Introduction
Perioperative myocardial ischemia is a common
condition associated with high mortality (1). Removal of the occlusion and
reperfusion of the ischemic area promotes additional cardiac injury
that exacerbates the process of infarction. This phenomenon is
termed ischemia/reperfusion (I/R) injury. Ischemic preconditioning
was first identified in 1986 by Murry as a phenomenon in which
repeated short episodes of ischemia protect the myocardium against
a subsequent I/R injury (2).
However, clinical application was unsatisfactory for the
unpredictability of ischemia. Ischemic postconditioning, a novel
cardioprotective intervention against I/R injury is performed
following ischemia, may offer a solution to this problem (3). In addition, a number of different
pharmacological agents have been reported to mimic ischemic
postconditioning to achieve cardioprotection (4,5).
Propofol is a widely used intravenous agent for the
induction and maintenance of anesthesia (6). Our previous study revealed that
propofol postconditioning (P-PostC) had protective effects against
hypoxia/reoxygenation (H/R)-induced apoptosis in cardiomyocytes
(7). However, the mechanism by
which P-PostC functions against I/R injury requires further
investigation. Autophagy is an evolutionarily conserved protein
degradation pathway that is essential for the intracellular
homeostasis between biosynthesis and catabolism (8,9). In
terminally differentiated cells, such as cardiomyocytes, autophagy
may serve an important role in cellular self-protection,
facilitating survival under stress (10). An increasing number of studies have
demonstrated that autophagy may be necessary for cardioprotection
conferred by preconditioning (11,12);
the mechanism may be associated with the upregulation of endogenous
protective mechanisms against myocardial I/R injury (13). The c-Jun NH2-terminal kinases (JNK)
were initially identified as the stress-activated protein kinases
(SAPK) which are activated by a number of stressors including I/R
injury and serve an important role in multiple stimulation-induced
autophagic events. Recent studies proved that JNK signal pathway
participated in the induction of p62 expression and its binding to
microtubule-associated protein 1A/1B-light chain 3 (LC3) (14). LC3 is an important protein involved
in autophagy, LC3-I is conjugated to phosphatidylethanolamine to
form LC3-II which is recruited to autophagosomal membranes
(15). p62 (also known as
sequestosome 1) is a ubiquitin- and LC3-binding protein involved in
cell autophagy; the induction of autophagy is accompanied by p62
protein degradation (16).
The present study hypothesized that propofol may
induce autophagy to protect the myocardium. To test this
hypothesis, the effects of P-PostC at various doses on the
induction of autophagy and the potential regulatory mechanisms were
investigated using an in vitro H/R model using rat
heart-derived H9c2 cells.
Materials and methods
Reagents
Propofol, 3-methyladenine (3-MA), acridine orange
(AO), monodansylcadaverine (MDC) and the c-Jun N-terminal kinase
(JNK) inhibitor SP600125 were purchased from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany) and were dissolved in dimethyl sulfoxide
(DMSO), and further diluted in PBS. The final DMSO concentration
was 0.1%, which did not affect cell function and the assay system.
Fetal bovine serum (FBS) and Dulbecco's modified Eagle's medium
(DMEM) were purchased from Gibco (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Primary antibodies used in the study included:
Rabbit anti-microtubule-associated protein 1A/1B-light chain 3B
(LC3B; cat. no. L7543; Sigma-Aldrich; Merck KGaA) and rabbit
anti-p62 (cat. no. P0067; Sigma-Aldrich; Merck KGaA); rabbit
anti-stress-activated protein kinase (SAPK)/JNK (cat. no. 9252;
Cell Signaling Technologies, Danvers, MA, USA) and rabbit
anti-phosphorylated (p)-SAPK/JNK (cat. no. 4668; Cell Signaling
Technologies) and mouse anti-β-actin (cat. no. sc130301; Santa Cruz
Biotechnology, Dallas TX, USA). Secondary antibodies included
horseradish peroxidase-conjugated goat anti-mouse (cat. no. sc2031;
Santa Cruz Biotechnology) and goat anti-rabbit immunoglobulin G
(cat. no. sc2007; Santa Cruz Biotechnology).
Cell line and culture conditions
The H9c2 embryonal rat heart-derived (cardiac
muscle) cell line was obtained from the American Type Culture
Collection (Manassas, VA, USA) and cultured in DMEM containing 4.5
g/l D-glucose, 1.5 g/l sodium bicarbonate and 110 mg/l sodium
pyruvate, supplemented with 10% FBS, penicillin (100 U/ml) and
streptomycin (100 Ag/ml), in a humidified incubator with 95% air
and 5% CO2 at 37°C.
H/R model and experimental groups
Hypoxia was established in Modular Incubator
Chambers (Billups-Rothenberg, Inc., Del Mar, CA, USA); the chambers
were flushed with a gas mixture of 95% N2 and 5%
CO2 at room temperature for 30 min at 10 l/min.
Following flushing, the chambers were sealed and maintained at 37°C
for 6 h. The concentration of O2 in each chamber was
monitored with an oxygen indicator (Mitsubishi Gas Chemical
Company, Inc., Tokyo, Japan). Hypoxia was terminated by exposing
the cells to fresh DMEM containing 10% FBS and incubating them in a
normal incubator (95% air; 5% CO2) for 4 h at 37°C to
simulate reperfusion (reoxygenation). Propofol of concentrations
between 12.5–100 µmol/l was added into the fresh medium at the
commencement of reperfusion/reoxygenation. SP600125 and 3-MA
treatments were administered at a final concentration of 10 µmol/l
and 10 mmol/l, respectively, according to previously published
methods (17,18), for 1 h prior to hypoxia exposure.
The cells were trypsinized with 0.25% trypsin at 37°C, harvested at
the centrifugation at 800 × g for 3 min at room temperature and
washed twice with PBS at the end of reoxygenation.
H9c2 cells were separated into the following
experimental groups: i) Control group of normally cultured H9c2
cells; ii) H/R group, which were subjected to H/R; iii) P-PostC
group, which received propofol treatment upon H/R; iv) P-PostC +
SP600125 group, which received SP600125 and P-PostC co-treatment
upon H/R (group); v) P-PostC+3-MA group, which were subjected to
3-MA and P-PostC co-treatment under H/R; vi) P-PostC + 3-MA +
SP600125 group, which received 3-MA, SP600125 and P-PostC
co-treatment upon H/R; vii) vehicle-treated group, which were H9c2
cells normally cultured with 0.1% DMSO in the culture medium; and
viii) propofol-treated group, which comprised vehicle-treated cells
subjected to propofol administration without H/R.
