Introduction
Congenital adrenal hyperplasia (CAH) is one of the
most common inherited metabolic disorders, and is associated with
substantial morbidity and mortality in affected children and adults
(1). Steroid 21-hydroxylase
deficiency (21-OHD), the most common variant of CAH, accounts for
~90% of CAH cases (2,3). Patients most commonly present with
symptoms associated with different extents of androgen
accumulation, a hallmark of CAH, with or without aldosterone
deficiency (1,3). Based on the broad spectrum of
presentations, CAH has been divided into two categories: Classical,
which is represented by salt wasting (SW; <2% 21-OH activity)
and simple virilizing (SV) forms (an increase of ~1–2% in 21-OH
activity compared with SW); and non-classical (NC; 20–50% 21-OH
activity).
Inherited in an autosomal recessive manner, 21-OHD
is caused by mutations in the cytochrome P450 family 21 subfamily A
member 2 (CYP21A2) gene, which is located on chromosome 6p21.3,
~3.1 kb from the highly homologous CYP21A1 pseudogene (CYP21A1P)
(4–7). At present, >200
CYP21A2-inactivating mutations are listed in the Human Gene
Mutation Database (HGMD; www.hgmd.org;
Table I). The majority of these
mutations are missense and nonsense mutations. However, splice site
mutations, deletions and insertions, in addition to other types of
mutations, have been detected. Due to complex mutation types and
the variety of clinical presentations, identification of the
genotype it is required to correlate with the phenotype.
 | Table I.Various types of CYP21A2 gene
mutation, according to the Human Gene Mutation Database. |
Table I.
Various types of CYP21A2 gene
mutation, according to the Human Gene Mutation Database.
| Mutation type | No. |
|---|
|
Missense/nonsense | 172 |
| Splicing | 18 |
| Regulatory | 2 |
| Small deletions | 22 |
| Small insertions | 13 |
| Small indels | 3 |
| Gross deletions | 13 |
| Gross insertions | 3 |
| Complex
rearrangements | 39 |
| Repeat
variations | 0 |
In the present study, molecular genetic analysis of
the CYP21A2 gene was performed in a patient with 21-OHD and other
family members. A novel compound heterozygous mutation, not
previously described, was identified and confirmed to be
responsible for 21-OHD in this Chinese pedigree. Managed with
hormone replacement therapy, the proband and the sister of the
proband succeeded in conceiving following 6 months of treatment
with dexamethasone. The results of the present study provided novel
information clarifying the CAH genotype-phenotype association,
offering evidence for patient counseling and improved prenatal
diagnosis.
Subjects and methods
Subjects
The present study was performed according to the
human research guidelines in the Declaration of Helsinki. The
research protocol was approved by the institutional review board of
Shandong Provincial Hospital, and written informed consent was
obtained from all subjects. All family members were examined and
blood was collected between September 2015-November 2015 for
genetic analysis. Subjects received a one-year follow-up.
Pedigree
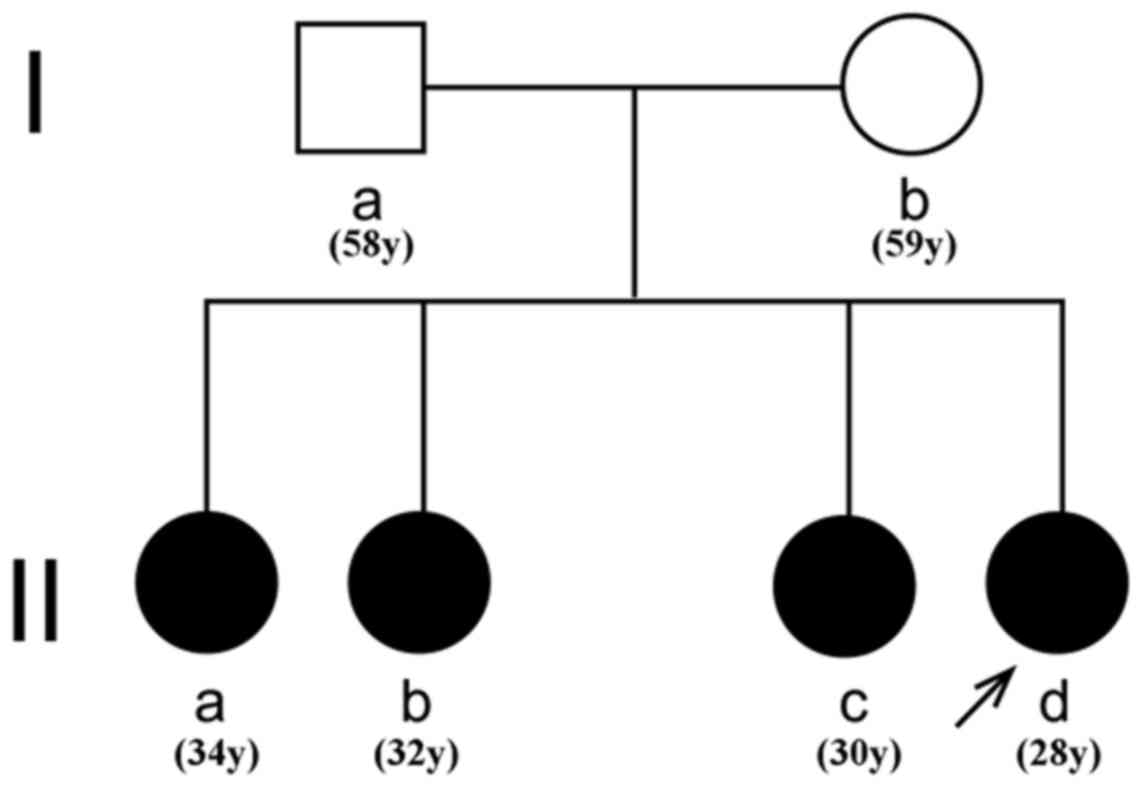
The pedigree, a two-generation family consisting of
six individuals, including the patient, and the father, mother and
three elder sisters of the patient, was referred to Shandong
Provincial Hospital (Jinan, China). The pedigree chart is displayed
in Fig. 1.
