Introduction
Clinical progression regarding pancreatic ductal
adenocarcinoma (PDAC) has been made in previous years, however the
5-year survival rate is still <10% (1). Due to the limited investigations of
the underlying molecular mechanisms and the lack of sensitive and
specific biomarkers for early diagnosis, the prognosis and
diagnosis for this disease are very poor. Therefore, identification
of the molecular mechanisms underlying PDAC are of primary concern
and will benefit the clinical treatment for patients.
Golgi protein-73 (GOLPH2) was firstly identified as
a type II Golgi glycoprotein (2).
The expression of GOLPH2 is predominantly observed in the
epithelial cells of biliary, gut, stomach and prostate (3). Aberrant expression of GOLPH2 has been
detected in various diseases. The pathological process underlying
the progression from hepatitis and cirrhosis to hepatocellular
carcinoma (HCC), reveals a gradual increase in the expression
levels of GOLPH2 (4). Furthermore,
the high expression of GOLPH2 in the serum of HCC patients is
considered a novel biomarker for the development of the disease
(4). In addition, the cancerous
tissues of non-small cell lung, renal cell and prostate cancers
demonstrate increased expression of GOLPH2 (5–7). In
addition, GOLPH2 has been reported to regulate the immune response
(8). However, the expression
pattern and the biological functions of GOLPH2 in PDAC remain
unknown.
Activation of protein kinase B (Akt) signaling is
typical in pancreatic cancer. Akt signaling is important in the
growth and migration of pancreatic cancer cells (9–11).
Akt is an effector downstream of the growth factor receptor,
therefore amplification of growth factor receptor leads to the
activation of Akt. Previously, activation of Akt has been observed
in the Golgi apparatus. GOLPH3 recruits Akt to the Golgi apparatus
and is responsible for its subsequent activation (12). However, the exact molecular
mechanisms underlying the activation of Akt in the Golgi apparatus
remain to be fully elucidated.
The present study investigated the expression
profile and the biological functions of GOLPH2 in pancreatic
cancer, and aimed to elucidate the underlying molecular mechanisms
responsible for the progression of the disease.
Materials and methods
Cell culture
Pancreatic cancer cells (Suit2, BXPC3), normal
pancreatic cell line HPDE, mouse fibroblast NIH3T3 and human
embryonic HEK293T cells were incubated in Dulbecco's modified
Eagle's medium (Invitrogen; Thermo Fisher Scientific, Inc. Waltham,
MA, USA) supplemented with 10% fetal bovine serum (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany). Cells were cultured in an
incubator at 37°C, in an environment containing 5%
CO2.
PDAC samples
The present study was approved by the ethics
committee of Fujian Medical University (Fuzhou, China) and written
informed consent was obtained from all patients (age, 50–60; 38
male and 14 female; all patients exhibited PDAC, and patients who
had received chemotherapy were excluded from the study). A total of
52 PDAC tissues and paired non-cancerous tissues were obtained from
the affiliated hospital of Fujian Medical University and were
stored at −80°C.
Plasmids and establishment of stable
cell lines
The DNA insert encoding GOLPH2 was cloned into the
plasmid pcDNA 3.1-myc (Clontech Laboratories, Inc., Mountainview,
CA, USA). Plasmids (1 µg) were transfected into BXPC3 and Suit2
cells using PolyJet (SignaGen, Rockville, MD, USA) according to the
manufacturer's protocols. Cells were selected with G418. The
expression of exogenous GOLPH2 was confirmed by western blotting
using the myc tag.
Reverse transcription-polymerase chain
reaction (RT-qPCR)
The RNA was extracted using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocols. The reverse transcription was performed according to the
manufacturer's protocol (Promega Corporation, Madison, WI, USA),
qPCR was conducted in a Bio-Rad PCR system (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) with a qPCR 2X SYBR Green Mixture (Takara
Biotechnology Co., Ltd., Dalian, China) according to the
manufacturer's protocol with the following conditions: 95°C for 30
sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 30 sec.
The primer sequences used were as follows: Forward,
5′-TGGAGAGCGTCAACAAGCTG-3′ and reverse, 5′-GACATCCTGCTAGCCTGC-3′
for human GOLPH2 gene; forward, 5′-GATCATTGCTCCTCCTGAGC-3′ and
reverse, 5′-ACTCCTGCTTGCTGATCCAC-3′ for human 18S which served as
an internal control. Dissociation was performed at the end of the
procedure to confirm that non-specific amplification had not
occurred. The relative levels of gene expression were calculated
using the 2−ΔΔCq method (13).
