Introduction
Left ventricular noncompaction (LVNC) is an
inherited cardiomyopathy that is characterized by a thick spongy
endocardial layer and a thin compacted epicardial layer of the left
ventricular myocardium, numerous prominent trabeculations and deep
intertrabecular recesses (1). With
the introduction of various diagnostic methods, LVNC has become a
common cardiomyopathy in population, but the exact prevalence of
LVNC has yet to be determined ranging from 0.01 to 0.3% (2–6).
Symptoms exhibited by patients with LVNC may vary from no symptoms
to severe heart failure, malignant arrhythmias, circulatory
embolism and sudden mortality (1,7). The
mortality rate of adult patients with LVNC is 5–12% per year
(4,8,9).
LVNC is widely considered to be a genetic cardiomyopathy in which
myocardial dysplasia is observed during embryogenesis. Despite the
diagnosis of LVNC having been improved following the introduction
of echocardiography, cardiac magnetic resonance imaging and
computed tomography (10), the
etiology and mechanism underlying LVNC remain undetermined. LVNC is
genetically heterogeneous in humans, and numerous genes have been
revealed to be associated with LVNC, including: Myosin heavy chain
7 (MYH7); tafazzin (TAZ); mindbomb E3 ubiquitin
protein ligase 1 (MIB1); sodium voltage-gated channel-α
subunit 5 (SCN5A); myosin binding protein C, cardiac
(MYBPC3); tropomyosin 1 (TPM1); tyrosine
3-monooxygenase/tryptophan 5-monooxygenase activation protein-ε
(YWHAE); actin, α-cardiac muscle 1 (ACTC1); and
troponin T2, cardiac type (TNNT2) (11–13).
Previous studies have revealed part of the pathogenesis of LVNC.
For example, Luxán, et al (14) found that MIB1 mutations
could prevent trabecular maturation and compaction and induce LVNC
by NOTCH signaling pathway. Finsterer (15) found that MYH7 was the most
important pathogenic genes belonging to sarcomere proteins gene
accounting for 22% of all LVNC mutations. The more causal genes and
pathogenic mechanisms are found, the more possible it is to find
effective treatments. Recently, a novel mutation in MYH7 [c.
cytosine (C) 1492 guanine (G)] was identified via whole exome
sequencing (WES) as a potential causal mutation in a Chinese family
suffering from LVNC (11).
However, the already established mutations associated with LVNC are
only exhibited in subset of patients suffering from LVNC, and thus
the genetic basis of LVNC has not been fully determined. The aim of
the present study was to further investigate the potential causal
mutations of LVNC by performing WES on a Chinese family with
LVNC.
Materials and methods
Patients and clinical
characteristics
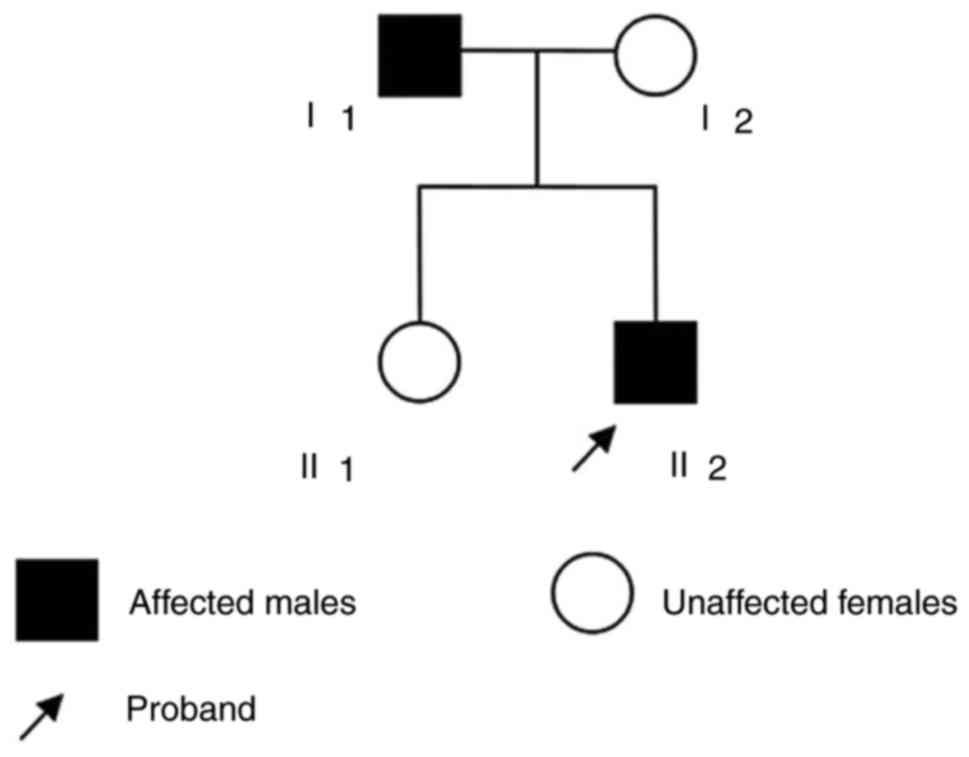
In the present study, the enrolled individuals (2
male and 2 female, age ranging between 19 and 50 years) were a
nuclear family from Jiangsu Province, China. A 19-year-old male
(patient II2) was the proband and exhibited no symptoms of chest
pain or dizziness; however, a heart murmur was detected during a
regular check-up in 2015. Following further physical examination,
the proband sought medical treatment at Huai'an First People's
Hospital and The First Affiliated Hospital of Nanjing Medical
University (Nanjing, China). During treatment, the patient
underwent echocardiography and 12-lead electrocardiography, and two
observers carefully and independently examined the results. The
patient met the following echocardiographic diagnostic criteria for
LVNC: (i) A noncompacted/compacted ratio for a two-layered
endocardium of >2; (ii) left ventricular deep endomyocardial
trabeculations; and (iii) deep recesses filled with blood
visualized on color Doppler imaging (16,17).