Assessment of lactate dehydrogenase
(LDH) activity and cell viability in culture medium
To measure the extent of cell injury, the LDH
activity was analyzed. Cells were plated in a 6-well plate at a
density of 5×105 cells/well. At the end of the
reoxygenation, the supernatants were collected. The LDH assay kit
(A020-1; Jiancheng Bioengineering Institute, Nanjing, China) was
employed according to the manufacturer's protocols. Briefly, 0.1 ml
culture medium was added into 3 ml LDH assay reaction mixture.
After 3 min incubation at 37°C, the optical density was measured at
a wavelength of 440 nm using a spectrophotometer. H9c2 cell
viability in the different treatment groups was analyzed by MTT
assay. Cells were plated at 1×104 cells/well in 96-well
microtiter plates. Each group was repeated in 10 wells. Following
treatment, MTT (5 mg/ml in PBS) was added to each well and
incubated for 3 h at 37°C. After careful removal of the medium, the
formazan crystals were dissolved in 200 µl DMSO and absorbance (A)
values were determined at 570 nm using a microplate reader (Model
550; Bio-Rad Laboratories, Inc., Hercules, CA, USA) within 30 min
following the dissolution of the formazan crystals.
Detection of myocyte apoptosis
Following treatment, myocytes were harvested with
0.25% trypsin and subjected to apoptosis analysis using an Annexin
V-fluorescein isothiocyanate (FITC) detection kit (Beyotime
Institute of Biotechnology, Haimen, China). Subsequently, the cell
density was adjusted to 1×106 cells/ml, 5 µl Annexin
V-FITC and 10 µl propidium iodide (PI) (1 µg/ml) were then added to
these cells and incubated for 10–20 min at room temperature
(20–25°C) in the dark. The rate of apoptosis was then analyzed with
a FACScalibur flow cytometer and calculated by CellQuest software
(Becton Dickinson, Franklin Lakes, NJ, USA). Early apoptotic cells
were positive for Annexin V-FITC and negative for PI.
MDC and AO staining of
autophagosomes
H9c2 cells were seeded at a density of
1×104 cells/well onto coverslips fixed in culture dishes
and allowed to reach 70–80% confluence. MDC, a selective
fluorescent marker preferentially accumulates in autophagic
vacuoles, exhibits alterations in fluorescence that can be observed
using a fluorescence microscope. Autophagy is characterized by
increased formation of lysosomes and autophagolysosomes which can
be stained with AO. Following H/R and P-PostC, cells were incubated
with 0.05 mmol/l MDC or 1 mg/ml AO for 15 min, fixed with 3.7%
paraformaldehyde in PBS for 30 min at 37°C, rinsed with PBS and
dried. Cells were observed under an IX70 fluorescence microscope
(Olympus Corporation, Tokyo, Japan) equipped with a filter system:
MDC, excitation wavelength, 380 nm and emission filter, 525 nm; AO,
excitation wavelength, 488 nm and emission filter, 525 nm. Five
randomly selected fields were observed per slide. Data were
obtained from ≥3 independent experiments.
Protein lysate preparation,
electrophoresis and immunoblot analysis
H9c2 cells were plated at a density of
1×106 cells/well in 6-well culture dishes. Following
treatment, cells were washed with PBS and lysed on ice for 30 min
in radioimmunoprecipitation assay buffer containing 50 mM Tris HCl
(pH 7.5), 250 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 1 mM PMSF, 1
mM DTT, 1 µg/ml leupeptin, 1 µg/ml aprotinin and 1 µg/ml pepstatin
(Sigma-Aldrich, Merck KGaA). Cell lysates were then centrifuged at
13,000 × g for 10 min at 4°C, and protein concentrations were
assayed using a Bicinchoninic Acid assay kit (Beyotime Institute of
Biotechnology). Protein samples were separated by 12% SDS-PAGE and
transferred to nitrocellulose membranes using an electro-blotting
apparatus (Bio-Rad Laboratories, Inc.). The membrane was blocked
with 5% nonfat milk in TBST solution, and incubated overnight with
primary antibodies (anti-LC3B, 1:1,000; anti-p62, 1:1,000;
anti-JNK, 1:1,000; anti-p-JNK, 1:1,000 and anti-β-actin, 1:500) in
the blocking solution at 4°C. After three washes with TBST
solution, the membrane was incubated at room temperature for 1 h,
with horseradish peroxidase-conjugated secondary antibody diluted
with TBST solution (1:3,000). The signals of detected proteins were
visualized by an enhanced chemiluminescence reaction (ECL) system
(Amersham, ECL kits). For semi-quantitative analysis, protein bands
detected by ECL were scanned into Adobe Photoshop CS6 (Adobe
Systems, Inc., San Jose, CA, USA) and analyzed using ImageJ
software, version 1.6 (National Institutes of Health, Bethesda, MD,
USA).
Statistical analysis
Data are reported as the mean ± standard deviation,
and statistical significance was assessed using SPSS 18.0 software
(SPSS, Inc., Chicago, IL, USA). Differences between groups were
evaluated with analysis of variance (ANOVA) followed by a
Bonferroni's post hoc test for multiple comparisons. Prior to
ANOVA, Levene's test for equality of variances was performed; data
were confirmed to be normally distributed. P<0.05 was considered
to indicate a statistically significant difference.