Western blot analysis
Cultures were harvested and the protein was
extracted using a radioimmunoprecipitation assay buffer (Cell
Signaling Technology, Inc., Danvers, MA, USA) containing protease
and phosphatase inhibitors. The concentration of proteins was
determined using the Bradford assay. Proteins (15 µg/lane) were
separated by 10% SDS-PAGE, transferred onto polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA) and probed
with specific primary antibodies at 4°C overnight and the secondary
antibodies at room temperature for 1 h. The immunoreactive protein
was examined using an enhanced chemiluminescence kit (Pierce;
Thermo Fisher Scientific, Inc.). GOLPH2 antibody (cat. no.
Ab109628; 1:1,000) was obtained from Abcam (Cambridge, MA, USA).
Akt (cat. no. 4685; 1:1,000), Cyclin D1 (cat. no. 2978; 1:1,000),
B-cell lymphoma (Bcl)-2 (cat. no. 15071; 1:1,000) and Bcl-2
associated X (cat. no. 2764; 1:1,000), apoptosis regulator (Bax;
cat. no. 5023; 1:1,000) antibodies were obtained from Cell
Signaling Technology, Inc., (Danvers, MA, USA) and the GAPDH
antibody (cat. no. sc-25778; 1:5,000) was obtained from Santa Cruz
Biotechnology (Dallas, TX, USA). HRP linked secondary antibodies
(anti-mouse IgG; cat. no. 7076 and anti-rabbit IgG; cat. no. 7074)
were obtained from Cell Signaling Technology, Inc. and used at
1:2,000.
GST pull-down assay
The fusion protein GST-AKT was purified using
Sepharose4B beads (GE Healthcare Life Sciences, Little Chalfont,
UK) according to the manufacturer's protocols. Cell lysates were
prepared using the lysis buffer. Following centrifugation (4°C,
10,000 × g, 20 min), the supernatant was incubated with 5 µg GST
fusion protein overnight at 4°C. The Sepharose4B beads were added
and incubated with cell lysate for another 4 h. Then, the beads
were washed and the protein pulled down and detected with western
blot analysis.
Knockdown of GOLPH2 expression
The commercial available RNAi lenti-virus short
hairpin (Sh) con and Sh GOLPH2 were supplied by Shanghai GenePharma
Co., Ltd (Shanghai, China). Virus was added to the BXPC3 and Suit2
cell culture medium and the transfection was performed according to
the manufacturer's protocols (MOI=1). Cells were then selected with
puromycine 24 h following transfection.
Immunoprecipitation assay
The immunoprecipitation method was conducted as
previously described (4). Cells
were washed with ice-cold PBS and lysed in Tris-buffered saline (pH
7.4), containing 50 mM Tris, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 1 mM
Na3VO4, 10 mM NaF, 2.5 mg/ml aprotinin and leupeptin, 1 mM
b-glycerophosphate and AEBSF (4-(2-aminoethyl) benzenesulfonyl
fluoride hydrochloride) and 10 mM iodoacetate. Lysates were
incubated on ice for 15 min prior to removal of cellular debris and
nuclei via centrifugation at 4°C and 10,000 × g for 20 min. Cell
lysates were incubated with the AKT antibody (cat. no. 4685;
1:1,000; Cell Signaling Technology, Inc.) overnight at 4°C. Protein
A-Sepharose (GE Healthcare Life Sciences) beads in a 50:50 mixture
in 50 mM Tris buffer, (pH 7.0), were added, and further incubation
occurred for another 4 h at 4°C. The immunoprecipitates were washed
4 times in Tris-buffered saline and boiled for 5 min in 40 µl
Laemmli buffer containing 0.02% blue bromophenol and 2%
b-mercaptoethanol.
Immunohistochemistry (IHC)
Tissues were fixed with 4% formalin at 4°C
overnight, embedded in paraffin, cut as 5 µm-thick consecutive
sections and then deparaffinized at room temperature using xylene
and ethanol. Antigen recovery was performed in sodium citrate
solution (pH 6.0, 20 min, 95°C). Following this, the sections were
washed 3 times with 0.01 mol/l PBS (8 mmol/l
Na2HPO4, 2 mmol/l
NaH2PO4 and 150 mmol/l NaCl) for 5 min each.