Subsequently, the parents of the proband, in addition to other
relatives, underwent a general medical history review, physical
examination and echocardiography. A brief questionnaire was
additionally administered in order to collect information on family
history. Finally, it was determined that the father (I1) of the
proband additionally matched the criteria of a LVNC diagnosis
(Fig. 1), whereas the mother of
the proband (I2) and the sister (II1) were revealed to be
healthy.
Ethics statement
The present study was supported and approved by the
Human Ethics Committee of The First Affiliated Hospital of Nanjing
Medical University, and written informed consent was provided by
the subjects prior to medical examination.
WES and mutation analysis
Samples collection and DNA
extraction
Blood samples from the proband and his parents were
collected during August 2015 in The First Affiliated Hospital of
Nanjing Medical University and analyzed. Genomic DNA was extracted
using a Genomic DNA Purification kit (Qiagen, Inc., Valencia, CA,
USA).
Genome sequencing
Exons of MYH7 and ACTC1 were sequenced
by the Department of Epidemiology and Biostatistics, School of
Public Health, Nanjing Medical University via Sanger sequencing to
identify whether the family trio harbored mutations in the already
established LVNC-associated causal genes; however, no mutations
were revealed in said genes. Thus, the sequencing results suggested
that unknown mutations may be responsible for the development of
LVNC. Therefore, WES was subsequently performed using the
TruSeq® DNA HS Sample Preparation Kit on the Illumina
TruSeq Exome Enrichment platform kit libraries (Illumina, Inc., San
Diego, CA, USA), which includes 20,794 target genes, with the aim
of targeting 62 Mb genomic regions using 95-base DNA probes. In
addition to covering the RefSeq and Ensembl coding sequences, the
enriched sequences also include 28 Mb of RefSeq untranslated
regions (UTR). Following this, genomic libraries were prepared,
according to the manufacturer's protocol. First, 100–500-bp DNA
fragments were prepared using genomic DNA (5 mg) in elution buffer
(80 ml) via sonication at 200 W and for 30 sec (10 cycles) at 4°C
using a Bioruptor (Diagenode Inc., Denville, NJ, USA). Following
this, DNA fragments ranging between 150 and 250 bp were isolated
via 2% gel electrophoresis. End repair was performed using T4 DNA
poly and Klenow poly cleave 3′ (supplied with the
TruSeq® DNA HS Sample Preparation kit) followed by a
size selection procedure (as set out in the TruSeq Enrichment Guide
and instructions in the MinElute Gel Extraction kit). An additional
adenosine base was then added to the 3′ end by Klenow 3′ to 5′ exo
minus (3′→5′ exo-), and following this, DNA fragments containing
the Illumina multiPE-adaptor (Illumina, Inc.) were isolated (PE
Adapters sequence: 5′P-GATCGGAAGAGCGGTTCAGCAGGAATGCCGAG,
5′ACACTCTTTCCCTACACGACGCTCTTCCGATCT; TruSeq® DNA HS
Sample Preparation kit). Following this, DNA products were
amplified via 12 cycles of polymerase chain reaction (PCR) using
Illumina multiPE primer #1 (PE-1.0:
5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC* T-3′;
1 ml; 25 mM; Illumina, Inc.), Illumina multiPE primer #2 (PE-2.0:
5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATC*
T-3′; 1 ml; 0.5 mM; Illumina, Inc.) and Illumina index primer (1
ml; 25 mM; Illumina, Inc.) to generate DNA libraries. The
thermocycling conditions were: 98°C for 30 sec followed by 10
cycles of 98°C (10 sec), 60°C (30 sec), 72°C (30 sec), 72°C (5 min)
and holding at 10°C.