Examinations
Circadian rhythm analysis of blood cortisol and a
low-dose dexamethasone suppression test were performed. A computed
tomography (CT) scan of the adrenal gland and magnetic resonance
imaging of pituitary gland were performed. Ultrasonic examination
of gynecopathy was also accomplished.
DNA extraction, polymerase chain
reaction (PCR) analysis and sequencing
Peripheral blood (4 ml) was collected from all
family members following 12 h of fasting using EDTA. DNA was
extracted from white blood cells using a DP304-03 genomic DNA kit
(Tiangen Biotech Co., Ltd., Beijing, China). PCR was performed as
previously reported using an Eppendorf Master Cycler 5333 system
(Eppendorf, Hamburg, Germany) (8).
A total of four pairs of primers were used to amplify CYP21A2 exons
1–3, 1–6, 3–10, and 6–10 without amplifying the neighboring
CYP21A1P gene (Table II). For
gene sequencing, capillary electrophoresis was implemented using
the ABI genetic analyzer 3,130×l (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) (9). The results
were analyzed using Mutation Surveyor_5.0-UG001 (www.softgenetics.com).
| Table II.Four pairs of primers of CYP21A2. |
Table II.
Four pairs of primers of CYP21A2.
| No | Sequence (5′-3′) | Base position | Exon | Fragment size
(bp) | Annealing
temperature |
|---|
| I | F:
TCGGTGGGAGGGTACCTGAAG | − 20–1,518 | 1→6 | 1,522 | 58 |
|
| R:
AGCTGCATCTCCACGATGTGA |
|
|
|
|
| II | F:
CCTGTCCTTGGGAGACTACTC | 918–3,189 | 3→10 | 2,370 | 52 |
|
| R:
GGGACATCCGGVTTGA |
|
|
|
|
| III | F:
AGGGATCACATCGTGGAGA | 1493–3,189 | 6→10 | 1,697 | 56 |
|
| R:
GGGACATCCGGCTTTGA |
|
|
|
|
| IV | F:
TTTTGTTCTTCAGGCGATTC | −310-940 | 1→3 | 1,150 | 52 |
|
| R:
GGGAGTAGTCTCCCAGGAC |
|
|
|
|
Multiplex ligation-dependent probe
amplification (MLPA) analysis
MLPA analysis was performed in all cases to identify
large gene deletions and conversions in the CYP21A2 gene using the
SALSA MLPA kit P050CAH probemix/P050-C1-0714 CAH-v31 (MRC-Holland
BV, Amsterdam, The Netherlands). Hybridization, ligation, and
amplification were performed according to the manufacturer's
protocol. Amplification products were detected using an ABI 3,130×l
Genetic Analyzer (Thermo Fisher Scientific, Inc.) with LIZ500 as an
internal size standard. The raw data were analyzed using Coffalyser
v9.4 software (MRC-Holland BV).
Analysis of CYP21A2 gene
mutations
The whole coding region of the CYP21A2 gene,
covering the intron-exon boundaries, was directly sequenced using a
dye terminator cycle-sequencing system on an ABI genetic analyzer
3,130×l (Thermo Fisher Scientific, Inc.). The resulting sequences
were compared to the corresponding wild-type sequences of CYP21A2
using Auto Assembler software (version 2.0; PerkinElmer, Inc.,
Waltham, MA, USA). The +1 numbering of CYP21A2 genomic DNA
corresponds to the A of the ATG translation initiation codon. The
mutations were designated using the recommendations of the
Nomenclature Working Group (10,11),
in which the genomic and cDNA sequence positions were designated by
the prefixes g. and c., respectively.
Results
Patient history and clinical
evaluation
The proband, a 28-year-old Chinese female, was
referred to the Shandong Provincial Hospital. The proband presented
with 14 years of enlarged prominentia laryngea and 7 years of
irregular menstruation. At age 14, the proband was tall compared
with her peers, with prominentia laryngea enlargement and excessive
pubic hair growth, although no menstrual cramps. However, by the
age of 16, the proband was of below-average height and at age 21,
the proband began to experience abnormal menstruation. The proband
did not present to the clinic until the age of 23. Due to the
non-classical symptoms and lack of genetic identification, the
diagnosis was not accurately made prior to referral to the Shandong
Provincial Hospital. Notably, three sisters of the proband
experienced similar symptoms, although they did not consult a
doctor. At the time of examination, the proband was 153 cm in
height and 60 kg in weight, with a blood pressure of 111/70 mmHg.
Physical examination revealed excessive hair growth (including
facial, axillary, leg and pubic hair), acne, prominentia laryngea,
breast hypoplasia, outer lip hypertrophy, clitoral hypertrophy, and
oligomenorrhea. Biochemical testing (Table III) revealed elevated
testosterone, 17α-hydroxyprogesterone and androstenedione levels,
normal dehydroepiandrosterone and aldosterone levels, and normal
sodium and potassium levels. Similar biochemical abnormalities were
detected in the eldest sister of the proband (data not shown).