Results
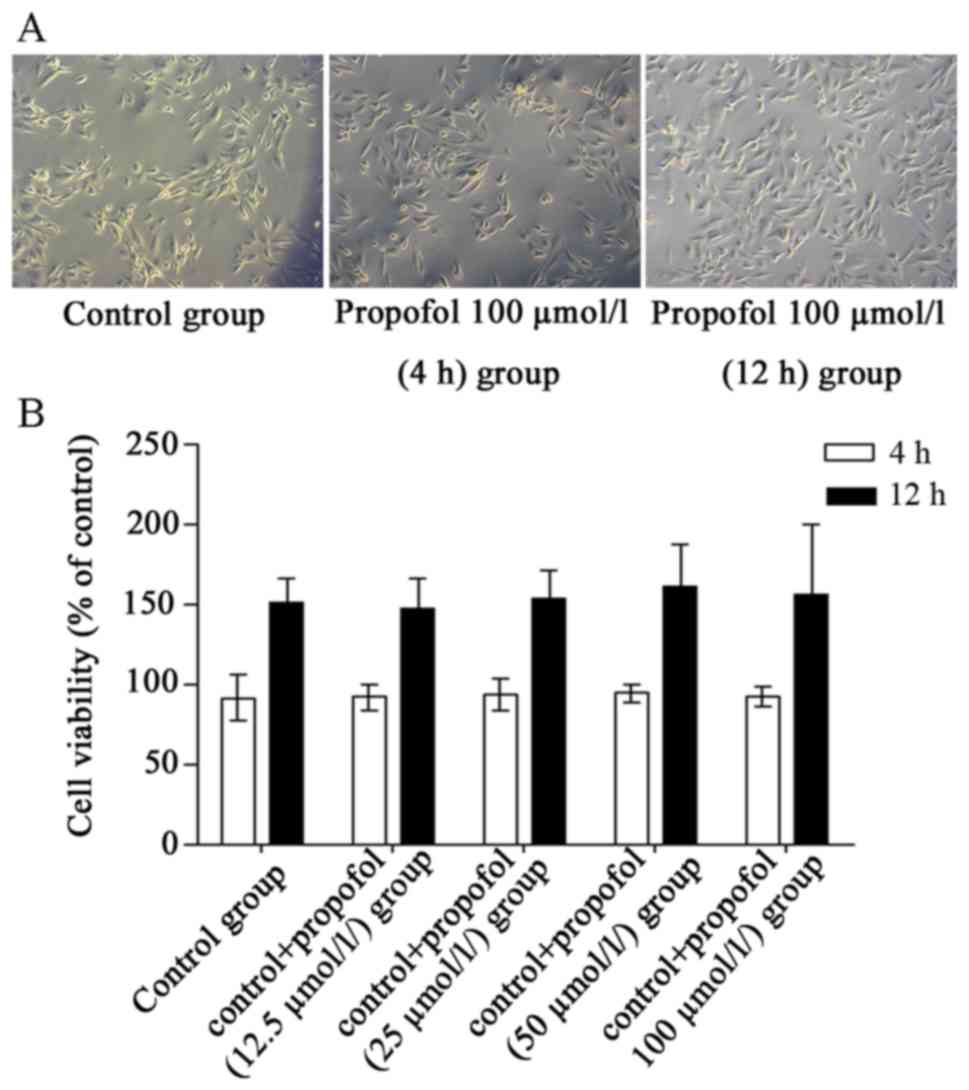
Propofol treatment alone exhibits no
effects on the survival and proliferation of cultured H9c2
cells
To exclude possible interference by using animals, a
heart-derived H9c2 cell line for in vitro observation was
used in the present study. H9c2 cells have been extensively used in
cardiological research, as they possess elements and properties of
the signaling pathways of adult myocytes (19), thus offering a suitable in
vitro experimental H/R model. To examine whether propofol
treatment alone affected the viability of cultured cardiomyocytes,
morphological observations and MTT cell viability assays were
performed, following treatment with propofol for 4 and 12 h.
Vehicle-treated cells (4 h) were considered to be 100% viable, the
viability of vehicle-treated cells (12 h) was 150% compared with
Vehicle-treated cells (4 h). No notable morphological alterations
were observed in propofol-treated cells (Fig. 1A) and no significant differences in
cell viability were detected (P>0.05; Fig. 1B) compared with the vehicle-treated
group.
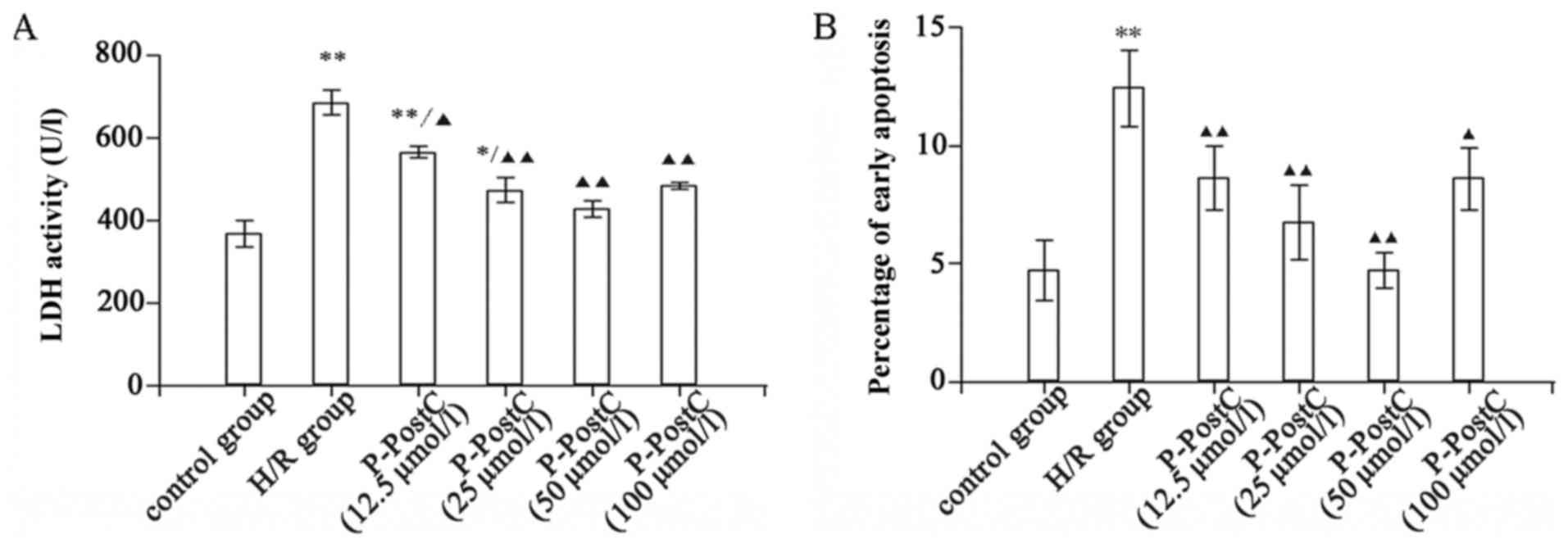
P-PostC protects H9c2 cells against
H/R injury by reducing LDH levels and the number of apoptotic
cells
Cellular damage was assessed by measuring the
activity levels of the cytosolic enzyme LDH released into the
medium, induced by cell membrane leakage (20). Following H/R exposure, cells
exhibited a significant increase in LDH activity in the culture
medium, 1.9-fold of the level in the control group (Fig. 2A). This increase was significantly
reduced by P-PostC treatments at various concentrations (P<0.01
vs. H/R; Fig. 2A). The
cardioprotection of P-PostC against H/R injury was further examined
by determining the rate of cardiomyocyte apoptosis. The percentage