Following this, the Illumina HiSeq1500 (Ilumina,
Inc.) was used to sequence the enriched DNA libraries according to
the manufacturer's protocol. Burrows-Wheeler Aligner (18) was subsequently utilized to align
the sequences to the human reference genome (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/).
Picard software (version 2.14.1, https://broadinstitute.github.io/picard/) was used to
exclude PCR duplicates, and following this, new regional
realignment and recalibration of quality scoring was performed
using the Genome Analysis Toolkit (GATK; v.2.8) (19). Variations were identified by GATK
using recommended parameters (https://gatkforums.broadinstitute.org/gatk/profile/GATK_Team).
Integrative Genomics Viewer (IGV; https://igv.org/),
a highly efficient and specialized tool for examining integrated
genomic datasets, was used to determine genetic variations based on
genomic position (20). Following
this, known variants listed by the HAPMAP (http://www.internationalgenome.org/category/hapmap/),
1000 Genomes Project (http://www.internationalgenome.org/) or Project of
dbSNP137 (http://varianttools.sourceforge.net/Annotation/DbSNP)
were excluded. In the following analysis, the remaining single
nucleotide variants (SNVs) were investigated further to identify
whether they were present only in the patients suffering from LVNC
and not in the healthy subjects in the family (the sister and
mother of the proband). Following this, the remaining variants of
nonsynonymous single nucleotides identified by SNV prioritization
via the integration of genomic data (SPRING) (21) were prioritized for further
analysis.
Statistical analysis
P-values were calculated by SPRING, combining six
deleterious scores (LRT, SIFT, PolyPhen2, PhyloP, GERP and Mutation
Taster) and five associated scores from multiple sources of genomic
data (including protein-protein interactions, gene ontology,
protein sequences, gene pathway annotations and protein domain
annotations). Co-expression analysis was performed based on data
obtained from Genotype-Tissue Expression (GTEx; gtexportal.org) on the ventricular myocardium. The R
package ‘clusterProfiler’ (22)
was used to perform Kyoto Encyclopedia of Genes and Genomes (KEGG)
enrichment analysis. P<0.05 was considered to indicate a
statistically significant result.
Results
Clinical characteristics
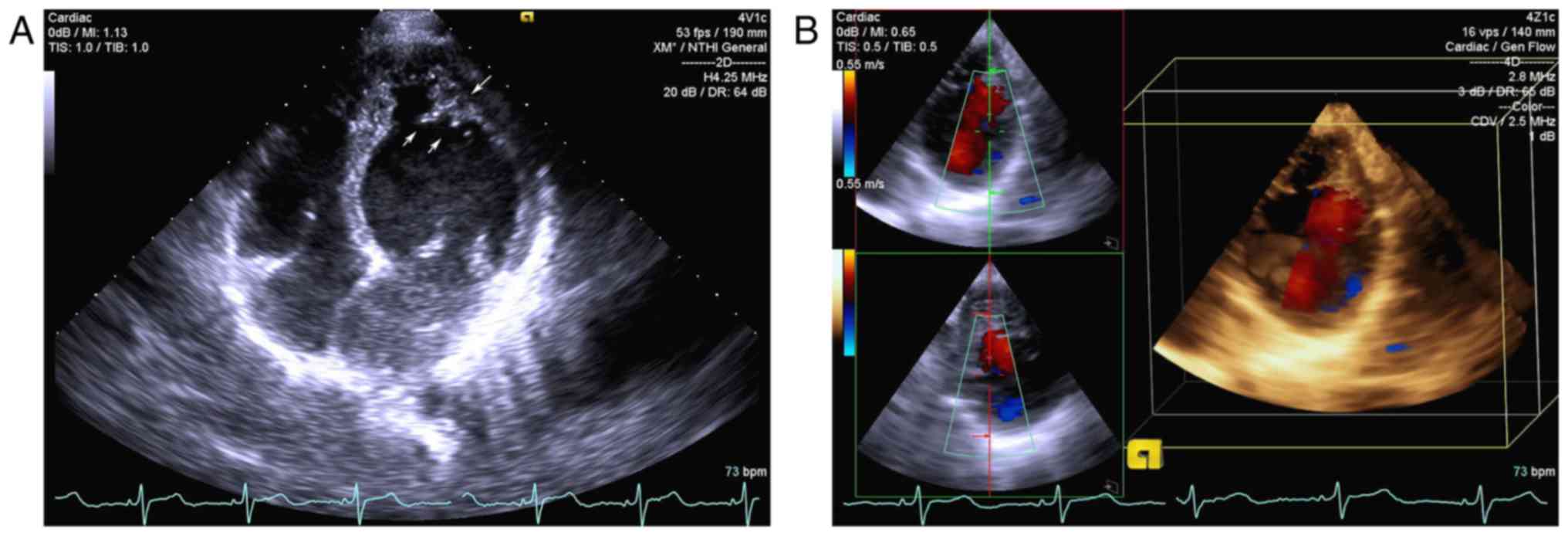
The proband (II2) exhibited no symptoms, and the
electrocardiogram results were normal. The echocardiogram of the
proband demonstrated prominent trabeculations associated with deep
recesses in the apex and bicuspid aortic valve. A thickened
endocardial layer and a thin epicardial layer were additionally
observed (Fig. 2). The father (I1)
of the proband was diagnosed with LVNC by echocardiography, which
revealed a noncompacted area in the lateral wall of the left
ventricle. The father of the proband additionally did not exhibit
any clinical symptoms, and the electrocardiogram results were
normal. The mother and sister of the proband were validated as
healthy by electrocardiographic and echocardiographic testing.
