Introduction
Silicosis is caused by long-term inhalation of
silicon dioxide dust in professional activities and its basic
pathological features include the formation of silicotic nodules
and lung fibrosis (1).
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a natural
tetrapeptide formed from its precursor, thymosin β4, released by
prolyl oligopeptides (2). Ac-SDKP
has antifibrosis effects and is expressed in human and animal
tissues (3,4). The downregulation of Ac-SDKP
expression is associated with organ fibrosis (5,6). The
renin-angiotensin system (RAS) is a key mediator of lung fibrosis
(7,8); the key enzyme of RAS,
angiotensin-converting enzyme (ACE), consists of 2 catalytic
domains, namely the C and N domains (9). The C domain catalyzes angiotensin
(Ang) I to form Ang II (10). Ang
II is the major active protein of RAS and its fibrotic effect is
primarily mediated by angiotensin II type 1 (AT1), which includes
the induction of inflammatory responses as well as increased
expression of transforming growth factor-β (TGF-β), proliferation
of fibroblasts and extracellular matrix (ECM) deposition (11–14).
Ac-SDKP is cleared by the N domain of ACE and its plasma and tissue
levels therefore significantly increase following ACE inhibition
(3,15). The role of ACE in producing the
pro-fibrotic molecule Ang II and degrading the anti-fibrotic
molecule Ac-SDKP suggests that the interaction between Ac-SDKP and
RAS may be important in silicosis. In the present study, changes in
Ac-SDKP and Ang II signaling during the development of silicosis
were investigated. The protective effect of Ac-SDKP and its
potential association with Ang II signals was also
investigated.
Materials and methods
Materials
A total of 100 specific pathogen-free male Wistar
rats (3 weeks of age) were obtained from the Vital River Laboratory
(Beijing, China). The rats were housed in a temperature-controlled
facility at 22±2°C, 50±10% humidity and 0.03–0.04% CO2,
with a 12 h light/dark cycle and received food and water according
to the guidelines established by the North China University of
Science and Technology (16). HOPE
MED 8050 exposure control apparatus was supplied by Tianjin Hope
Industry and Trade Co., Ltd. (Tianjin, China). SiO2 was
obtained from Sigma Aldrich (Merck KGaA, Darmstadt, Germany). The
silica content of SiO2 was >99% and its dust particle
size distribution was as follows: 97% <5 µm diameter, 80% <3
µm diameter (according to the supplier). Ac-SDKP was supplied by
Bachem (Bubendorf, Switzerland). Ang II and valsartan were
purchased from Sigma-Aldrich (Merck KGaA). Ac-SDKP (cat. no.
CSB-E0925r) and Ang II (cat. no. CSB-E04494r) ELISA kits were
obtained from CUSABIO (Wuhan, Hubei, China). The PV-6000 1-step
polymer detection system for mouse, rabbit and rat antibodies were
acquired from Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.
(Beijing, China). Enhanced chemiluminescence reagent (cat. no.
RPN2232) was supplied by GE Healthcare (Chicago, IL, USA). Masson's
trichrome stain was supplied by Baso Diagnostics, Inc. (Wuhan,
China). Polyclonal rabbit anti-collagen (Col) I (cat. no. ab34710),
anti α-smooth muscle actin (SMA; cat. no. ab32575),
anti-Fibronectin (Fn) (cat. no. ab45688), rabbit monoclonal
anti-AT1 (cat. no. ab124734) and mouse monoclonal anti-ACE (cat.
no. ab117334) were purchased from Abcam (Cambridge, UK). Polyclonal
rabbit anti-matrix metalloproteinase (MMP)-1 (cat. no. EL800296-50)
and anti-tissue inhibitor of metalloproteinases-1 (TIMP-1; cat. no.
EL800628-50) were obtained from Eterlife, Ltd. (Birmingham, UK).