Circadian rhythm analysis of blood cortisol and a low-dose
dexamethasone suppression test demonstrated that the circadian
rhythm of cortisol secretion was lost and was not suppressed by
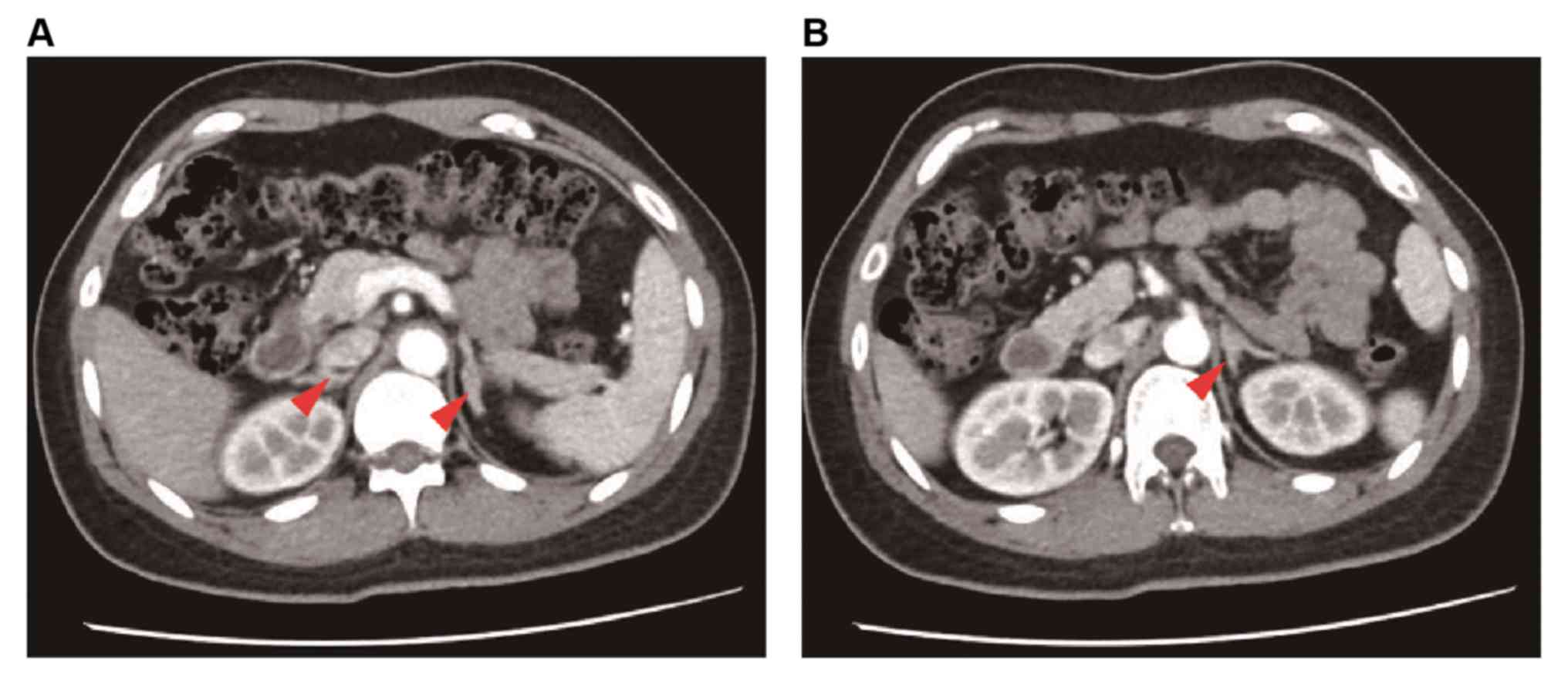
low-dose dexamethasone (data not shown). A computed tomography (CT)
scan of the adrenal gland revealed bilateral hyperplasia (Fig. 2), and magnetic resonance imaging
revealed an abnormal pituitary gland. Ultrasonic examination of
gynecopathy indicated decreased uterus volume. The chromosomal
karyotype of the proband was 46XX. The three sisters experienced
similar symptoms.
| Table III.Biochemical parameter analysis in the
proband. |
Table III.
Biochemical parameter analysis in the
proband.
| Parameter | Results | Reference
range |
|---|
| Aldosterone,
pmol/l | 203.13 | 40–310 |
| Follicle
stimulating hormone, mIU/ml | 5.61 | 1.27–19.26 |
| Luteinizing
hormone, mIU/ml | 2.76 | 1.24–8.62 |
| Progesterone,
ng/ml | 6.13 | 0.1–0.2 |
| Testosterone,
ng/ml | 1.59 | 0–0.75 |
| Testosterone, ng/ml
(post-treatment) | 0.22 | 0–0.75 |
|
17α-Hydroxyprogesterone, ng/ml | 8.97 | 0.07–1.53 |
|