Mutation screening
A total of 8,354 MB high-quality reads were obtained
for patient II2, 2,689 M reads for patient I1 and 7,408 M reads for
healthy control I2. A total of 21,337 variants or indels were
detected in the proband, 18,230 were detected in the father of the
proband additionally suffering from LVNC (I1) and 17,865 were
detected in the healthy mother of the proband (I2). Following
exclusion of variants revealed in multiple databases (HAPMAP,
dbSNP137 and 1000 Genomes Project), 1,633, 1,750 and 1,524 variants
or indels were retained, respectively. For subsequent analysis,
synonymous variants and non-frameshift indels were excluded.
Finally, a total of 43 non-silent coding variants (LIN7C, PDCD6,
FLT3, NALCN, HMCN1, TLL2, NUBPL, LIM2, OSBPL8, SH3PXD2A, ATP13A5,
SPTB, NUP188, C16orf71, FKBP9, TP53BP2, MVP, LRRC3B, ARID1B, TXNIP,
NR5A2, MUC5B, ANKAR, MGAT4C, USP24, DOK1, HMGN2, ANKRD20A4, SSC5D,
ATP9A, DLG5, TAX1BP1, ZNF814, SMC1B, ELAVL3, PLEKHH2, REPIN1,
TCEANC2, PLEKHA4, DOK3, PCDHA5, ZNF707 and MUC12) were
revealed to be present in the two patients suffering from LVNC and
not in the healthy control. SPRING was then used to prioritize the
disease-causing nonsynonymous SNVs, and seven SNVs with P<0.05
were subsequently identified: hemicentin 1 (HMCN1) [c.
thymine (T)9776C], programmed cell death 6 (PDCD6) [c.
adenine (A)418T], tolloid like 2 (TLL2) (c.C2615T), lin-7
homolog C, crumbs cell polarity complex component (LIN7C)
(c.C91T), fms related tyrosine kinase 3 (FLT3) (c.G976A),
sodium leak channel, non-selective (NALCN) (c.G4975A) and
nucleotide binding protein like (NUBPL) (c.G91T; Table I). In addition, the results
demonstrated that TLL2 (c.C2615T) and FLT3 (c.G976A)
are located in conserved regions and are annotated as deleterious
by the SIFT, PolyPhe2, LRT and MutationTaster databases.
Furthermore, NUBPL (c.G91T) and HMCN1 (c.T9776C) were
revealed to be located in conserved regions and annotated as
deleterious by the PolyPhe2, LRT and MutationTaster databases.
However, these variants (Table I)
have not been previously reported in the Human Gene Mutation
Database (www.hgmd.org). Following this, the
reliability of the seven identified mutations was further validated
by investigating the mapping quality around these mutations using
IGV (data not shown; are available from the corresponding author on
reasonable request).
 | Table I.Single nucleotide variants with
P<0.05 and their categorization, as determined by single
nucleotide variants prioritization via the integration of genomic
data analysis. |
Table I.
Single nucleotide variants with
P<0.05 and their categorization, as determined by single
nucleotide variants prioritization via the integration of genomic
data analysis.