They were then blocked for 1 h in 0.01 mol/l PBS containing 0.3%
Triton X-100 and 5% BSA (Shanghai Shenggong Biology Engineering
Technology Service, Ltd., Shanghai, China) at room temperature,
followed by addition of anti-GOLPH2 (cat. no. Ab109628; 1:100)
antibody at 4°C for at least 8 h. Following brief washing with 0.01
mol/l PBS, sections were incubated with 0.01 mol/l PBS containing
horseradish peroxidase-conjugated rabbit anti-goat IgG (1:500; cat.
no. 7074; Cell Signaling Technology, Inc.) for 2 h at room
temperature, followed by development with 0.003%
H2O2 and 0.03% 3,30-diaminobenzidine in 0.05
mol/l Tris-HCl (pH 7.6). Immunohistochemistry for each sample was
repeated three times. Slides were then developed with DAB and
counterstained with hematoxylin (room temperature, 2 min). The
slides were evaluated using a light microscope.
Crystal violet assay
BXPC3 and Suit2 cells (1,000 cells/well) were
cultured for 14 days in the incubator as detailed above. Then, the
cells were examined using 1% crystal violet staining solution at
room temperature for 5 min. Following extensive washing, cells were
imaged, dissolved using 1% SDS solution and the optical density
(OD) was measured at 600 nm. The experiments were repeated three
times. In the AKT inhibitor treatment group, cells were treated
with the inhibitor of Akt (AZD5363; MEC, Shanghai, China; 10 nM)
for 24 h.
Over-expression of KrasV12 in NIH3T3
cells
The coding sequence of KrasV12 was inserted into the
pBabe vector. The helper plasmid and KrasV12 expression vector were
co-transfected into 293T cells to produce the retrovirus. The
supernatant of the 293T cells was collected and added to the NIH3T3
culture and incubated overnight. Cells were selected with
puromycine. The resistant cells were collected and examined the
expression of exogenous KrasV12 using the antibody for HA tag.
Boyden chamber assay
The upper chamber was loaded with BXPC3 and Suit2
cells (2×105) and 0.05 ml DMEM (1% FBS), and 0.152 ml
DMEM (10% FBS) was placed in the lower chamber. Cells were
incubated for 12 h. Then, the migratory cells were examined using
hematoxylin and eosin staining at room temperature for 3 min. The
migratory cells were quantified under an inverted microscope
(Olympus Corporation, Tokyo, Japan).
Anchorage-independent growth
assay
For the soft agar assay, 5,000 cells/well were
suspended in the upper layer (0.35% agarose and 10% FBS in DMEM) in
6-well plates. The plates were coated with a bottom layer (0.5%
agarose and 10% FBS in DMEM). After 14 days of incubation, the
colonies were counted and measured. All the experiments were
performed at least three times.
PDAC mouse model (Pdx-Cre;
Loxp-Stop-Loxp KrasG12D)
Pdx-Cre; Loxp-Stop-Loxp KrasG12D mice were obtained
by crossing the Pdx-Cre mice with the Loxp-Stop-Loxp KrasG12Dmice.
Pdx-Cre-mediated recombination removes the transcriptional STOP
cassette present in the K-RasG12D allele to allow bicistronic
expression of K-RasG12D and induced the PanIN formation 8 month
after birth. c57 mice were bred in house at Fujian Medical
University (Fuzhou, China). A total of three 8-week old male mice
(body weight ~20–22 g) were included in each group. There were two
groups in total. Mice were housed at 25°C with a 12-h light/dark
cycle with sufficient food and water. After 8 months (the diameter
of the tumors was ~1 mm), the mice were sacrificed with anesthesia
(isoflurane) and the pancreatic tissues obtained.
Tumorigenesis assay
The pcDNA6 plasmids containing the coding sequence
for the luciferase gene were transfected into the BXPC3 cells using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocols. Then, the expression of
GOLPH2 was knocked down in BXPC3 cells (BXPC3/si GOLPH2). The cells
(1×106) were subcutaneously injected into the mice.