Polyclonal mouse anti-β-actin (cat. no. sc-47778) and polyclonal
rabbit anti-GAPDH (cat. no. sc-25778) were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA). Dulbecco's modified
Eagle's medium (DMEM) was obtained from Biological Industries USA
(Cromwell, CT, USA). Fetal bovine serum (FBS) was purchased from
Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Animal studies
All animal studies were approved by the
Institutional Animal Care and Use Committee of North China
University of Science and Technology. HOPE MED 8050 exposure
control apparatus was used to create the silicosis model. The
settings were as follows: Cabinet temperature, 20–25°C; humidity,
70–75%; pressure, −50 to +50 Pa; oxygen concentration, 20%; flow
rate of SiO2: 3.0–3.5 m/min; dust mass concentration,
2,000 mg/m3. Each rat inhaled SiO2 for 3 h
per day for 0, 2, 4, 8, 12 and 16 weeks. The rats were randomly
divided into 6 groups (n=10) and inhaled dust for 0, 2, 4, 8, 12
and 16 weeks to observe dynamic changes during the development of
silicosis. To determine the anti-silicotic effect of Ac-SDKP, rats
were randomly divided into 3 groups (n=10): i) 16 week control
(treated with 0.9% saline for 16 weeks); ii) 16 week silicosis
(treated with 0.9% saline 48 h before SiO2 inhalation
and thereafter for 16 weeks); and iii) Ac-SDKP post-treatment
(inhaled SiO2 and then treated with 0.9% saline for 8
weeks and Ac-SDKP for another 8 weeks). Ac-SDKP (800 mg/kg/day) or
control (0.9% saline) was administered via a mini-osmotic pump
implanted into the abdominal cavity as previously described
(17). The rats were anesthetized,
and sera and lung samples were collected and frozen at −80°C until
subsequent analysis.
Cell culture
Rats (n=10) were anaesthetized and lung tissue was
removed. Lung fibroblasts were isolated from minced tissue, as
previously described (18) and
plated on 25-cm2 plates containing DMEM supplemented
with 10% FBS and 1% penicillin-streptomycin. The cells were
cultured in a humidified atmosphere containing 5% CO2 at
37°C. Cells at 80% confluence were cultured in FBS-free DMEM for 24
h, when most cells were quiescent. The induction effect of Ang II
(10−7 mol/l) on the transformation of lung fibroblasts
into myofibroblasts was studied at different time points (0, 5, 15
and 30 min, and 1, 3, 6, 12, 24 and 48 h) at 37°C. Primary lung
fibroblasts were divided into four groups and cultured for 24 h at
37°C: i) Control (FBS-free DMEM); ii) Ang II (10−7
mol/l); iii) Ang II + Ac-SDKP (10−8 mol/l); and iv) Ang
II + valsartan (10−6 mol/l).
Lung histopathology
The right lower lungs were fixed in 4%
paraformaldehyde at 4°C for 24 h, embedded in paraffin, sectioned
at 5 µm and subjected to hematoxylin and eosin (H&E) and Masson
trichrome staining at room temperature for pathophysiological
observation. For H&E stainng, the tissues sections were stained
with hematoxylin for 30 sec and with stain 1% eosin solution for 1
min. For Masson trichrome staining, the tissue sections were
stained with Weigert's working hematoxylin for 10 min.
Subsequently, the sections were washed in running tap water for 5
min. Biebrich scarlet staining was subsequently performed for 5
min. Phosphomolybdic acid was added for 10 min and then the
solution was discarded. Tissue samples were transferred directly
into Aniline blue for 5 min and rinsed in distilled water. Images
in each rat were obtained from 5 randomly selected fields of view
using Olympus DP80 light microscope (magnification, ×200; Olympus
Corporation, Tokyo, Japan).