Dehydroepiandrosterone, µg/dl | 271.70 | 98.8–340 |
| Androstenedione,
nmol/l | 8.25 | 4–6.6 |
| Potassium,
mmol/l | 3.70 | 3.5–5.5 |
| Sodium, mmol/l | 139.00 | 135–155 |
| Adrenocorticotropic
hormone (8:00), pg/ml | 118.20 | 7.2–63.3 |
| Cortisol (8:00),
nmol/l | 562.80 | 171–536 |
| Adrenocorticotropic
hormone (8:00), pg/ml (post-treatment) | 26.60 | 7.2–63.3 |
| Cortisol (8:00),
nmol/l (post-treatment) | 510.70 | 171–536 |
Mutation analysis
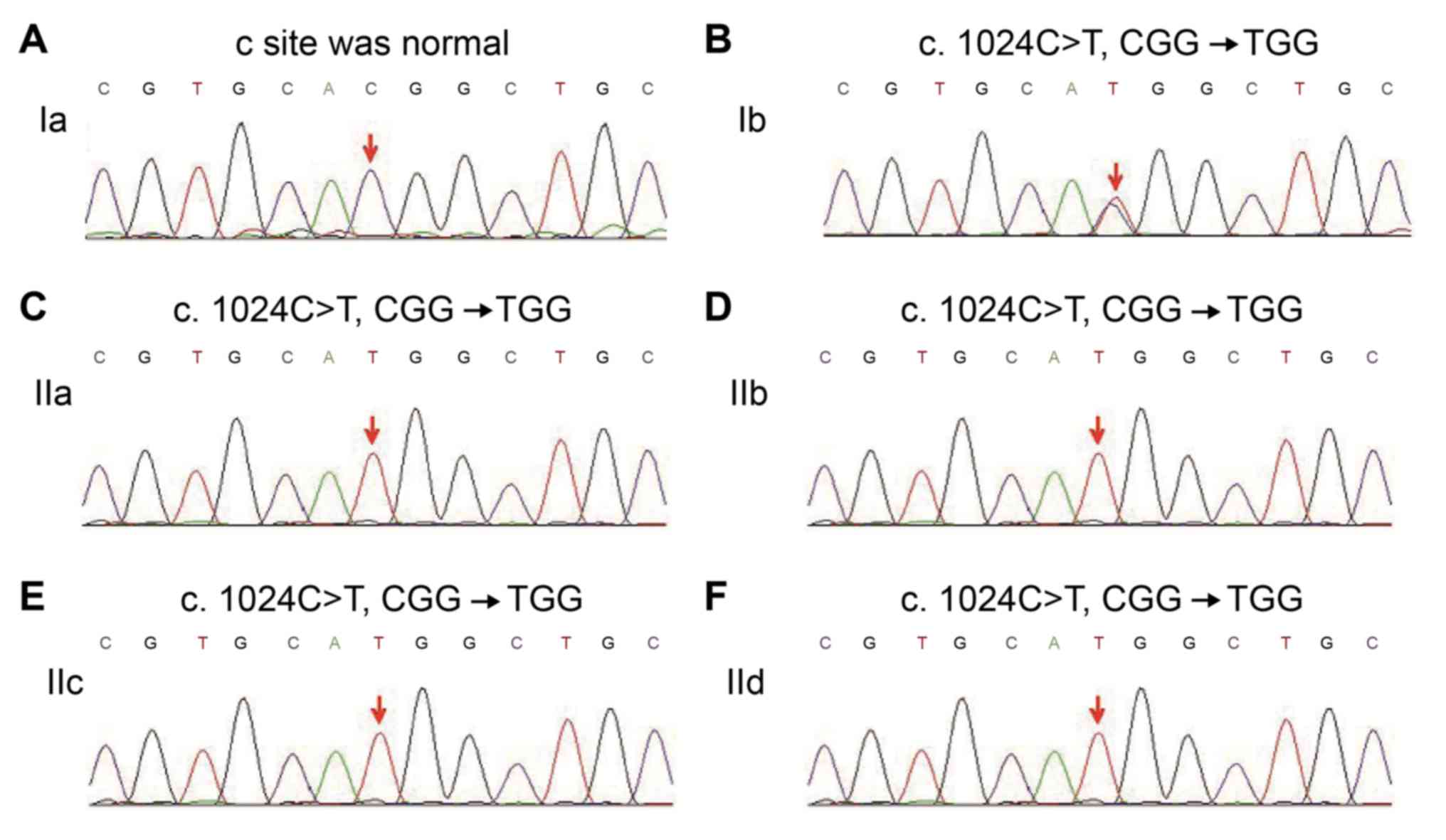
The DNA fragments of the parents and four sisters
were sequenced in the forward and reverse orientations and the
resulting sequences were compared with the corresponding wild-type
sequences of CYP21A2. A missense mutation, C-T (Fig. 3; indicated by an arrow), at
nucleotide 1,024 in exon 8 of the CYP21A2 gene, which led to the
predicted amino acid residue change from Arg to Trp at codon342,
was identified in the mother, the proband and the three sisters of
the proband. In the present study a previously reported point
mutations in exons based on the HGMD, including the missense
variant identified in the present study was summarized (Fig. 4). Additionally, MLPA analysis was