There were two groups, and four nude mice in each group (SLACK,
Shanghai, ~20 g per mouse). The mice were kept in the SPF housing
at 25°C with a 12-h light/dark cycle with sufficient food and
water. The growth of the tumor cells was monitored by an in
vivo imaging system (PerkinElmer, Inc., Waltham, MA, USA) after
administration of the luciferin, the substrate for the luciferase.
The tumor cells were traced by the luciferase gene which
metabolized luciferin and generated photons. The photons were
detected by the CCD camera and the survival rates of the mice
analyzed.
Statistical analysis
All data are presented as the mean ± standard
deviation, and statistical comparisons were performed using
Student's t-test. Multiple comparisons regarding cell growth and
migration were performed using One- or two-way analysis of variance
followed by Bonferroni's post hoc test was used for multiple
comparisons of data with normal distribution and equal variance
(D'Agostino-Pearson omnibus normality test). SPSS software, version
15.0 (SPSS, Inc., Chicago, IL, USA) was used to analyze data and
each experiment was repeated at least three times. P<0.05 was
considered to indicate a statistically significant difference.
Results
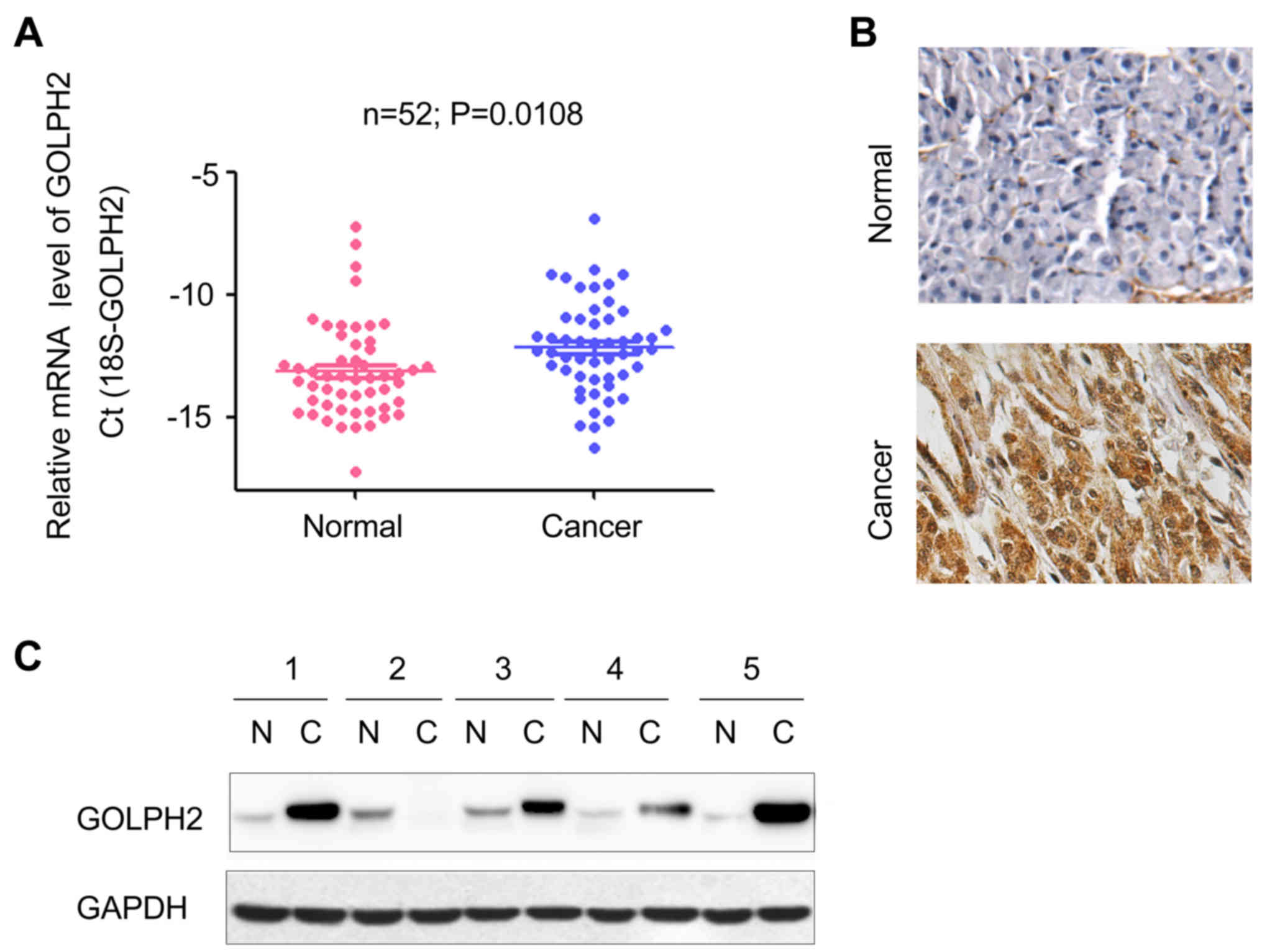
GOLPH2 is upregulated in PDAC clinical
samples
To determine the mRNA level of GOLPH2 in pancreatic
cancer tissue, RT-qPCR was performed. Compared with the adjacent
normal tissues, GOLPH2 was observed to be upregulated in PDAC
clinical samples (Fig. 1A).