Immunohistochemistry (IHC)
IHC of lung tissue sections (5 µm) and cultured
fibroblasts (passage 9) was performed. Paraffin embedded sections
were deparaffinized, rehydrated and underwent removal of endogenous
peroxidase with 3% H2O2 and antigen retrieval
was performed using 0.01 M citrate buffer. Following blocking with
5% bovine serum albumin (Sigma-Aldrich, Merck KGaA) in 0.1 M
phosphate-buffer saline (PBS) for 30 min at room temperature to
reduce nonspecific binding, samples were incubated with a primary
antibody against α-SMA (1:200; cat. no. ab32575; Abcam) overnight
at 4°C, followed by incubation with a goat anti-mouse/rabbit
secondary antibody (cat. no. PV-6000, ready to use solution;
OriGene Technologies, Inc., Beijing, China) at 37°C for 1 h.
Immunoreactivity was visualized with 5% 3,3′-diaminobenzidene (cat.
no. ZLI-9018; OriGene Technologies, Inc., Beijing, China) at room
temperature for 5 min. Brown staining was considered positive. PBS,
instead of primary antibody, was used as a negative control and
α-SMA staining of vascular smooth muscle cells was used as a
positive control. Images were obtained from 5 separate randomly
selected fields of view using the Olympus DP80 (magnification,
×200).
Western blotting
Total proteins were extracted from lung tissues or
cells. Lung or cultured fibroblasts were lysed with
radioimmunoprecipitation assay (RIPA) buffer (1% NP-40, 0.5% sodium
deoxycholate, 0.1% SDS, 150 mM NaCl, 1 mM EDTA, and 50 mM Tris-HCl,
pH 7.5) and centrifuged at 14,000 × g for 10 min at 4°C. Protein