performed to determine large gene deletions. As indicated by the
red arrows in Fig. 5, a 35–50%
reduction in the relative peak height of the amplification product
of exons 1, 3, 4, 6 and 7 was identified in the father, the proband
as well as the three sisters (data not shown), indicating paternal
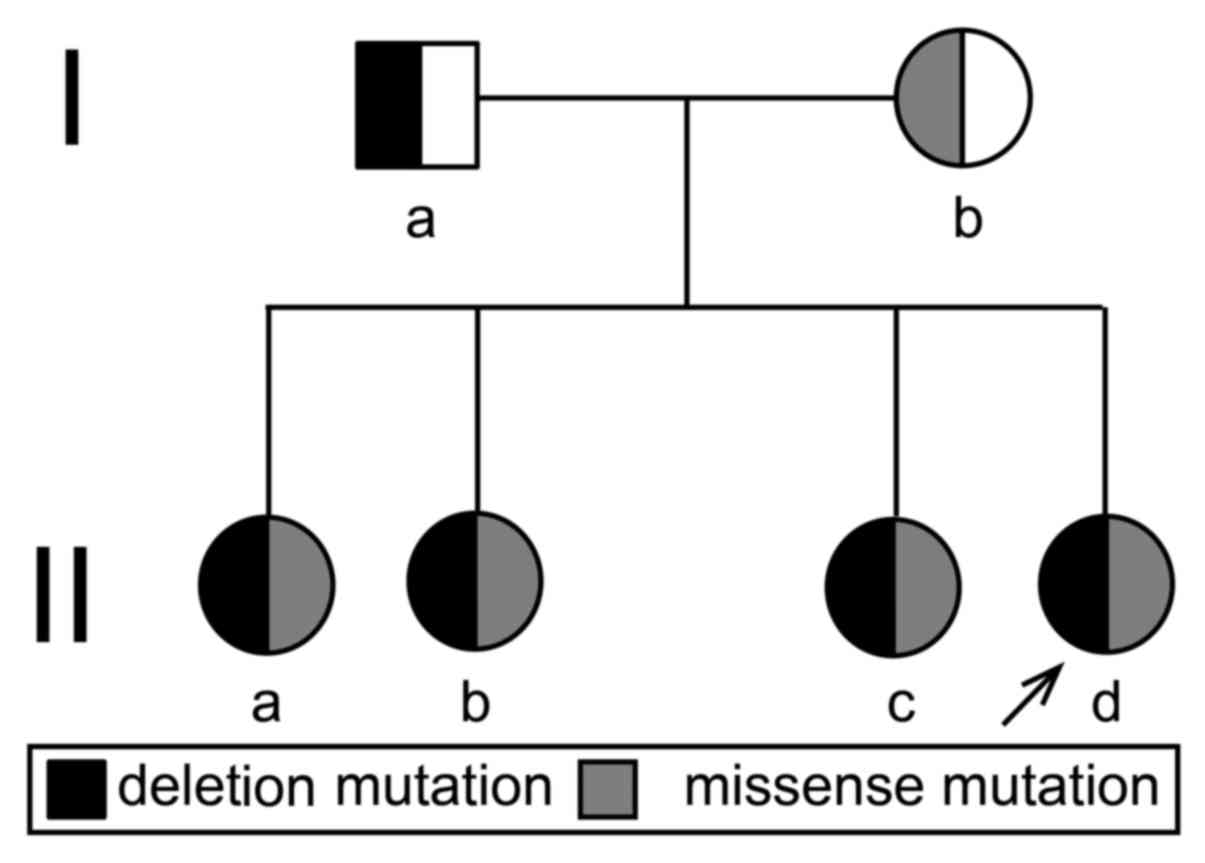
heterozygous deletion mutations. Therefore, the four sisters in the
present study all exhibited a compound heterozygous mutation in the
CYP21A2 gene, including a missense mutation c.1024C>T (p.
Arg342Trp) in exon 8 of maternal origin and heterozygous deletion
mutations of exons 1, 3, 4, 6 and 7 of paternal origin (Fig. 6).
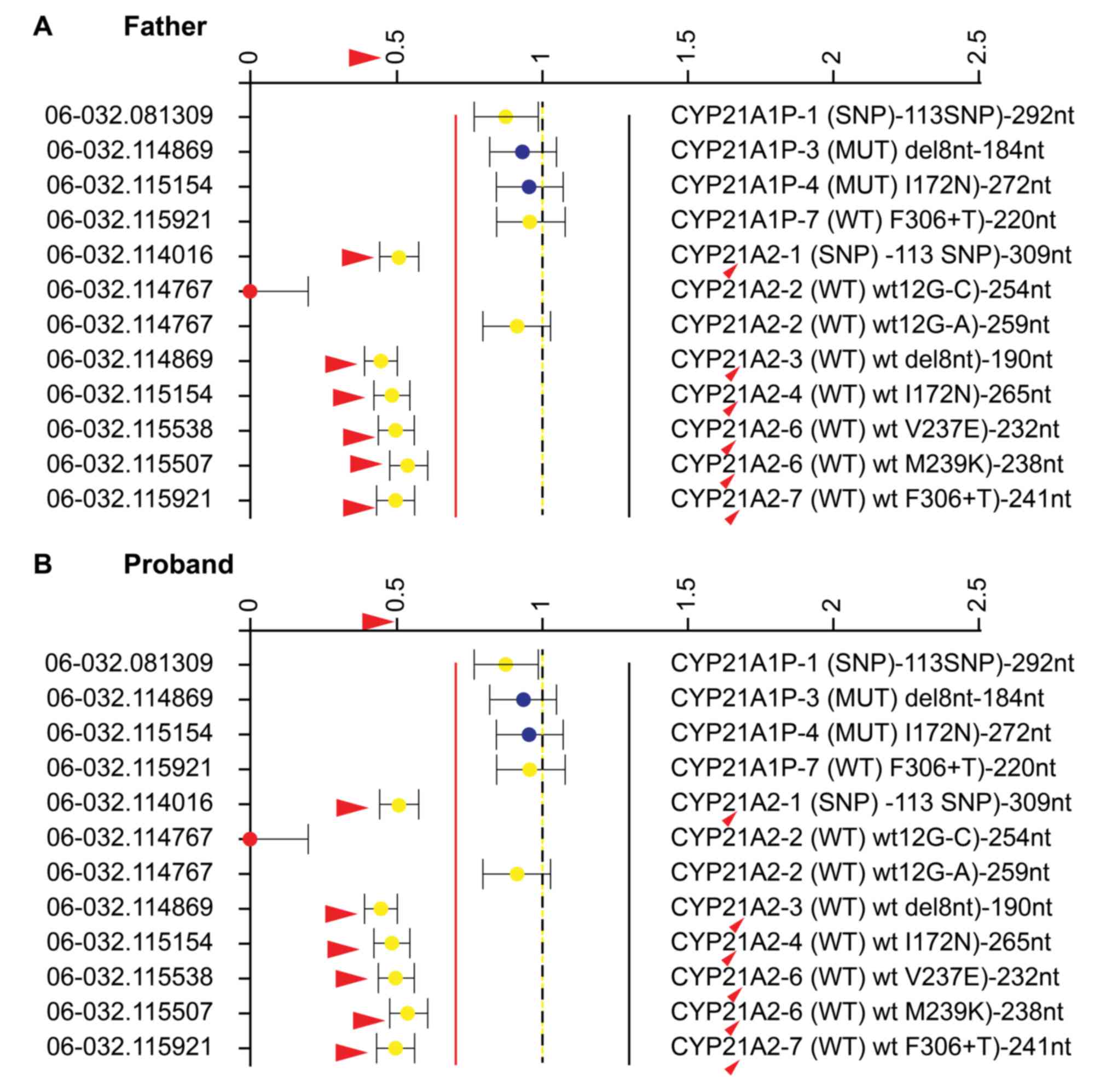 | Figure 5.Map view of the multiplex
ligation-dependent probe amplification analysis. (A) Relative peak
ratios of probes of the father, who exhibited heterozygous deletion
mutations of CYP21A2 exons 1,3,4,6 and 7 (red arrows). (B) Relative
peak ratios of probes of the proband, who presented with the same
heterozygous loss of CYP21A2 exons 1, 3, 4, 6 and 7 (red arrows).
