalloproteinase, which in turn increased the invasiveness of
cancer cells (17). The present
study confirmed that pancreatitis induces EMT of acinar cells.
Therefore, pancreatitis not only promotes carcinogenesis of
pancreatic cancer, it may also lead to acinar cells under malignant
transformation becoming highly invasive. It is therefore
hypothesized that pancreatic cancer cells gained invasiveness
predominantly during carcinogenesis. This was different to that of
the traditional view, which suggests that cancer cells gain
invasiveness step by step by accumulation of mutations and
stimulation of inflammation.
For two reasons, the present study suggested that
luteolin could be an effective agent to inhibit pancreatic
carcinogenesis. Firstly, luteolin inhibits ADM formation induced by
pancreatitis. ADM gives rise to PanIN and has been confirmed as an
early step in Kras-dependent pancreatic carcinogenesis (4). Furthermore, the IL6-STAT3 pathway,
EGFR-ras pathway and SOX9 protein, all of which are inhibited by
luteolin, serve essential roles in pancreatic carcinogenesis
(8,11,12,15,16).
Due to the limitation of the therapeutic methods, pancreatic cancer
remains almost untreatable once it occurs. The interference of
carcinogenesis in people with significant risk factors may become
an important treatment for pancreatic cancer. With anti-ADM
formation and anti-EMT properties, luteolin may serve an important
role in anti-pancreatic cancer strategies in the future.
Acknowledgements
The current study was sponsored by grants from the
National Natural Science Foundation of China (grant no. 81070372)
and the Provincial Department of Construction Project (grant no.
WKJ2012-2-033).
Glossary
Abbreviations
Abbreviations:
|
ADM
|
acinar-ductal metaplasia
|
|
CK19
|
cytokeratin-19
|
|
EGFR
|
epithelial growth factor receptor
|
|
EMT
|
epithelial-mesenchymal transition
|
|
H&E
|
hematoxylin and eosin
|
|
PanIN
|
pancreatic intra-epithelial
neoplasia
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
References
|
1
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ekbom A, McLaughlin JK and Nyrén O:
Pancreatitis and the risk of pancreatic cancer. N Engl J Med.
329:1502–1503. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guerra C, Schuhmacher AJ, Cañamero M,
Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP and
Barbacid M: Chronic pancreatitis is essential for induction of
pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice.
Cancer Cell. 11:291–302. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Morris JP IV, Cano DA, Sekine S, Wang SC
and Hebrok M: Beta-catenin blocks Kras-dependent reprogramming of
acini into pancreatic cancer precursor lesions in mice. J Clin
Invest. 120:508–520. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hruban RH, Wilentz RE and Maitra A:
Identification and analysis of precursors to invasive pancreatic
cancer. Methods Mol Med. 103:1–13. 2005.PubMed/NCBI
|
|
6
|
De La OJP, Emerson LL, Goodman JL, Froebe
SC, Illum BE, Curtis AB and Murtaugh LC: Notch and Kras reprogram
pancreatic acinar cells to ductal intraepithelial neoplasia. Proc
Natl Acad Sci USA. 105:pp. 18907–18912. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shih HP, Kopp JL, Sandhu M, Dubois CL,
Seymour PA, Grapin-Botton A and Sander M: A Notch-dependent
molecular circuitry initiates pancreatic endocrine and ductal cell
differentiation. Development. 139:2488–2499. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kopp JL, von Figura G, Mayes E, Liu FF,
Dubois CL, Morris JP IV, Pan FC, Akiyama H, Wright CV, Jensen K, et
al: Identification of Sox9-dependent acinar-to-ductal reprogramming
as the principal mechanism for initiation of pancreatic ductal
adenocarcinoma. Cancer Cell. 22:737–750. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Prévot PP, Simion A, Grimont A, Colletti
M, Khalaileh A, Van den Steen G, Sempoux C, Xu X, Roelants V, Hald
J, et al: Role of the ductal transcription factors HNF6 and Sox9 in
pancreatic acinar-to-ductal metaplasia. Gut. 61:1723–1732. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grimont A, Pinho AV, Cowley MJ, Augereau
C, Mawson A, Giry-Laterrière M, Van den Steen G, Waddell N, Pajic
M, Sempoux C, et al: SOX9 regulates ERBB signalling in pancreatic
cancer development. Gut. 64:1790–1799. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ardito CM, Grüner BM, Takeuchi KK,
Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE,
Carpenter ES, Halbrook CJ, Hall JC, et al: EGF receptor is required
for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 22:304–317.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Navas C, Hernández-Porras I, Schuhmacher
AJ, Sibilia M, Guerra C and Barbacid M: EGF receptor signaling is
essential for k-ras oncogene-driven pancreatic ductal
adenocarcinoma. Cancer Cell. 22:318–330. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Collins MA, Yan W, Sebolt-Leopold JS and
Pasca di Magliano M: MAPK signaling is required for
dedifferentiation of acinar cells and development of pancreatic
intraepithelial neoplasia in mice. Gastroenterology.