concentrations in supernatants were measured with a Bradford assay
(Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China). Protein samples (20 µg/lane) were separated by 10% SDS-PAGE
and transferred onto polyvinylidene fluoride membranes. Membranes
were blocked with 5% non-fat milk for 2 h at room temperature and
incubated overnight at 4°C with the primary antibody against Col I,
α-SMA, Fn, ACE and AT1 (all 1:500) followed by a peroxidase-labeled
affinity-purified secondary antibody to rabbit/mouse IgG (1:5,000,
cat. no. 074-1506, Kirkegaard & Perry Laboratories, Inc.,
Gaithersburg, MD, USA) for 2 h at 37°C. Target bands were
visualized by the addition of an Enhanced Chemiluminescence Prime
Western Blotting Detection reagent (GE Healthcare, Chicago, IL,
USA). The density of specific bands was analyzed using Image-Pro
Plus image processing software (version 6.0; Media Cybernetics,
Inc., Rockville, MD, USA) and the results were normalized to
β-actin or GAPDH (both 1:500).
ELISA assay
Lung tissues were lysed with RIPA and the levels of
Ac-SDKP and Ang II were determined by using the corresponding ELISA
kits according to the manufacturer's protocol.
Statistical analysis
All experiments were performed in triplicate. The
results are expressed as the mean ± standard error of the mean.
Comparisons between multiple independent groups were performed
using one-way analysis of variance followed by a post hoc analysis
with the Bonferroni test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Morphological alterations in lungs of
rats exposed to silica