Furthermore, the immunohistochemistry staining revealed that the
protein level of GOLPH2 was significantly increased in tumor
tissues (Fig. 1B), which was
consistent with the observations from Fig. 1A. The protein level of GOLPH2 in 5
PDAC tissues and paired adjacent normal tissues was examined.
Increased GOLPH2 protein levels were observed in 80% (4 out of 5)
of the PDAC tissues (Fig. 1C).
Notably, the majority of non-cancerous samples demonstrated a low
protein expression of GOLPH2, however the mRNA levels of GOLPH2
were detectable (Fig. 1A and C).
These data indicated increased expression levels of GOLPH2 in
pancreatic cancer, compared with normal healthy tissue.
GOLPH2 promotes growth and motility of
pancreatic cancer cells
To further study the role of GOLPH2 in the
progression of pancreatic cancer, GOLPH2 was overexpressed in BXPC3
and Suit2 cells (Fig. 2A). Then,
the function of increased GOLPH2 on the growth and motility of
BXPC3 and Suit2 cells was examined. As presented in Fig. 2B, GOLPH2 significantly promoted
cell growth. In addition, GOLPH2 enhanced cell migratory activity
of pancreatic cancer cells in the Boyden chamber assay (Fig. 2C). Collectively, these observations
demonstrated that GOLPH2 promoted cell growth and migration in
pancreatic cancer cells.
The effects of exogenous GOLPH2 on the growth and
migration of the pancreatic cancer cells led to examination of the
role of endogenously expressed GOLPH2. The expression of endogenous
GOLPH2 was knocked down using two independent siRNA sequences
(Fig. 3A). Knockdown of GOLPH2
expression in BXPC3 and Suit2 cells inhibited cell growth, as
demonstrated by the crystal violet staining (Fig. 3B). In addition, downregulation of
GOLPH2 attenuated the motility of pancreatic cancer cells (Fig. 3C). These observations were
consistent with the findings in the gain-of-function assay, and
further confirmed the tumor-promoting functions of GOLPH2 in
pancreatic cancer.
GOLPH2 binds with Akt and enhances Akt
activity
To explore the underlying molecular mechanism, mass
spectrometry was used to identify the binding proteins of GOLPH2
(data not shown). Akt was demonstrated to be the most efficient
candidate. The present study firstly examined the effects of GOLPH2
on the Akt signaling. Inhibiting GOLPH2 expression impaired the
phosphorylation of Akt Thr473 (Fig.
4A). Furthermore, downregulation of GOLPH2 promoted the
expression of Bax, and decreased the protein levels of Cyclin D1
and Bcl2, which are downstream effectors of Akt signaling (Fig. 4B). The inhibitor of Akt (AZD5363,
MEC, 10 nM, treated for 24 h) abolished the cell growth induced by
GOLPH2 (Fig. 4C). These findings
demonstrated the regulation of Akt signaling by GOLPH2. The
interaction between GOLPH2 and Akt was further confirmed by a
GST-pull down assay and immunoprecipitation assay. As presented in
Fig. 4D, the interaction between
GST-Akt and GOLPH2 was detected in BXPC3 cells. Furthermore, the
endogenously expressed GOLPH2 was demonstrated to form a complex
with Akt (Fig. 4E). Collectively,
these results suggested that GOLPH2 interacted with and positively
regulated Akt signaling.