CYP21A2, cytochrome P450 family 21 subfamily A member 2; SNP,
single-nucleotide polymorphism. |
Bioinformatics analysis
The missense mutation c.1024C>T (p. Arg342Trp) in
exon 8 results in an alteration in amino acid composition by
replacing an arginine with a tryptophan, thereby affecting the
function of the protein. Heterozygous deletion mutations in exons
1, 3, 4, 6 and 7 lead to mutations that alter a codon specific for
one amino acid to one specific for another amino acid, thus
affecting the amino acid composition and the polypeptide chain
sequence. Since the polypeptide chains lose their appropriate
primary amino sequence, they fail to form functional proteins.
Therefore, these mutations may severely affect protein function and
induce a series of pathologies.
Therapy and 1-year-follow-up
Following 4 months of treatment with dexamethasone,
the elder sister of the proband conceived. The proband received
periodic review, following the recommendations of a doctor.
Following treatment, the results gradually improved, as
testosterone, adrenocorticotropic hormone (ACTH) and cortisol
levels were within normal range (Table II). Following 6 months of
treatment with dexamethasone, the proband conceived. The results of
the present study suggested that an annual clinical follow-up maybe
recommended to observe the recovery of the patient.
Discussion
In the present study, it was demonstrated that a
compound heterozygous mutation, a missense mutation c.1024C>T
(p. Arg342Trp) and heterozygous deletion mutations of exons 1, 3,
4, 6 and 7 in the CYP21A2 gene may serve an important role in a
Chinese pedigree of four sisters affected by 21-OHD. To the best of
the authors' knowledge, this compound heterozygous mutation has not
yet been reported elsewhere. The confirmation of a CAH diagnosis by
genetic analysis is important to correlate genotype with phenotype.
In addition, the patient presented with SV-CAH, and a detailed
molecular analysis may provide useful insights into the functions
of the CYP21A2 gene.
The 21-OH locus includes a real gene, functional
CYP21A2, which encodes a protein of 495 amino acids, and the
non-functional CYP21A1P, which is a pseudogene and hence does not
code for a protein. CYP21A2 and CYP21A1 Peach consist of 10 exons
and 9 introns, with 98% homology among exons and 96% homology among
introns (5). Due to their high
homology, CYP21A2 and CYP21A1P are prone to complex and diverse
genetic recombinations, which is one of the reasons for the
relatively high incidence of mutations. A total of ~95% of CYP21A2
mutations result from intergenic recombination between CYP21A2 and
the CYP21A1P pseudogene. The complete deletion of neighboring
complement C4B and a net deletion of CYP21A1, or apparent gene
conversion events that transfer deleterious mutations normally
present in CYP21A1P to CYP21A2, may occur due to unequal crossing
over during meiosis (12). These
defects or repeated fragments stimulate RCCX unit homologous genes
to form different chimeras (13).
The sequence variants identified in the present
study are hypothesized to be disease-causing mutations in this
Chinese pedigree. The missense mutation c.1024C>T (p. Arg342Trp)
detected in the mother and four sisters is expected to exert a
substantial impact on the structure and function of the CYP21A2
protein due to the replacement of an arginine with a tryptophan.
Arginine residues that exist in the cytochrome P450 enzymes of
different mammalians are highly conserved in the family and are
important for enzymatic activity (14). Arginine is a positively-charged
polar amino acid with a long hydrophobic side chain, whereas
tryptophan is a non-polar aromatic amino acid and thus is not
conducive to the formation of an α-helix. In the present study, the
patient and mother of the patient presented with the same hybrid
genetic mutations in CYP21A2, indicating that these mutations are
of maternal inheritance. The loss of the larger pieces of the gene
adversely influences its functions, altering a number of essential
amino acids (15,16). In the present study, heterozygous
deletion mutations of exons 1, 3, 4, 6 and 7 of paternal origin
were additionally detected, leading to early termination of protein
translation and the creation of a truncated protein. The remaining
amino acids were unable to form the four important structural
domains, thereby deactivating CYP21A2.
Clinical manifestations of 21-OHD are complex and
various. 21-OHD is primarily characterized by cortisol deficiency
and the increased accumulation of progesterone,
17α-hydroxyprogesterone, and androgen, with or without aldosterone
deficiency (3,12). In general, 21-OHD may be divided
into SV, SW and NC types, according to the level of enzyme
activity. The SV form is characterized by virilization of the
external genitalia in newborn females and by hypocortisolism and
precocious pseudopuberty due to reactive androgen overproduction in
the two sexes. In addition to hyperandrogenism signs, the SW type
manifests with an insufficient secretion of aldosterone,
hyponatremia and hyperkalemia. The symptoms of the NC form are
variable and patients are asymptomatic at birth, manifesting
symptoms that include hirsutism, oligomenorrhea and anovulation
later in life. In the present study, the proband presented clitoral
hypertrophy, and biochemical tests indicated increased
17α-hydroxyprogesterone, ACTH and testosterone. Adrenal CT revealed
clear bilateral adrenal hyperplasia. The proband was diagnosed with
CAH with normal blood pressure and no symptoms of salt loss.