of early apoptotic cells in the untreated control and H/R groups
were 4.72±1.3 and 12.45±1.6%, respectively (P<0.05). The
proportion of Annexin V-FITC-stained cells in the 12.5–50 µmol/l
P-PostC-treated groups decreased significantly and in a
dose-dependent manner, compared with the H/R group (Fig. 2B) There was a marked reduction in
the number of early apoptotic cells, ranging between 4.7±0.8 and
8.6±1.3%, with different concentrations (12.5–50 µmol/l) of
propofol compared with H/R. This indicated that 12.5–50 µmol/l
P-PostC conferred dose-dependent protection against H/R injury to
cardiomyocytes. However, the percentage of early apoptotic cells in
the 100 µmol/l P-PostC group was 8.5±1.1%, compared with in the 50
µmol/l P-PostC-treated group (4.7±0.8%, P<0.05); the optimal
concentration of propofol for P-PostC was demonstrated to be 50
µmol/l. P-PostC (12.5–50 µmol/l) protected the H/R cardiomyocytes
from apoptosis, whereas propofol concentrations >100 µmol/l
exhibited less positive effects on cell apoptosis. These data were
consistent with a previous study and may be due to the known
inhibitory effect of calcium channels at high concentrations
(21).
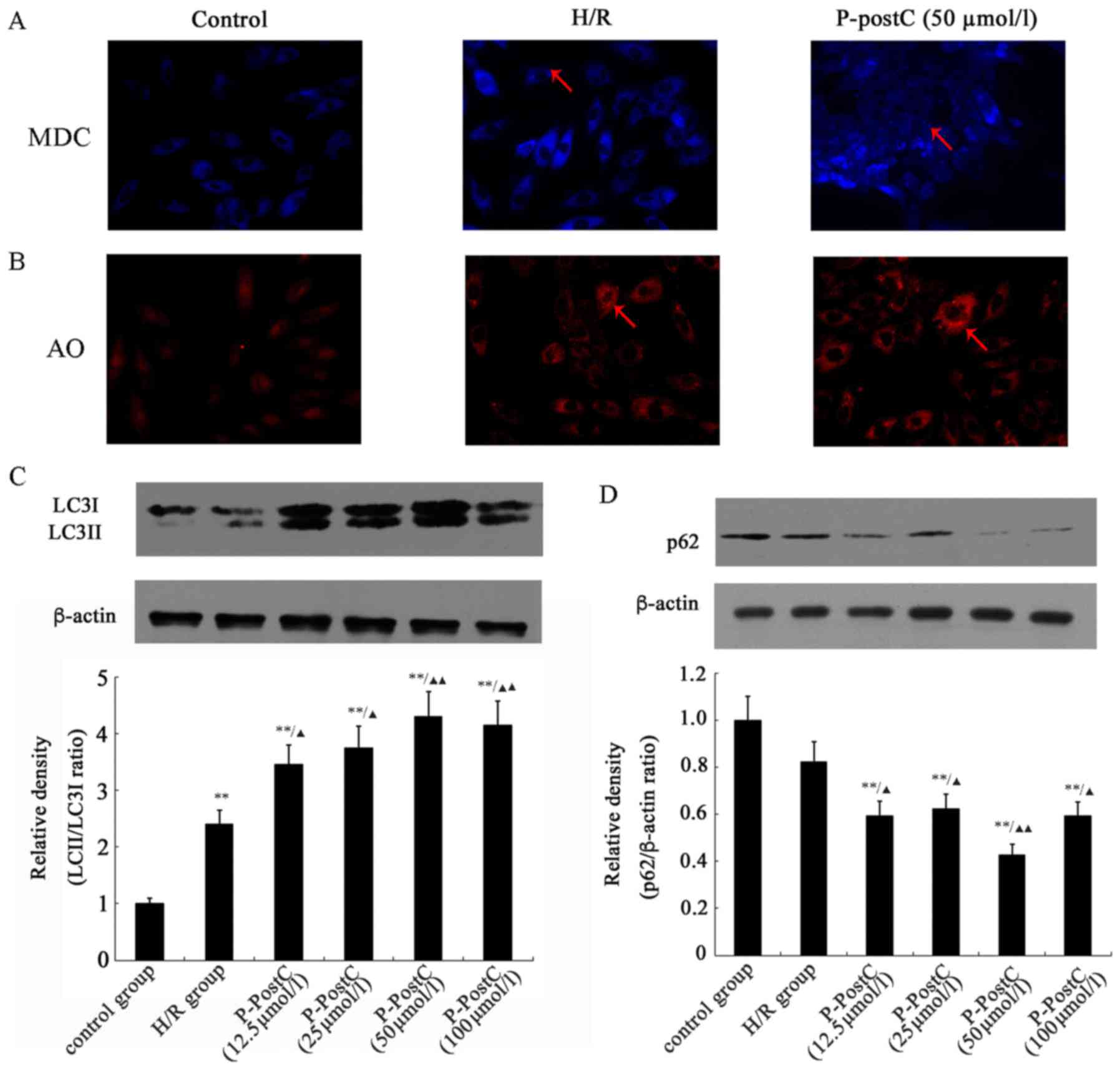
P-PostC induces autophagy in H9c2
cardiac cells
The fluorescent dye MDC is an autofluorescent marker
that accumulates in autophagic vacuoles, and is used to detect
autophagic vesicle formation (22). Autophagy was induced by incubating
H9c2 cells for 6 h in hypoxic conditions followed by 4 h of
reoxygenation. The level of autophagic vacuole formation was
evaluated by staining the cells with MDC. The results demonstrated
that autophagosome formation was enhanced under H/R conditions
(Fig. 3A), and P-PostC (50 µmol/l)
further induced autophagy compared with untreated control cells.