146:822–834.e7. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu CY, Carpenter ES, Takeuchi KK, Halbrook
CJ, Peverley LV, Bien H, Hall JC, DelGiorno KE, Pal D, Song Y, et
al: PI3K regulation of RAC1 is required for KRAS-induced pancreatic
tumorigenesis in mice. Gastroenterology. 147:1405–1416.e7. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Corcoran RB, Contino G, Deshpande V,
Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA
and Bardeesy N: STAT3 plays a critical role in KRAS-induced
pancreatic tumorigenesis. Cancer Res. 71:5020–5029. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lesina M, Kurkowski MU, Ludes K, Rose-John
S, Treiber M, Klöppel G, Yoshimura A, Reindl W, Sipos B, Akira S,
et al: Stat3/Socs3 activation by IL-6 transsignaling promotes
progression of pancreatic intraepithelial neoplasia and development
of pancreatic cancer. Cancer Cell. 19:456–469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fukuda A, Wang SC, Morris JP IV, Folias
AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ, et
al: Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma
initiation and progression. Cancer Cell. 19:441–455. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, Morris JP IV, Yan W, Schofield
HK, Gurney A, Simeone DM, Millar SE, Hoey T, Hebrok M and Pasca di
Magliano M: Canonical wnt signaling is required for pancreatic
carcinogenesis. Cancer Res. 73:4909–4922. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pasca di Magliano M, Sekine S, Ermilov A,
Ferris J, Dlugosz AA and Hebrok M: Hedgehog/Ras interactions
regulate early stages of pancreatic cancer. Genes Dev.
20:3161–3173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brabletz S, Bajdak K, Meidhof S, Burk U,
Niedermann G, Firat E, Wellner U, Dimmler A, Faller G, Schubert J
and Brabletz T: The ZEB1/miR-200 feedback loop controls Notch
signalling in cancer cells. EMBO J. 30:770–782. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang F, Sloss C, Zhang X, Lee SW and
Cusack JC: Membrane-bound heparin-binding epidermal growth factor
like growth factor regulates E-cadherin expression in pancreatic
carcinoma cells. Cancer Res. 67:8486–8493. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rhim AD, Mirek ET, Aiello NM, Maitra A,
Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK,
Vonderheide RH, et al: EMT and dissemination precede pancreatic
tumor formation. Cell. 148:349–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Middleton E Jr, Kandaswami C and
Theoharides TC: The effects of plant flavonoids on mammalian cells:
Implications for inflammation, heart disease, and cancer. Pharmacol
Rev. 52:673–751. 2000.PubMed/NCBI
|
|
24
|
Kandaswami C, Lee LT, Lee PP, Hwang JJ, Ke
FC, Huang YT and Lee MT: The antitumor activities of flavonoids. In
Vivo. 19:895–909. 2005.PubMed/NCBI
|
|
25
|
Jia Z, Nallasamy P, Liu D, Shah H, Li JZ,
Chitrakar R, Si H, McCormick J, Zhu H, Zhen W and Li Y: Luteolin
protects against vascular inflammation in mice and
TNF-alpha-induced monocyte adhesion to endothelial cells via
suppressing IΚBα/NF-κB signaling pathway. J Nutr Biochem.
26:293–302. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi RX, Ong CN and Shen HM: Luteolin
sensitizes tumor necrosis factor-alpha-induced apoptosis in human
tumor cells. Oncogene. 23:7712–7721. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hong Z, Cao X, Li N, Zhang Y, Lan L, Zhou
Y, Pan X, Shen L, Yin Z and Luo L: Luteolin is effective in the
non-small cell lung cancer model with L858R/T790M EGF receptor
mutation and erlotinib resistance. Br J Pharmacol. 171:2842–2853.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang X, Dai S, Dai J, Xiao Y, Bai Y, Chen
B and Zhou M: Luteolin decreases invasiveness, deactivates STAT3
signaling, and reverses interleukin-6 induced
epithelial-mesenchymal transition and matrix metalloproteinase
secretion of pancreatic cancer cells. Onco Targets Ther.
8:2989–3001. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Seymour PA, Freude KK, Tran MN, Mayes EE,
Jensen J, Kist R, Scherer G and Sander M: SOX9 is required for
maintenance of the pancreatic progenitor cell pool. Proc Natl Acad
Sci USA. 104:pp. 1865–1870. 2007; View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rau B, Paszkowski A, Lillich S, Baumgart
K, Möller P and Beger HG: Differential effects of
caspase-1/interleukin-1beta-converting enzyme on acinar cell
necrosis and apoptosis in severe acute experimental pancreatitis.
Lab Invest. 81:1001–1013. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reid LE and Walker NI: Acinar cell
apoptosis and the origin of tubular complexes in caerulein-induced
pancreatitis. Int J Exp Pathol. 80:205–215. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bagli E, Stefaniotou M, Morbidelli L,
Ziche M, Psillas K, Murphy C and Fotsis T: Luteolin inhibits
vascular endothelial growth factor-induced angiogenesis: Inhibition
of endothelial cell survival and proliferation by targeting
phosphatidylinositol 3′-kinase activity. Cancer Res. 64:7936–7946.
2004. View Article : Google Scholar : PubMed/NCBI
|