 | Figure 4.GOLPH2 activates Akt signaling in
BXPC3 and Suit2 cells. (A) Knocking down the expression of GOLPH2
decreased the phosphorylation level of Akt. (B) Knocking down the
expression of GOLPH2 inhibited the expression of target genes
downstream of Akt signaling. (C) An Akt inhibitor abolished the
growth advantage of BXPC3 cells induced by GOLPH2. (D) GST-pull
down assay to verify the interaction between GOLPH2 and Akt. (E)
Interaction between Akt and GOLPH2 in BXPC3 cells was demonstrated
by an immunoprecipitation assay. GOLPH2, Golgi protein-73; si con,
small interfering RNA control; si GOLPH2, RNAi lenti-virus
targeting GOLPH2; p, phosphorylated; Akt, protein kinase B;
pcDNA3.1, control expression vector; myc-GOLPH2, myc-tagged
pcDNA3.1 GOLPH2 expression vector; Bcl-2, B-cell lymphoma; Bax,
Bcl-2 associated X, apoptosis regulator; IP, immunoprecipitation;
IgG, immunoglobin G. |
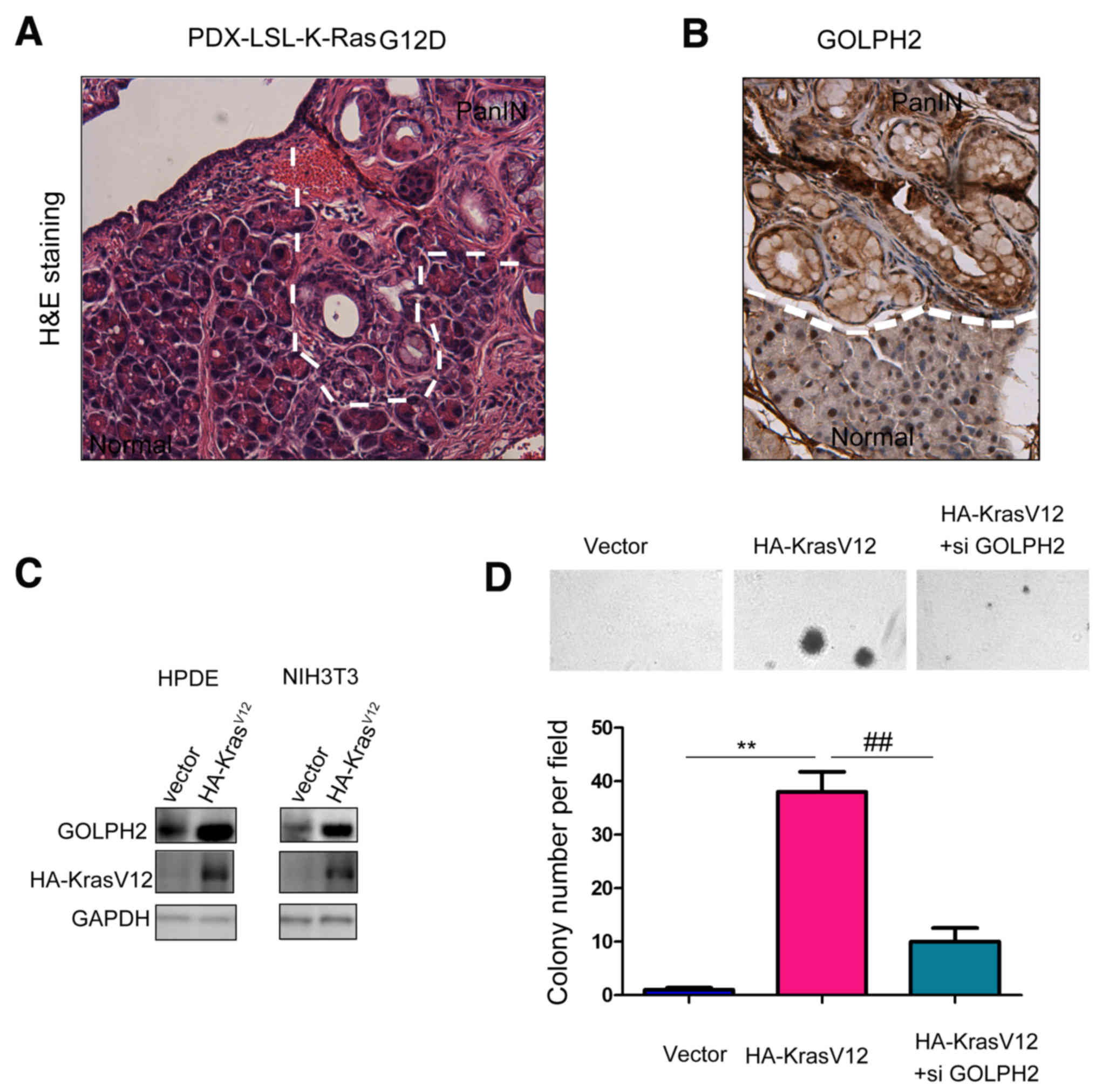
GOLPH2 is a downstream gene of Kras
signaling and promotes transformation of normal pancreatic
cells
The oncogenic functions of GOLPH2 in pancreatic
cancer cells led to an examination of its role in the early stage
of pancreatic carcinogenesis. The present study used a pancreatic
cancer mouse model Pdx-Cre; KrasG12D in which pancreatic
intraepithelial neoplasia (PanINs), precursors of PDAC, were formed
at the age of 10 months (Fig. 5A).