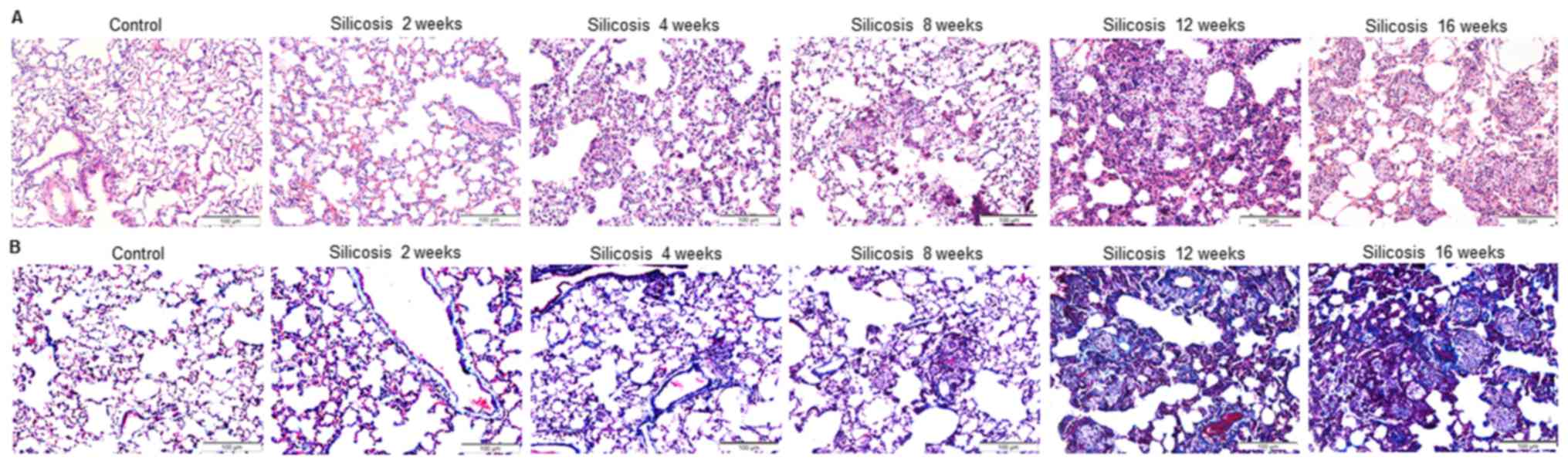
The results of HE staining revealed that in the
normal controls, the alveolar walls were thin and there was no
obvious inflammatory cell infiltration (Fig. 1A). In the 2-week silicosis group,
macrophages were observed in the alveoli and the alveolar wall was
wider in appearance. The 4-week silicosis group exhibited isolated
cell nodules that contained macrophages. The 8- and 12-week
silicosis groups had significantly wider alveolar walls and a
greater number of silicotic nodules. The 16-week silicosis group
exhibited an increase in silicotic nodule volume, nodule fusion,
interstitial fibrosis and cell fibrous nodules (Fig. 1A). Blue purple Masson trichrome
staining was observed in the nodules and interstitial fibrosis
area, indicating collagen deposition, which gradually increased
with model preparation time and most distinct in the 16-week
silicosis group (Fig. 1B).
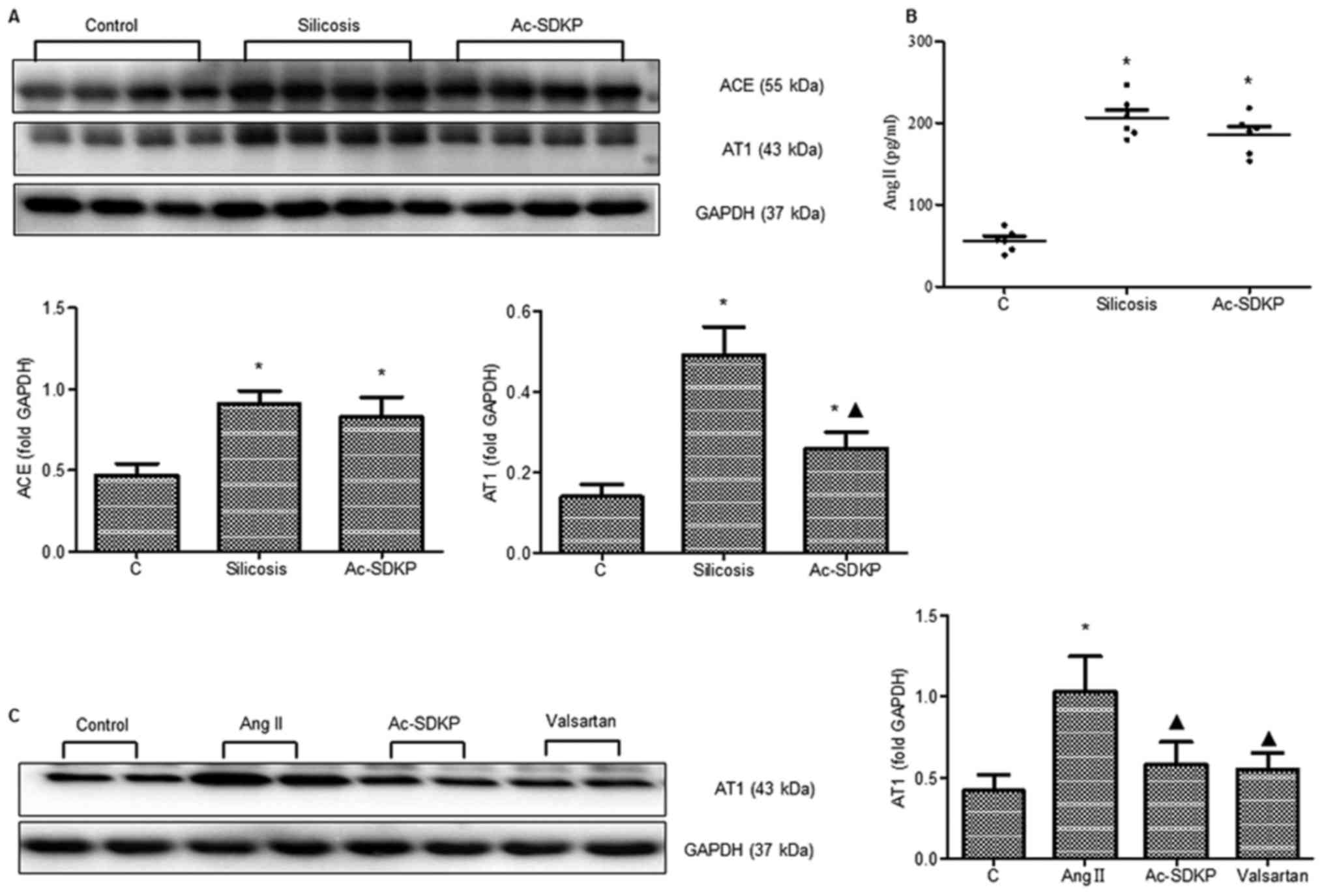
Expression of ACE, Ang II, AT1 and
Ac-SDKP in the lungs of rats exposed to silica
To investigate whether ACE/Ang II/AT1 proteins are
involved in the pathogenesis of silicotic fibrosis, the expression
of ACE, Ang II and AT1 in rat lungs was assessed. The expression of
ACE, Ang II and AT1 was increased in the 2-, 4-, 8-, 12- and
16-week silicosis groups compared with the control group (Fig. 2A and B). Ac-SDKP expression in the
2-week silicosis group was significantly higher compared with the
control group, whereas expression in the 4-, 8-, 12- and 16-week
silicosis groups decreased (Fig.
2C).