| Location of
mutation | Gene | Amino acid
change | P-value | PhyloP | SIFT | PolyPhen2 | LRT | Mutation
taster |
|---|
| Chr1: 186059938
T>C | HMCN1 | p.L3259P |
2.05×10−2 | Conserved | Tolerate | Deleterious | Deleterious | Deleterious |
| Chr5: 311458
A>T | PDCD6 | p.R140W |
7.57×10−3 | Neutral | Deleterious | Deleterious | Neutral | Deleterious |
| Chr10: 98133400
G>A | TLL2 | p.S872L |
4.44×10−2 | Conserved | Deleterious | Deleterious | Deleterious | Deleterious |
| Chr11: 27523414
G>A | LIN7C | p.P31S |
5.40×10−3 | Conserved | Tolerate | Deleterious | Neutral | Deleterious |
| Chr13: 28623581
C>T | FLT3 | p.G326R |
1.13×10−2 | Conserved | Deleterious | Deleterious | Deleterious | Deleterious |
| Chr13: 101710339
C>T | NALCN | p.D1659N |
1.73×10−2 | Conserved | Tolerate | Possibly
damaging | Deleterious | Deleterious |
| Chr14: 32295867
G>T | NUBPL | p.A31S |
4.86×10−2 | Conserved | Tolerate | Deleterious | Deleterious | Deleterious |
Co-expressed gene enrichment
analysis
Co-expression analysis on the seven identified genes
and previously revealed LVNC-associated genes was performed using
on data from the ventricular myocardium in GTEx (https://www.gtexportal.org/home/). The results
demonstrated that NUBPL was co-expressed with almost all of
the established LVNC-associated genes, including SCN5A, MYH7,
ACTC1, YWHAE, MYBPC3, TAZ, MIB1 and TPM1. A further
gene, PDCD6, was revealed to be co-expressed with the
established LVNC-associated genes SCN5A, MYH7, ACTC1, YWHAE,
MYBPC3, TAZ and MIB1, although not TPM1. KEGG
pathway analysis revealed that genes co-expressed with NUBPL
and PDCD6 were enriched in the Notch signaling pathway (data
not shown; are available from the corresponding author on
reasonable request).
Discussion
WES is an important tool for genetic research;
however, investigations regarding LVNC using WES have not been
extensively performed. In the present study, WES was performed on a
Chinese family with members suffering from LVNC and seven novel
mutations were revealed that, to the best of our knowledge, have
not previously been reported to be associated with LVNC. Among
them, TLL2 (c.C2615T) and FLT3 (c.G976A) are located
in conserved regions and were revealed to be deleterious by SIFT,
PolyPhe2, LRT and MutationTaster database analyses. Thus, these
mutations may represent potential causal mutations of LVNC.
TLL2 encodes an astacin-like zinc-dependent
metalloprotease that is a subfamily member of the bone
morphogenetic protein-1 (BMP-1)/tolloid (TLD) protein family. The
BMP-1/TLD protein family is conserved from Drosophila to
Homo sapiens. and includes BMP-1, TLD, TLL-1 and TLL-2
proteins, which are encoded for by the BMP1, TLL1 and
TLL2 genes, respectively (23). TLL-1 and TLL-2 are important for
normal morphogenesis and extracellular matrix deposition (24). Previous studies have revealed that
TLL-1 is highly expressed in the endocardium and developing septa
of the mouse heart and is involved in the regulation of heart
morphogenesis via activation of transforming growth factor-β
(TGF-β)-like molecules (25,26).
In addition, mutations in TLL1 have been demonstrated to be
associated with atrial septal defects in humans (27). Despite the structure of TLL-2 being
similar to that of TLL-1, TLL-2 is predominantly expressed in the
developing skeletal muscle, where it activates latent myostatin and
regulates muscle growth (23,25,28).
Therefore, it may be suggested that the novel SNV (c.C2615T) in
TLL2 revealed in the present study may be associated with
heart development in humans; however, further research is required
to investigate this result.
FLT3 encodes for a membrane-bound receptor
tyrosine kinase that is associated with hematopoietic cell
proliferation and differentiation (29,30).
In addition, a further study revealed that FLT3 expression is
upregulated following myocardial infarction in mouse hearts and
that the activation of FLT3 by FLT3 ligand protects cardiomyocytes
from oxidative stress-induced apoptosis via a protein kinase
B-dependent mechanism (31).
However, whether mutations in FLT3 lead to abnormal
myocardial development remains undetermined.
Despite other novel mutations in conserved regions
of genes not being annotated as deleterious by SIFT, PolyPhe2, LRT
and the MutationTaster database analyses, it was revealed that a
number of said mutations are associated with cardiac function. In
the present study, a mutation in HMCN1 (c.T9776C), which
encodes for an extracellular protein belonging to the
immunoglobulin superfamily, was annotated as deleterious by
PolyPhe2, LRT and the MutationTaster databases (32). Furthermore, HMCN1 has previously
been revealed to be predominantly expressed in the vascular
endothelial cells of coronary arteries and sparsely expressed in
the endocardial endothelium in the mouse heart, which may have an
important role in myocardial remodeling following myocardial
infarction by affecting cardiac fibroblast migration via TGF-β1
signaling (33). Furthermore, a
previous study using animal models and induced pluripotent stem
cell-derived cardiomyocytes demonstrated that abnormal regulation
of TGF-β may represent a potential mechanism underlying LVNC
(34). Thus, it may be suggested
that there may be an association between HMCN1 and LVNC.
The mutation in NUBPL (c.G91T) was revealed
to be deleterious by PolyPhe2, LRT and MutationTaster database
analyses. The results of the present study revealed that
NUBPL is co-expressed with genes that have been previously
demonstrated to be associated with LVNC, including SCN5A, MYH7,
ACTC1, YWHAE, MYBPC3, TAZ, MIB1 and TPM1, although not
TNNT2, based on GTEx. In addition, enrichment analysis
revealed that NUBPL is enriched in the Notch pathway.