Therefore, the proband was diagnosed with SV CAH.
The molecular diagnosis of CAH is complex due to its
unusual features. At present, genetic diagnosis of 21-OHD for
patients with CAH is associated with two principal difficulties.
CYP21A2 is composed of 10 exons, and each exon is likely to present
genetic mutations, loss or amplification of small fragments. Thus,
it is recommended that the full-length 21-OHD be sequenced for each
patient, which is time-consuming (17). The conventional method used for the
detection of a 21-OHD mutation is as follows: A specific primer is
designed to amplify the gene segments of CYP21A2, the different
amplified fragments are directly sequenced via fragment
purification, and the full-length sequence of the CYP21A2 gene
as-obtained is used to determine the genetic mutation site
(18). The all-gene sequencing
method is used to identify the mutation site (19). The second difficulty is the
deficiency or amplification of large pieces in the 21-OHD gene of
patients with CAH, and such complexity and diversity further
complicate the detection. The most commonly used method is Southern
imprinting (20), although this is
not suitable for a large-scale population or for use in the clinic.
In addition, certain conventional methods have been adopted,
including long PCR-enzyme digestion analysis, PCR product T-A
cloning analysis, and quantitative PCR-based methods for
determining CYP21A2 gene copy number (21,22).
MLPA maybe used for copy number detection of multiple CYP21A2 gene
sites simultaneously (23). This
technology combined with gene sequencing is therefore commonly used
for CYP21A2 gene diagnosis. Additionally, when a single missing
exon is detected by MLPA inspection, it maybe verified by
sequencing and by long-segment PCR.
The phenotype of 21-OHD is associated with its
genotype, including point mutations, frame shift mutations and
deletion mutations of small DNA fragments. SW-, SV- or NC-type
21-OHD maybe predicted by clinical gene sequencing. Concolino et
al (24) demonstrated that the
consensus between genotype and SW CAH patients was 81.8%.
Baumgartner-Parzer et al (25) reported that the consistency between
genotype and clinical phenotype was 80%. However, genotype and
clinical phenotype are not always completely consistent, and the
same 21-OHD gene mutation in patients with CAH may result in
different clinical phenotypes (26,27).
New et al (28) reported
that certain mutations, including the P30L, I2Gand I172N mutations,
were prone to yield different CAH phenotypes. In SW and NC CAH, a
phenotype maybe attributed to a genotype. However, in SV CAH, wide
phenotypic variability may be observed, particularly with the exon
4 I172N mutation (28). Janjanin
et al (29) described five
patients with 21-OHD belonging to three generations of the same
family (grandmother, parents and two children), whose genotype
corresponded to NC CAH. Notably, in spite of one minorCYP21
mutation, the proband presented with the SV form of 21-OHD and
required glucocorticoid replacement therapy from the age of 4
years. In the present study, the CYP21A2 gene sequencing of the
patient, in addition to the clinical symptoms and gene sequencing
results, revealed c.1024C>T (p. Arg342Trp) and heterozygous
deletion mutations, which supported the clinical diagnosis of SV
CAH.
The present study provided strong evidence for the
presence of a novel compound heterozygous mutation in the CYP21A2
gene that is predicted to result in the partial loss of enzymatic
activity of the resulting protein. This mutation caused SV 21-OHD
in a Chinese pedigree. The results of the present study provided
information for the prenatal diagnosis of CAH caused by 21-OHD
using amniotic cells or chorionic villi. The in vitro
expression analysis of residual enzymatic activity may be performed
to analyze the functional consequences of this newly identified
mutation, thereby proving its relevance to the clinical
presentation and analyzing its impact on CYP21A2 function. By
investigating the molecular genetic mechanisms of CAH, the
identification and diagnosis may be optimized, which has
substantial clinical value for diagnosis and genetic counseling of
carriers in the family of a patient.
Acknowledgements
The present study was supported by the grants from
the National Natural Sciences Foundation of China (grant nos.
81100593, 81770785, 81370891 and 81670720), special funds for
Taishan Scholar Project (grant no. tsqn20161071) and the Provincial
key research and development plan (2017GSF18154).
References
|
1
|
Bachelot A, Grouthier V, Courtillot C,
Dulon J and Touraine P: Management of endocrine disease: Congenital
adrenal hyperplasia due to 21-hydroxylase deficiency: Update in
management of adult patients and prenatal treatment. Eur J
Endocrinol. 176:R167–R181. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Forest MG: Recent advances in the
diagnosis and management of congenital adrenal hyperplasia due to
21-hydroxylase deficiency. Hum Reprod Update. 10:469–485. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Speiser PW, Azziz R, Baskin LS, Ghizzoni
L, Hensle TW, Merke DP, Meyer-Bahlburg HF, Miller WL, Montori VM,
Oberfield SE, et al: Congenital adrenal hyperplasia due to steroid
21-hydroxylase deficiency: An endocrine society clinical practice
guideline. J Clin Endocrinol Metab. 95:4133–4160. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
White PC, Grossberger D, Onufer BJ,
Chaplin DD, New MI, Dupont B and Strominger JL: Two genes encoding
steroid 21-hydroxylase are located near the genes encoding the
fourth component of complement in man. Proc Natl Acad Sci USA.
82:pp. 1089–1093. 1985; View Article : Google Scholar : PubMed/NCBI
|
|
5
|
White PC, New MI and Dupont B: Structure
of human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA.
83:pp. 5111–5115. 1986; View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bruque CD, Delea M, Fernández CS, Orza JV,
Taboas M, Buzzalino N, Espeche LD, Solari A, Luccerini V, Alba L,
et al: Structure-based activity prediction of CYP21A2 stability
variants: A survey of available gene variations. Sci Rep.
6:390822016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee HH: Mutational analysis of CYP21A2
gene and CYP21A1P pseudogene: Long-range PCR on genomic DNA.