Fluorescence microscopic analysis identified MDC-positive
structures in the H/R and P-PostC groups, particularly in the
P-PostC group, whereas MDC-labeled structures were almost
undetectable in normal cells. In addition, the acidic vesicular
organelles in cells were also stained with AO. Fluorescence
microscopic analysis demonstrated that the H/R and P-PostC treated
cells exhibited abundant cytoplasmic acidic vesicular organelle
formation, compared with the Control group (Fig. 3B).
The induction of autophagy was confirmed by
detecting increased protein expression of LC3-II and the enhanced
ratio of LC3-II/LC3-I expression. LC3-II expression was enhanced in
H9c2 cells in the H/R group (Fig.
3C), and preconditioning with different concentrations of
propofol (12.5–100 µmol/l) significantly increased the expression
of LC3-II in a dose-dependent manner, compared with expression in
the H/R group (Fig. 3C). The
levels of p62 expression in H9c2 cells were reduced following H/R
exposure and decreased in response to P-PostC. Accompanied by
increased LC3 lipidation, the steady-state levels of p62 were
reduced following H/R exposure and P-PostC, which confirmed the
autophagy within H9c2 cells (Fig.
3D).
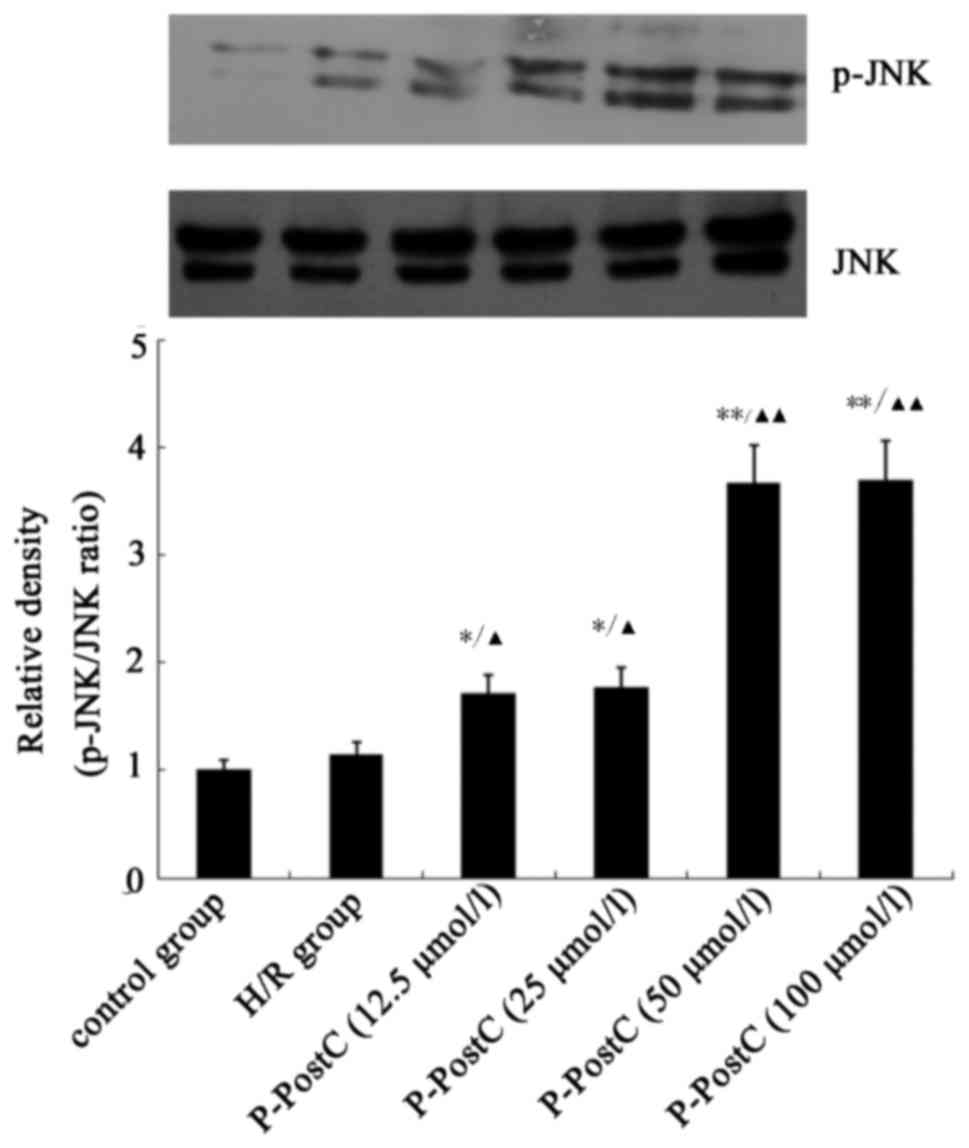
P-PostC activates phosphorylation of
JNK
To study the role of the SAPK/JNK signaling pathway
in P-PostC-induced autophagy, the activation of JNK in the H/R and
H/R + P-PostC groups was detected by western blotting (Fig. 4). H/R exposure slightly increased
JNK phosphorylation compared with in control; Fig. 4, and P-PostC co-treatment further
enhanced the phosphorylation of JNK (P<0.05 vs. H/R; Fig. 4).