Notably, Luxán et al (14)
demonstrated that Notch pathway dysfunction may lead to LVNC.
Furthermore, NUBPL encodes for a protein that is required
for nicotinamide adenine dinucleotide dehydrogenase (human complex
I) assembly in the respiratory chain (35). Loeffen et al (36) and Benit et al (37) demonstrated that human complex I
deficiency is associated with hypertrophic cardiomyopathy.
Therefore, it may be suggested that mutations in NUBPL may
induce human complex I deficiency and abnormal myocardial
development. Based on the previous evidence and the results of the
present study, it may be hypothesized that NUBPL represents
a potential causal gene of LVNC.
Furthermore, the present study identified
PDCD6 as an additional gene that is enriched in the Notch
signaling pathway. PDCD6 is a calcium-binding modulator
associated with cell proliferation and death (38). Numerous studies have revealed that
PDCD6 is associated with tumors; however, the underlying
mechanism remains unclear (39–42).
Furthermore, one study demonstrated that overexpression of
PDCD6 suppressed angiogenesis in vitro via the
phosphatidylinositol 3-kinase/protein kinase mTOR/ribosomal protein
S6 kinase B1 pathway (43).
However, as PDCD6 was annotated by the LRT database analysis
as neutral, it is less likely to be a causal gene of LVNC.
In conclusion, the results of the present study
revealed numerous novel mutations [TLL2 (c.C2615T),
FLT3 (c.G976A), NUBPL (c.G91T) and PDCD6
(c.A418T)] that may be associated with LVNC in a Chinese family.
Among them, mutations in NUBPL are more likely to represent
causal SNVs for LVNC. Considering that this was an exploratory
study using a small sample population, additional investigations,
including expanding the sample size and performing reverse
transcription-PCR, may be required to verify the preliminary
results of the present study and to determine the mechanisms
underlying the pathogenesis of LVNC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the
Cardiovascular disease clinical medical research center of Jiangsu,
China (grant no. KFSN201403-05).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ, ZQ, JY and JZ performed the study and were major
contributors to writing the manuscript. MZ analyzed and interpreted
the patient data regarding LVNC. XH, YW and HW collected clinical
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was supported and approved by the
Human Ethics Committee of The First Affiliated Hospital of Nanjing
Medical University, and written informed consent was provided by
the subjects prior to medical examination.
Consent for publication
Written informed consent was provided by the
subjects prior to medical examination.
Competing interests
All authors declare that there are no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
LVNC
|
left ventricular noncompaction
|
|
WES
|
whole exome sequencing
|
|
GATK
|
Genome Analysis Toolkit
|
|
SNVs
|
single nucleotide variants
|
|
MYH7
|
myosin heavy chain 7
|
|
TAZ
|
tafazzin
|
|
MIB1
|
mindbomb E3 ubiquitin protein ligase
1
|
|
SCN5A
|
sodium voltage-gated channel α-subunit
5
|
|
MYBPC3
|
myosin binding protein C, cardiac
|
|
TPM1
|
tropomyosin 1
|
|
YWHAE
|
tyrosine 3-monooxygenase/tryptophan
5-monooxygenase activation protein-ε
|
|
ACTC1
|
actin, α-cardiac muscle 1
|
|
TNNT2
|
troponin T2, cardiac type
|
|
SPRING
|
single nucleotide variant
prioritization via the integration of genomic data
|
|
PCR
|
polymerase chain reaction
|
|
IGV
|
integrative genomics viewer
|
|
GTEx
|
genotype-tissue expression
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
BMP-1/TLD
|
bone morphogenetic
protein-1/tolloid
|
|
TGF-β
|
transforming growth factor-β
|
References
|
1
|
Sarma RJ, Chana A and Elkayam U: Left
ventricular noncompaction. Prog Cardiovasc Dis. 52:264–273. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cheng TO: Left ventricular noncompaction
cardiomyopathy: Three decades of progress. Int J Cardiol.
174:227–229. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ronderos R, Avegliano G, Borelli E,
Kuschnir P, Castro F, Sanchez G, Perea G, Corneli M, Zanier MM,
Andres S, et al: Estimation of prevalence of the left ventricular
noncompaction among adults. Am J Cardiol. 118:901–905. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ritter M, Oechslin E, Sütsch G, Attenhofer
C, Schneider J and Jenni R: Isolated noncompaction of the
myocardium in adults. Mayo Clin Proc. 72:pp. 26–31. 1997;
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ozkutlu S, Ayabakan C, Celiker A and
Elshershari H: Noncompaction of ventricular myocardium: A study of
twelve patients. J Am Soc Echocardiogr. 15:1523–1528. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stollberger C, Blazek G, Winkler-Dworak M
and Finsterer J: Sex differences in left ventricular noncompaction
in patients with and without neuromuscular disorders. Rev Esp
Cardiol. 61:130–136. 2008.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oechslin EN, Attenhofer Jost CH, Rojas JR,
Kaufmann PA and Jenni R: Long-term follow-up of 34 adults with
isolated left ventricular noncompaction: A distinct cardiomyopathy
with poor prognosis. J Am Coll Cardiol. 36:493–500. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stollberger C, Blazek G, Wegner C and
Finsterer J: Neurological comorbidity affects prognosis in left
ventricular hypertrabeculation/noncompaction. Heart Lung.