Methods Mol Biol. 1167:275–287. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li C, Zhou X, Han W, Jiang X, Liu J, Fang
L, Wang H, Guan Q, Gao L, Zhao J, et al: Identification of two
novel mutations in SLC12A3 gene in two Chinese pedigrees with
Gitelman syndrome and review of literature. Clin Endocrinol (Oxf).
83:985–993. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Menassa R, Tardy V, Despert F,
Bouvattier-Morel C, Brossier JP, Cartigny M and Morel Y: p.H62L, a
rare mutation of the CYP21 gene identified in two forms of
21-hydroxylase deficiency. J Clin Endocrinol Metab. 93:1901–1908.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Antonarakis SE: Recommendations for a
nomenclature system for human gene mutations. Nomenclature Working
Group. Hum Mutat. 11:1–3. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
den Dunnen JT and Antonarakis SE: Mutation
nomenclature extensions and suggestions to describe complex
mutations: A discussion. Hum Mutat. 15:7–12. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
White PC: Congenital adrenal hyperplasias.
Best Pract Res Clin Endocrinol Metab. 15:17–41. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bánlaki Z, Szabó JA, Szilágyi Á, Patócs A,
Prohászka Z, Füst G and Doleschall M: Intraspecific evolution of
human RCCX copy number variation traced by haplotypes of the
CYP21A2 gene. Genome Biol Evol. 5:98–112. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barbaro M, Soardi FC, Östberg LJ, Persson
B, de Mello MP, Wedell A and Lajic S: In vitro functional studies
of rare CYP21A2 mutations and establishment of an activity gradient
for nonclassic mutations improve phenotype predictions in
congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 82:37–44.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu Y, Wang J, Huang X, Wang Y, Yang P, Li
J, Tsuei SH, Shen Y and Fu Q: Molecular characterization of 25
Chinese pedigrees with 21-hydroxylase deficiency. Genet Test Mol
Biomarkers. 15:137–142. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Coeli FB, Soardi FC, Bernardi RD, de
Araújo M, Paulino LC, Lau IF, Petroli RJ, de Lemos-Marini SH,
Baptista MT, Guerra-Júnior G and de-Mello MP: Novel deletion
alleles carrying CYP21A1P/A2 chimeric genes in Brazilian patients
with 21-hydroxylase deficiency. BMC Med Genet. 11:1042010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Concolino P, Mello E, Zuppi C and
Capoluongo E: Molecular diagnosis of congenital adrenal hyperplasia
due to 21-hydroxylase deficiency: An update of new CYP21A2
mutations. Clin Chem Lab Med. 48:1057–1062. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee HH, Chao HT, Ng HT and Choo KB: Direct
molecular diagnosis of CYP21 mutations in congenital adrenal
hyperplasia. J Med Genet. 33:371–375. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Concolino P, Mello E, Toscano V, Ameglio
F, Zuppi C and Capulongo E: Multiplex ligation-dependent probe
amplification (MLPA) assay for the detection of CYP21A2 gene
deletions/duplications in congenital adrenal hyperplasia: First
technical report. Clin Chim Acta. 402:164–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
White PC, Vitek A, Dupont B and New MI:
Characterization of frequent deletions causing steroid
21-hydroxylase deficiency. Proc Natl Acad Sci USA. 85:pp.
4436–4440. 1988; View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Parajes S, Quinterio C, Domínguez F and
Loidi L: A simple and robust quantitative PCR assay to determine
CYP21A2 gene dose in the diagnosis of 21-hydroxylase deficiency.
Clin Chem. 53:1577–1584. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee HH, Lee YJ and Lin CY: PCR-based
detection of the CYP21 deletion and TNXA/TNXB hybrid in the RCCX
module. Genomics. 83:944–950. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Krone N, Reisch N, Idkowiak J, Dhir V,
Ivison HE, Hughes BA, Rose IT, O'Neil DM, Vijzelaar R, Smith MJ, et
al: Genotype-phenotype analysis in congenital adrenal hyperplasia
due to P450 oxidoreductase deficiency. J Clin Endocrinol Metab.
97:E257–E267. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Concolino P, Mello E, Patrosso MC, Penco S
and Zuppi C: p.H282N and p.Y191H: 2 novel CYP21A2 mutations in
Italian congenital adrenal hyperplasia patients. Metabolism.
61:519–524. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baumgartner-Parzer SM, Fischer G and
Vierhapper H: Predisposition for de novo gene aberrations in the
offspring of mothers with a duplicated CYP21A2 gene. J Clin
Endocrinol Metab. 92:1164–1167. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Leccese A, Longo V, Dimatteo C, De
Girolamo G, Trunzo R, D'Andrea G, Bafunno V, Margaglione M and
Santacroce R: Lack of genotype-phenotype correlation in congenital
adrenal hyperplasia due to a CYP21A2-like gene. Clin Chim Acta.
437:48–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Anastasovska V and Kocova M:
Genotype-phenotype correlation in CAH patients with severe CYP21A2
point mutations in the Republic of Macedonia. J Pediatr Endocrinol
Metab. 23:921–926. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
New MI, Abraham M, Gonzalez B, Dumic M,
Razzaghy-Azar M, Chitayat D, Sun L, Zaidi M, Wilson RC and Yuen T:
Genotype-phenotype correlation in 1,507 families with congenital
adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl
Acad Sci USA. 110:pp. 2611–2616. 2013; View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Janjanin N, Dumic M, Skrabic V, Kusec V,
Grubic Z and Spehar Uroic A: Five patients with congenital adrenal
hyperplasia due to 21-hydroxylase deficiency (one with associated
neuroblastoma) discovered in three generations of one family. Horm
Res. 67:111–116. 2007.PubMed/NCBI
|