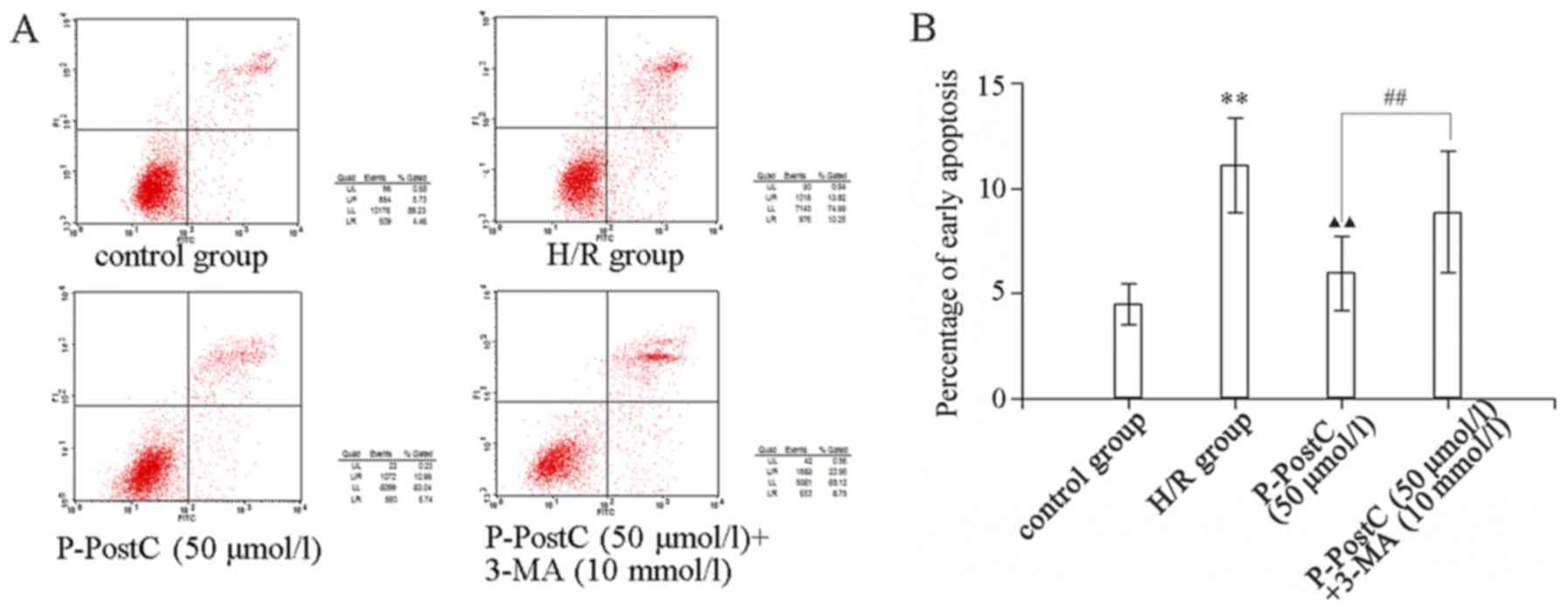
P-PostC mediated-autophagic induction
is negatively associated with the induction of apoptosis
As presented in Fig.
5, H/R induced the apoptosis of cardiomyocytes compared with in
the control group, while P-PostC (50 µmol/l) markedly reduced the
percentage of apoptosis. To investigate the association between the
apoptosis and autophagy induced in the myocardiocytes, cells of the
P-PostC+3-MA group were pre-treated with the autophagy inhibitor
3-MA (10 mmol/1) for 1 h prior to hypoxia, and then treated with
H/R and P-PostC (50 µmol/l). The results revealed that
pre-treatment with 3-MA markedly increased the percentage of
apoptotic cells compared with the P-PostC (50 µmol/l) group
(P<0.01 vs. P-PostC 50 µmol/l; Fig.
5). As autophagy was inhibited in the 3-MA-treated
cardiomyocytes, the cardioprotection of P-PostC (50 µmol/l) was
also eliminated.
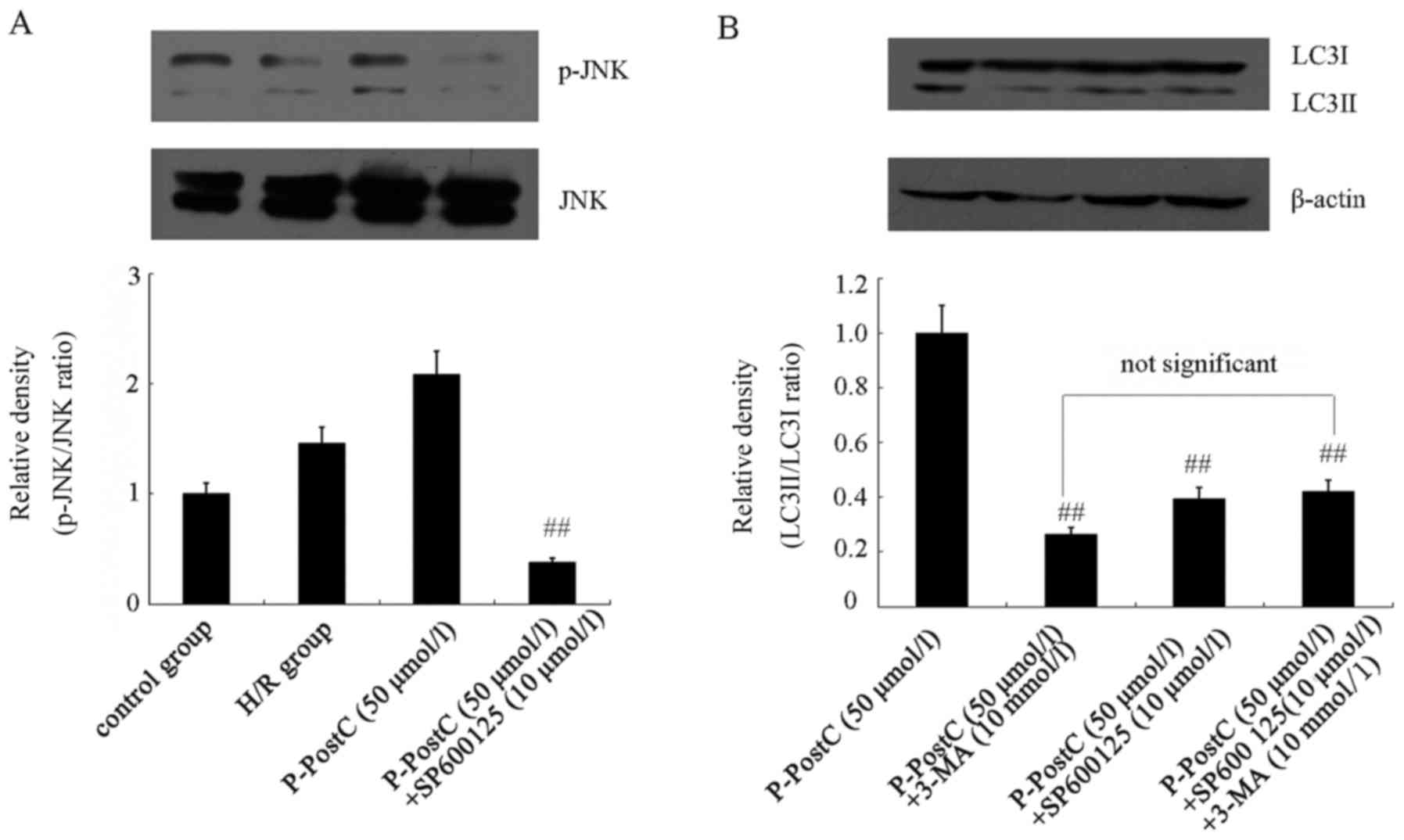
Activation of JNK is required for
P-PostC-induced autophagy
To further investigate the role of JNK in
P-PostC-mediated autophagy, cells were pretreated with 10 µmol/l
SP600125, a JNK activity-specific inhibitor, for 1 h, followed by
exposure to H/R or P-PostC (50 µmol/l) treatment. SP600125
significantly eliminated the phosphorylation of JNK induced by
P-PostC (50 µmol/l) on H/R cells (Fig.