Next, the expression of GOLPH2 in the PanINs was examined.
Upregulation of GOLPH2 was observed in PanINs, suggesting the
regulation of GOLPH2 by the oncogenic Kras signaling (Fig. 5B). Consistent with these
observations, overexpression of KrasV12 in NIH3T3 and normal
pancreatic cells HPDE upregulated the expression of GOLPH2
(Fig. 5C). Furthermore, knocking
down the expression of GOLPH2 abolished cell transformation induced
by KrasV12 in the anchorage-independent growth assay (Fig. 5D), indicating the important
function of GOLPH2 in the tumorigenesis of pancreatic cancer.
Knockdown of GOLPH2 expression
inhibits tumorigenesis of pancreatic cancer cells in nude mice
Next, the present study investigated the effects of
GOLPH2 on the tumorigenesis of pancreatic cancer cells. BXPC3 cells
underwent forced expression of the luciferase gene, which enabled
the tracing of tumor cells in vivo. Knocking down the
expression of GOLPH2 inhibited the tumor growth in the nude mice,
which was demonstrated by the weaker photon counts (Fig. 6A and B). Furthermore,
down-regulation of GOLPH2 improved the survival of the nude mice
(Fig. 6C). These results further
emphasized the oncogenic role of GOLPH2 in pancreatic cancer.
Discussion
The present study demonstrated that expression
levels of GOLPH2 were increased in pancreatic cancer tissues
compared with adjacent normal tissue. The expression level of
GOLPH2 in the serum of the pancreatic cancer patients was not
examined, therefore further investigation of the expression of
GOLPH2 in serum of patients would clarify the importance of GOLPH2
in the diagnosis of this disease. Furthermore, GOLPH2 was
demonstrated to promote the growth and motility of cancer cells in
the present study. Notably, GOLPH2 interacted with Akt, promoted
its phosphorylation and regulated the expression of a panel of
genes involved in cell growth and apoptosis, which explained the
role of GOLPH2 in pancreatic cancer. In addition, GOLPH2 expression
was revealed to be induced by the oncogenic Kras signaling and
transformed the normal pancreatic cells. The present study revealed
the oncogenic role of GOLPH2 in pancreatic cancer, and suggested
that GOLPH2 may act as a potential target for treatment.
A previous study identified the progression from
pancreatitis to PanIN, followed by development of PDAC (14). It has previously been demonstrated
that inflammation is a high-risk factor for the development of PDAC
(15). Previous studies
demonstrated that the expression of GOLPH2 is induced under
inflammatory conditions (16). The
present study did not examine the expression of GOLPH2 in the
pancreatitis tissue, however it is possible that pancreatitis
tissues may demonstrate an upregulation of GOLPH2, and further
investigation is required to verify this. Therefore, GOLPH2 may be
upregulated at the early stage of tumorigenesis.