41:594–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peters F, Khandheria BK, Botha F, Libhaber
E, Matioda H, Dos Santos C, Govender S, Meel R and Essop MR:
Clinical outcomes in patients with isolated left ventricular
noncompaction and heart failure. J Card Fail. 20:709–715. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paterick TE, Umland MM, Jan MF, Ammar KA,
Kramer C, Khandheria BK, Seward JB and Tajik AJ: Left ventricular
noncompaction: A 25-year odyssey. J Am Soc Echocardiogr.
25:363–375. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang J, Zhu M, Wang Y, Hou X, Wu H, Wang
D, Shen H, Hu Z and Zou J: Whole-exome sequencing identify a new
mutation of MYH7 in a Chinese family with left ventricular
noncompaction. Gene. 558:138–142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zaragoza MV, Arbustini E and Narula J:
Noncompaction of the left ventricle: Primary cardiomyopathy with an
elusive genetic etiology. Curr Opin Pediatr. 19:619–627. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Klaassen S, Probst S, Oechslin E, Gerull
B, Krings G, Schuler P, Greutmann M, Hurlimann D, Yegitbasi M, Pons
L, et al: Mutations in sarcomere protein genes in left ventricular
noncompaction. Circulation. 117:2893–2901. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luxán G, Casanova JC, Martinez-Poveda B,
Prados B, D'Amato G, MacGrogan D, Gonzalez-Rajal A, Dobarro D,
Torroja C, Martinez F, et al: Mutations in the NOTCH pathway
regulator MIB1 cause left ventricular noncompaction cardiomyopathy.
Nat Med. 19:193–201. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Finsterer J: Cardiogenetics,
neurogenetics, and pathogenetics of left ventricular
hypertrabeculation/noncompaction. Pediatr Cardiol. 30:659–681.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jenni R, Oechslin E, Schneider J,
Attenhofer Jost C and Kaufmann PA: Echocardiographic and
pathoanatomical characteristics of isolated left ventricular
non-compaction: A step towards classification as a distinct
cardiomyopathy. Heart. 86:666–671. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tian T, Liu Y, Gao L, Wang J, Sun K, Zou
Y, Wang L, Zhang L, Li Y, Xiao Y, Song L and Zhou X: Isolated left
ventricular noncompaction: Clinical profile and prognosis in 106
adult patients. Heart Vessels. 29:645–652. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Thorvaldsdottir H, Robinson JT and Mesirov
JP: Integrative Genomics Viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu J, Li Y and Jiang R: Integrating
multiple genomic data to predict disease-causing nonsynonymous
single nucleotide variants in exome sequencing studies. PLoS Genet.
10:e10042372014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
R Core Team: R: A language and environment
for statistical computingR Foundation for Statistical Computing.
Vienna: 2014
|
|
23
|
Lee SJ: Genetic analysis of the role of
proteolysis in the activation of latent myostatin. PLoS One.
3:e16282008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bayley CP, Ruiz Nivia HD, Dajani R, Jowitt
TA, Collins RF, Rada H, Bird LE and Baldock C: Diversity between
mammalian tolloid proteinases: Oligomerisation and non-catalytic
domains influence activity and specificity. Sci Rep. 6:214562016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clark TG, Conway SJ, Scott IC, Labosky PA,
Winnier G, Bundy J, Hogan BL and Greenspan DS: The mammalian
Tolloid-like 1 gene, Tll1, is necessary for normal septation and
positioning of the heart. Development. 126:2631–2642.
1999.PubMed/NCBI
|
|
26
|
Sieron AL and Stanczak P: ASD-lessons on
genetic background from transgenic mice with inactive gene encoding
metalloprotease, Tolloid-like 1 (TLL1). Med Sci Monit.