6A). In addition, SP600125 could reverse the autophagy-induced
protective effects of P-PostC (50 µmol/l). Compared with P-PostC
treatment, pretreatment with SP600125 inhibited the increase of
LC3-II induced by P-PostC (50 µmol/l; Fig. 6B) and had similar effects as the
3-MA pretreatment (Fig. 6B). These
results indicated that the JNK-specific inhibitor could block
autophagy induced by P-PostC in H9c2 cardiac cells.
Discussion
The present study demonstrated that P-PostC
treatment significantly alleviated cardiomyocyte H/R injury and
reduced the percentage of apoptotic cells induced by H/R, which was
consistent with our previous study (7). Furthermore, the present results
suggested that the cardioprotection of P-PostC may be conferred
through autophagy enhancement activated by the SAPK/JNK signaling
pathway in H/R injury.
Autophagy is a self-clearing process that removes
damaged organelles and protein aggregates, which occurs at low
basal levels under normal conditions in the myocardium; however,
autophagy rapidly increases in response to stress conditions, such
as ischemia or hypoxia (23).
Enhanced autophagy is often observed in dying cardiac myocytes
(24); however, the functional
significance of autophagy under these conditions remains unclear.
Previous studies have demonstrated that the upregulation of
autophagy may protect cardiac cells against I/R injury and may also
promote cell survival (25,26).
As the cardioprotection of P-PostC treatment may be due to its
ability to activate survival signaling pathways, the present study
aimed to determine the ability of P-PostC to induce autophagy
during simulated in vitro myocardial I/R injury, and to
explore the signaling pathways that may be involved in this
process. The results indicated that P-PostC treatment induced
cardiac autophagy and generated survival signaling in H9c2 cardiac
myoblast cells, and that the inhibition of autophagy diminished the
cardioprotection of P-PostC. Furthermore, it was revealed that
P-PostC treatment induced autophagy through the activation of
SAPK/JNK survival signaling pathways to confer
cardioprotection.
Previous studies on the underlying mechanisms of
autophagy regulation in I/R injury have revealed that autophagy is
regulated by multiple signaling pathways. However, the detailed
mechanism by which I/R induces autophagy, as well as the direct
targets of the autophagic signaling cascade, remained unknown. JNK
as a ‘stress-responsive’ member of the mitogen-activated protein
kinase family (27) was considered
to serve a role in cardiomyocyte death, including apoptosis and
autophagy, during I/R injury (28,29).
A previous study reported that autophagy was inhibited in cells
treated with a JNK inhibitor, indicating that the JNK pathway may
be required to activate autophagy (30). Results from the present study
demonstrated that P-PostC treatment was able to activate the JNK
pathway and consequently mediate autophagy. It was also revealed
that P-PostC co-treatment enhanced the phosphorylation levels of
JNK in H9c2 cells. In addition, inhibition of the JNK signaling
pathway by the specific inhibitor SP600125 was able to reverse the
cardioprotective effects of P-PostC against H/R injury.
Apoptosis and autophagy are two types of programmed
cell death (31); they are not
always separate, and may be triggered by similar stimuli (32). A previous study presented
conflicting views on the roles of autophagy in myocardial I/R
injury; it was demonstrated that autophagy is enhanced during I/R
(33). Another study reported that
autophagy was an important element of the endogenous defense
mechanisms activated by ischemic preconditioning (34). Consistent with this finding, the
present results indicated that autophagy may alleviate the H/R
injury and reduce apoptosis in the H/R model of H9c2 cells during
P-PostC. By contrast, the pharmacological suppression of autophagy
by 3-MA treatment increased the apoptotic rate in H/R exposed cells
that were co-treated with P-PostC. Furthermore, SP600125 treatment
effectively inhibited the phosphorylation of SAPK/JNK, and the
induction of autophagy was also suppressed. These data indicated
that the activation of autophagy may serve a crucial role in
cardioprotection, and appears to be negatively correlated with the
induction of apoptosis.
All data was obtained from in vitro
experimentation; therefore, there are several limitations to the
present study. H9c2 embryonal rat heart-derived cells were used in
the H/R model. In contrast to the non-proliferating nature of
primary cardiomyocytes, H9c2 cells are able to proliferate, but
exhibit multiple similarities to primary cardiomyocytes including
membrane morphology and electrophysiological properties; however
in vivo studies are required further investigation within
model animals. Transmission electron microscopy has been the gold
standard to identify autophagosome formation, which is
characterized by their double-membrane structure (35); however, owing to the limitations of
experiment conditions and funding, transmission electron microscopy
was not used in the present study. Alternative methods of
detection, such as AO and MDC staining, the ratio of LC3-II/I and
the expression of protein p62, were used to confirm autophagy.
In conclusion, the results of the present study
indicated that P-PostC treatment promoted cell survival through the
induction of autophagy in H9c2 cardiac myoblast cells, and that the
SAPK/JNK survival pathway may be partly involved in P-PostC-induced
autophagy. These results may explain the induction of autophagy in
response to H/R injury, and also provided a novel mechanism for the
endogenous defensive cardioprotection activated by autophagy in
P-PostC.
Acknowledgements
This study was partially supported by the National
Nature Science Foundation of China (grant no. 81601209), the Miaopu
Fund of General Hospital of People's Liberation Army (grant no.