A previous study identified the spatial regulation
of Akt signaling, and GOLPH3 has been reported to regulate
Akt-mechanistic target of rapamycin signaling in the Golgi
apparatus (13). The results of
the aforementioned study, in combination with the results of the
present study, reveal the mechanism of the spatial regulation of
Akt signaling in the Golgi apparatus. Considering the importance of
GOLPH2 in Akt signaling, further experiments would involve
elucidating the physiological outcomes and specific molecular
mechanisms underlying the regulation of GOLPH2.
In conclusion, the results of the study revealed the
tumor-promoting role of GOLPH2 in the progression of pancreatic
cancer. GOLPH2 may serve as the therapeutic target and as a tumor
marker for PDAC. Further study using the GOLPH2 mouse model will
elucidate the novel functions of GOLPH2.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Fujian Province (grant no. 2013J05048) and
the PhD Research Startup Foundation of Fujian Medical University
(grant no. 2011BS001).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wei S, Dunn TA, Isaacs WB, De Marzo AM and
Luo J: GOLPH2 and MYO6: Putative prostate cancer markers localized
to the golgi apparatus. Prostate. 68:1387–1395. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Riener MO: Diagnosis of tumours of the
liver and the biliary tract: New tissue and serum markers.
Pathologe. 2 32 Suppl:S304–S309. 2011.(In German). View Article : Google Scholar
|
|
4
|
Riener MO, Stenner F, Liewen H,
Hellerbrand C, Bahra M and Kristiansen G: Alpha-fetoprotein and
serum golgi phosphoprotein 2 are equally discriminative in
detecting early hepatocellular carcinomas. Hepatology. 50:3262009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fritzsche FR, Riener MO, Dietel M, Moch H,
Jung K and Kristiansen G: GOLPH2 expression in renal cell cancer.
BMC Urol. 8:152008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang F, Gu Y, Li X, Wang W, He J and Peng
T: Up-regulated golgi phosphoprotein 2 (GOLPH2) expression in lung
adenocarcinoma tissue. Clin Biochem. 43:983–991. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kristiansen G, Fritzsche FR, Wassermann K,
Jäger C, Tölls A, Lein M, Stephan C, Jung K, Pilarsky C, Dietel M
and Moch H: GOLPH2 protein expression as a novel tissue biomarker
for prostate cancer: Implications for tissue-based diagnostics. Br
J Cancer. 99:939–948. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Laurent-Puig P, Lievre A and Blons H:
Mutations and response to epidermal growth factor receptor
inhibitors. Clin Cancer Res. 15:1133–1139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sharma N, Nanta R, Sharma J, Gunewardena
S, Singh KP, Shankar S and Srivastava RK: PI3K/AKT/mTOR and sonic
hedgehog pathways cooperate together to inhibit human pancreatic
cancer stem cell characteristics and tumor growth. Oncotarget.
6:32039–32060. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buck E, Eyzaguirre A, Haley JD, Gibson NW,
Cagnoni P and Iwata KK: Inactivation of Akt by the epidermal growth
factor receptor inhibitor erlotinib is mediated by HER-3 in
pancreatic and colorectal tumor cell lines and contributes to
erlotinib sensitivity. Mol Cancer Ther. 5:2051–2059. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu C, Hu DM and Zhu Q: eEF1A2 promotes
cell migration, invasion and metastasis in pancreatic cancer by
upregulating MMP-9 expression through Akt activation. Clin Exp
Metastasis. 30:933–944. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zeng Z, Lin H, Zhao X, Liu G, Wang X, Xu
R, Chen K, Li J and Song L: Overexpression of GOLPH3 promotes
proliferation and tumorigenicity in breast cancer via suppression
of the FOXO1 transcription factor. Clin Cancer Res. 18:4059–4069.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bardeesy N and DePinho RA: Pancreatic
cancer biology and genetics. Nat Rev Cancer. 2:897–909. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guerra C, Collado M, Navas C, Schuhmacher
AJ, Hernández-Porras I, Cañamero M, Rodriguez-Justo M, Serrano M
and Barbacid M: Pancreatitis-induced inflammation contributes to
pancreatic cancer by inhibiting oncogene-induced senescence. Cancer
cell. 19:728–739. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang F, Long Q, Gong Y, Hu L, Zhang H,
Oettgen P and Peng T: Epithelium-Specific ETS (ESE)-1 upregulated
GP73 expression in hepatocellular carcinoma cells. Cell Biosci.
4:762014. View Article : Google Scholar : PubMed/NCBI
|