12:RA17–RA22. 2006.PubMed/NCBI
|
|
27
|
Stanczak P, Witecka J, Szydlo A,
Gutmajster E, Lisik M, Augusciak-Duma A, Tarnowski M, Czekaj T,
Czekaj H and Sieron AL: Mutations in mammalian tolloid-like 1 gene
detected in adult patients with ASD. Eur J Hum Genet. 17:344–351.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Scott IC, Blitz IL, Pappano WN, Imamura Y,
Clark TG, Steiglitz BM, Thomas CL, Maas SA, Takahara K, Cho KW and
Greenspan DS: Mammalian BMP-1/Tolloid-related metalloproteinases,
including novel family member mammalian Tolloid-like 2, have
differential enzymatic activities and distributions of expression
relevant to patterning and skeletogenesis. Dev Biol. 213:283–300.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stirewalt DL and Radich JP: The role of
FLT3 in haematopoietic malignancies. Nat Rev Cancer. 3:650–665.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Garg M, Nagata Y, Kanojia D, Mayakonda A,
Yoshida K, Keloth SH, Zang ZJ, Okuno Y, Shiraishi Y, Chiba K, et
al: Profiling of somatic mutations in acute myeloid leukemia with
FLT3-ITD at diagnosis and relapse. Blood. 126:2491–2501. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pfister O, Lorenz V, Oikonomopoulos A, Xu
L, Häuselmann SP, Mbah C, Kaufmann BA, Liao R, Wodnar-Filipowicz A
and Kuster GM: FLT3 activation improves post-myocardial infarction
remodeling involving a cytoprotective effect on cardiomyocytes. J
Am Coll Cardiol. 63:1011–1019. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Omer WH, Narita A, Hosomichi K, Mitsunaga
S, Hayashi Y, Yamashita A, Krasniqi A, Iwasaki Y, Kimura M and
Inoue I: Genome-wide linkage and exome analyses identify variants
of HMCN1 for splenic epidermoid cyst. BMC Med Genet. 15:1152014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chowdhury A, Herzog C, Hasselbach L,
Khouzani HL, Zhang J, Hammerschmidt M, Rudat C, Kispert A, Gaestel
M, Menon MB, et al: Expression of fibulin-6 in failing hearts and
its role for cardiac fibroblast migration. Cardiovasc Res.
103:509–520. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kodo K, Ong SG, Jahanbani F, Termglinchan
V, Hirono K, InanlooRahatloo K, Ebert AD, Shukla P, Abilez OJ,
Churko JM, et al: iPSC-derived cardiomyocytes reveal abnormal TGF-β
signalling in left ventricular non-compaction cardiomyopathy. Nat
Cell Biol. 18:1031–1042. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Calvo SE, Tucker EJ, Compton AG, Kirby DM,
Crawford G, Burtt NP, Rivas M, Guiducci C, Bruno DL, Goldberger OA,
et al: High-throughput, pooled sequencing identifies mutations in
NUBPL and FOXRED1 in human complex I deficiency. Nat Genet.
42:851–858. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Loeffen J, Elpeleg O, Smeitink J, Smeets
R, Stockler-Ipsiroglu S, Mandel H, Sengers R, Trijbels F and van
den Heuvel L: Mutations in the complex I NDUFS2 gene of patients
with cardiomyopathy and encephalomyopathy. Ann Neurol. 49:195–201.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Benit P, Beugnot R, Chretien D, Giurgea I,
De Lonlay-Debeney P, Issartel JP, Corral-Debrinski M, Kerscher S,
Rustin P, Rötig A and Munnich A: Mutant NDUFV2 subunit of
mitochondrial complex I causes early onset hypertrophic
cardiomyopathy and encephalopathy. Human Mutat. 21:582–586. 2003.
View Article : Google Scholar
|
|
38
|
Park SH, Lee JH, Lee GB, Byun HJ, Kim BR,
Park CY, Kim HB and Rho SB: PDCD6 additively cooperates with
anti-cancer drugs through activation of NF-κB pathways. Cell
Signal. 24:726–733. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
He YQ, Zhou B, Shi SQ, Zhang L and Li WM:
Genetic variation in PDCD6 and susceptibility to lung cancer. Asian
Pac J Cancer Prev. 13:4689–4693. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang K, Zhou B, Shi S, Song Y and Zhang
L: Variations in the PDCD6 gene are associated with increased
uterine leiomyoma risk in the Chinese. Genet Test Mol Biomarkers.
17:524–528. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou B, Zhang P, Tang T, Zhang K, Wang Y,
Song Y, Liao H and Zhang L: Prognostic value of PDCD6 polymorphisms
and the susceptibility to bladder cancer. Tumour Biol.
35:7547–7554. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou B, Bai P, Xue H, Zhang Z, Shi S,
Zhang K, Wang Y, Wang K, Quan Y, Song Y and Zhang L: Single
nucleotide polymorphisms in PDCD6 gene are associated with the
development of cervical squamous cell carcinoma. Fam Cancer.
14:1–8. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rho SB, Song YJ, Lim MC, Lee SH, Kim BR
and Park SY: Programmed cell death 6 (PDCD6) inhibits angiogenesis
through PI3K/mTOR/p70S6K pathway by interacting of VEGFR-2. Cell
Signal. 24:131–139. 2012. View Article : Google Scholar : PubMed/NCBI
|