2015YW29) and the Sanya Medical and Health Innovation Program
(grant no. 15KMM42).
References
|
1
|
Badner NH, Knill RL, Brown JE, Novick TV
and Gelb AW: Myocardial infarction after noncardiac surgery.
Anesthesiology. 88:572–578. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murry CE, Jennings RB and Reimer KA:
Preconditioning with ischemia: A delay of lethal cell injury in
ischemic myocardium. Circulation. 74:1124–1136. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F,
Wang NP, Guyton RA and Vinten-Johansen J: Inhibition of myocardial
injury by ischemic postconditioning during reperfusion: comparison
with ischemic preconditioning. Am J Physiol Heart Circ Physiol.
285:H579–H588. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vinten-Johansen J, Zhao ZQ, Jiang R and
Zatta AJ: Myocardial protection in reperfusion with
postconditioning. Expert Rev Cardiovasc Ther. 3:1035–1045. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang JK, Yu LN, Zhang FJ, Yang MJ, Yu J,
Yan M and Chen G: Postconditioning with sevoflurane protects
against focal cerebral ischemia and reperfusion injury via PI3K/Akt
pathway. Brain Res. 1357:142–151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Marik PE: Propofol: Therapeutic
indications and side-effects. Curr Pharm Des. 10:3639–3649. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li H, Tan J, Zou Z, Huang CG and Shi XY:
Propofol post-conditioning protects against cardiomyocyte apoptosis
in hypoxia/reoxygenation injury by suppressing nuclear factor-kappa
B translocation via extracellular signal-regulated kinase
mitogen-activated protein kinase pathway. Eur J Anaesthesiol.
28:525–534. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Levine B and Klionsky DJ: Development by
self-digestion: Molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Powell SR: The ubiquitin-proteasome system
in cardiac physiology and pathology. Am J Physiol Heart Circ
Physiol. 291:H1–19H. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dosenko VE, Nagibin VS, Tumanovska LV and
Moibenko AA: Protective effect of autophagy in anoxia-reoxygenation
of isolated cardiomyocyte? Autophagy. 2:305–306. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gurusamy N, Lekli I, Gherghiceanu M,
Popescu LM and Das DK: BAG-1 induces autophagy for cardiac cell
survival. Autophagy. 5:120–121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan WJ, Dong HL and Xiong LZ: The
protective roles of autophagy in ischemic preconditioning. Acta
Pharmacol Sin. 34:636–643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou YY, Li Y, Jiang WQ and Zhou LF:
MAPK/JNK signalling: A potential autophagy regulation pathway.
Biosci Rep. 35:pii: e001992015.
|
|
15
|
Tanida I, Ueno T and Kominami E: LC3 and
autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shvets E, Fas E, Scherz-Shouval R and
Elazar Z: The N-terminus and Phe52 residue of LC3 recruit
p62/SQSTM1 into autophagosomes. J Cell Sci. 121:2685–2695. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo C, Wang SL, Xu ST, Wang JG and Song
GH: SP600125 reduces lipopolysaccharide-induced apoptosis and
restores the early-stage differentiation of osteoblasts inhibited
by LPS through the MAPK pathway in MC3T3-E1 cells. Int J Mol Med.
35:1427–1434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song L, Liu H, Ma L, Zhang X, Jiang Z and
Jiang C: Inhibition of autophagy by 3-MA enhances endoplasmic
reticulum stress-induced apoptosis in human nasopharyngeal
carcinoma cells. Oncol Lett. 6:1031–1038. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hoch B, Haase H, Schulze W, Hagemann D,
Morano I, Krause EG and Karczewski P: Differentiation-dependent
expression of cardiac delta-CaMKII isoforms. J Cell Biochem.
68:259–268. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Danpure CJ: Lactate dehydrogenase and cell
injury. Cell Biochem Funct. 2:144–148. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mizushima N: Methods for monitoring
autophagy. Int J Biochem Cell Biol. 36:2491–2502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Makoto S, Hajime I and Takashi N: Propofol
protects the immature rabbit heart against ischemia and reperfusion
injury: Impact on functional recovery and histopathological
changes. Biomed Res Int. 2014:6012502014.PubMed/NCBI
|
|
23
|
Dong Y, Undyala VV, Gottlieb RA, Mentzer
RM Jr and Przyklenk K: Autophagy: Definition, molecular machinery,
and potential role in myocardial ischemia-reperfusion injury. J
Cardiovasc Pharmacol Ther. 15:220–230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gustafsson AB and Gottlieb RA: Eat your
heart out: Role of autophagy in myocardial ischemia/reperfusion.
Autophagy. 4:416–421. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gustafsson AB and Gottlieb RA: Recycle or
die: The role of autophagy in cardioprotection. J Mol Cell Cardiol.
44:654–661. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matsui Y, Kyoi S, Takagi H, Hsu CP,
Hariharan N, Ago T, Vatner SF and Sadoshima J: Molecular mechanisms
and physiological significance of autophagy during myocardial
ischemia and reperfusion. Autophagy. 4:409–415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Uehara T, Bennett B, Sakata ST, Satoh Y,
Bilter GK, Westwick JK and Brenner DA: JNK mediates hepatic
ischemia reperfusion injury. J Hepatol. 42:850–859. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shimizu S, Konishi A, Nishida Y, Mizuta T,
Nishina H, Yamamoto A and Tsujimoto Y: Involvement of JNK in the
regulation of autophagic cell death. Oncogene. 29:2070–2082. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptotic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Valentim L, Laurence KM, Townsend PA,
Carroll CJ, Soond S, Scarabelli TM, Knight RA, Latchman DS and
Stephanou A: Urocortin inhibits Beclin1-mediated autophagic cell
death in cardiac myocytes exposed to ischaemia/reperfusion injury.
J Mol Cell Cardiol. 40:846–852. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang C, Yitzhaki S, Perry CN, Liu W,
Giricz Z, Mentzer RM Jr and Gottlieb RA: Autophagy induced by
ischemic preconditioning is essential for cardioprotection. J
Cardiovasc Transl Res. 3:365–373. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zakeri Z, Melendez A and Lockshin RA:
Detection of autophagy in cell death. Methods Enzymol. 442:289–306.
2008. View Article : Google Scholar : PubMed/NCBI
|