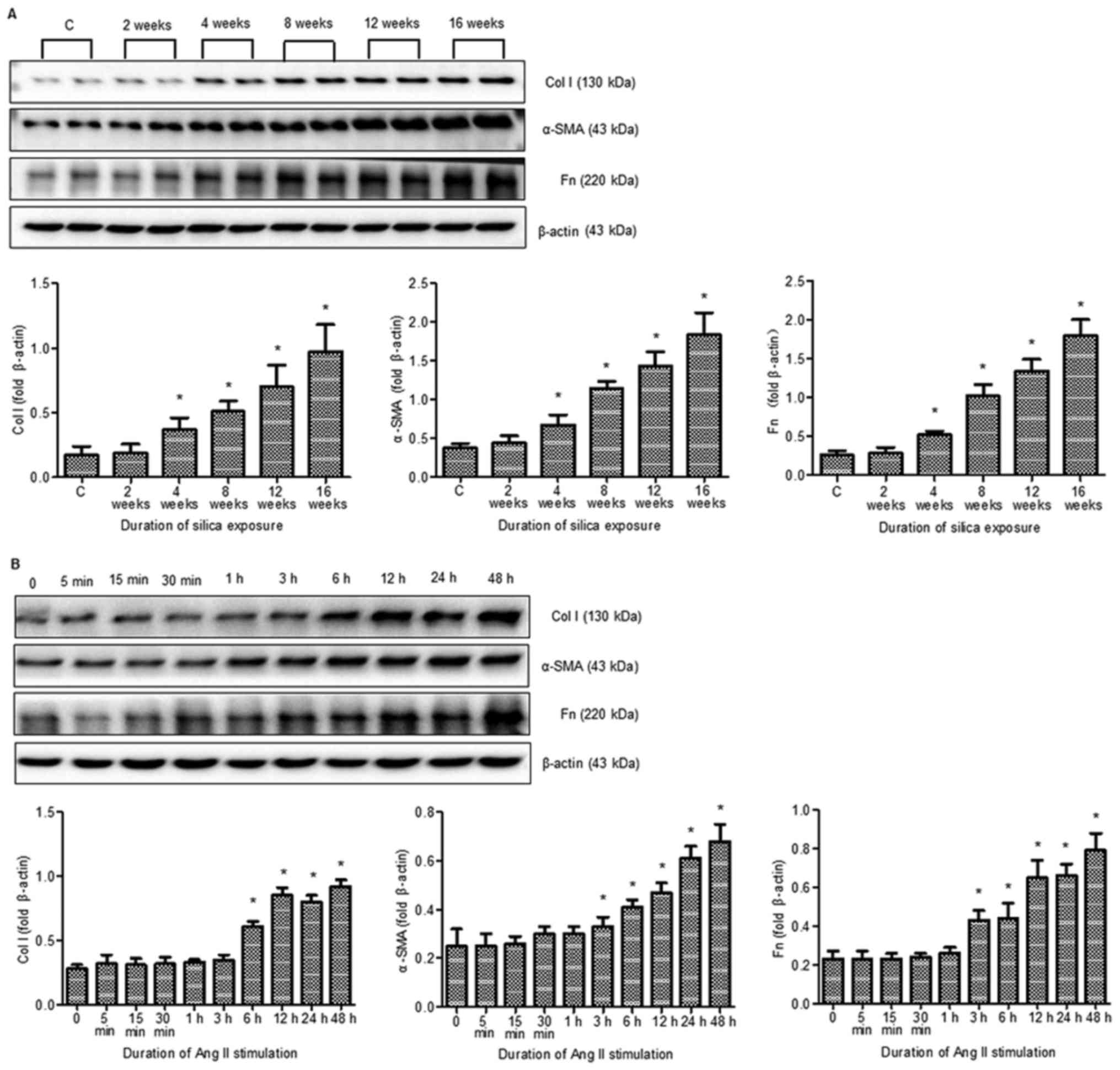
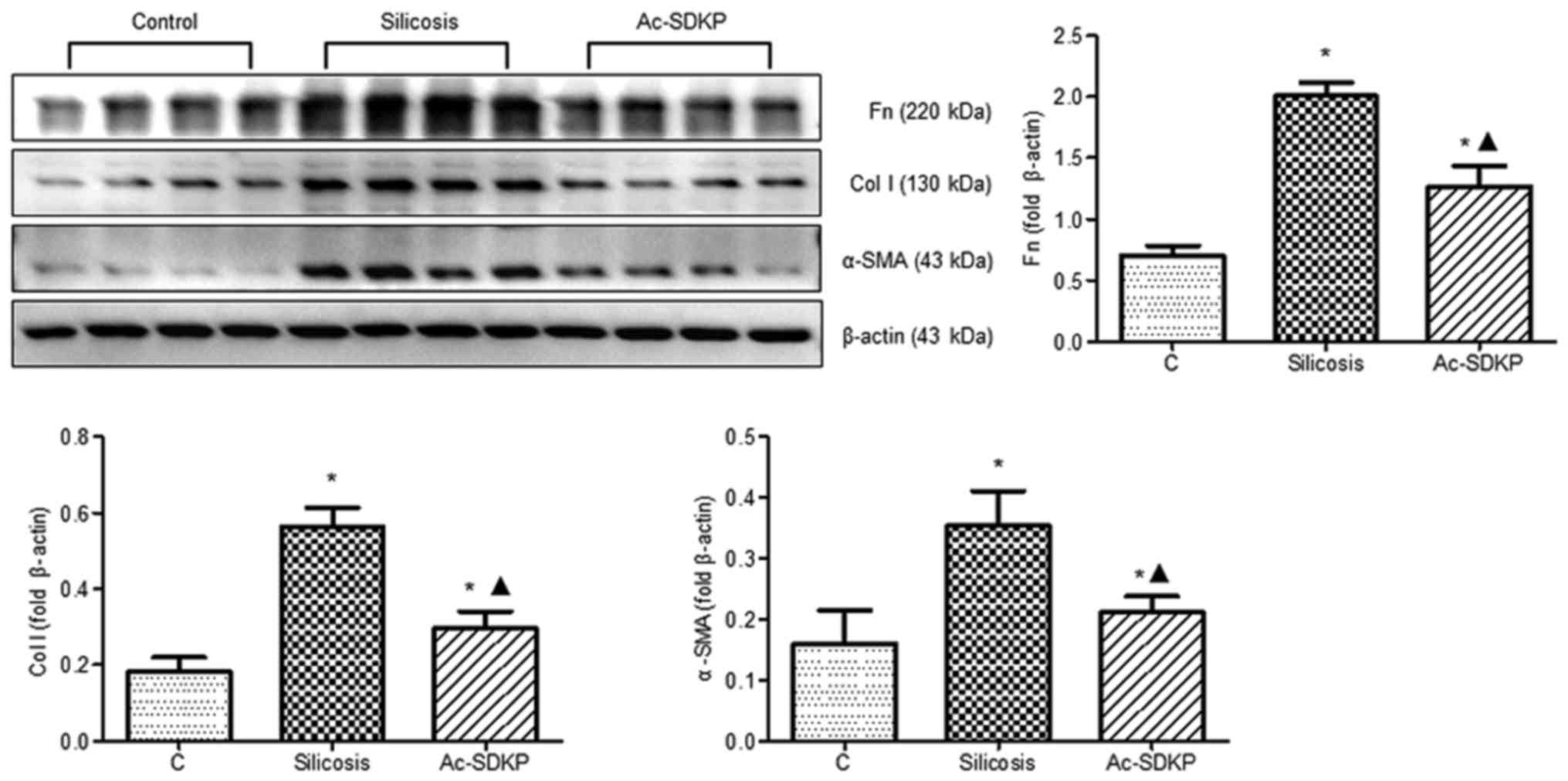
Expression of α-SMA, Col I and Fn in
silicosis and in fibroblasts induced by Ang II
Western blotting revealed that, compared with the
control group, α-SMA, Col I and Fn expression was gradually
increased in the 4-, 8-, 12- and 16-week silicosis groups (Fig. 3A). In response to Ang II stimuli,
the cultured rat lung fibroblasts underwent differentiation and
produced excessive amounts of ECM in vitro. Ang II
stimulated the expression of α-SMA, Col I and Fn, which increased
with treatment duration (Fig. 3B).
Based on these findings and results of a previous study (19), 24 h was selected as the optimal
treatment time for subsequent studies.
Ac-SDKP regulates ACE, Ang II and AT1
during silicosis and the expression of AT1 in fibroblasts induced
by Ang II
Compared with the 16-week silicosis group, treatment
with Ac-SDKP resulted in decreased AT1 expression. The expression
of ACE and Ang II (Fig. 4A and B)
in the Ac-SDKP treatment group decreased, but was not statistically
significant. The Ang II-dependent increase in AT1 expression was
also inhibited following Ac-SDKP and valsartan treatment (Fig. 4C).
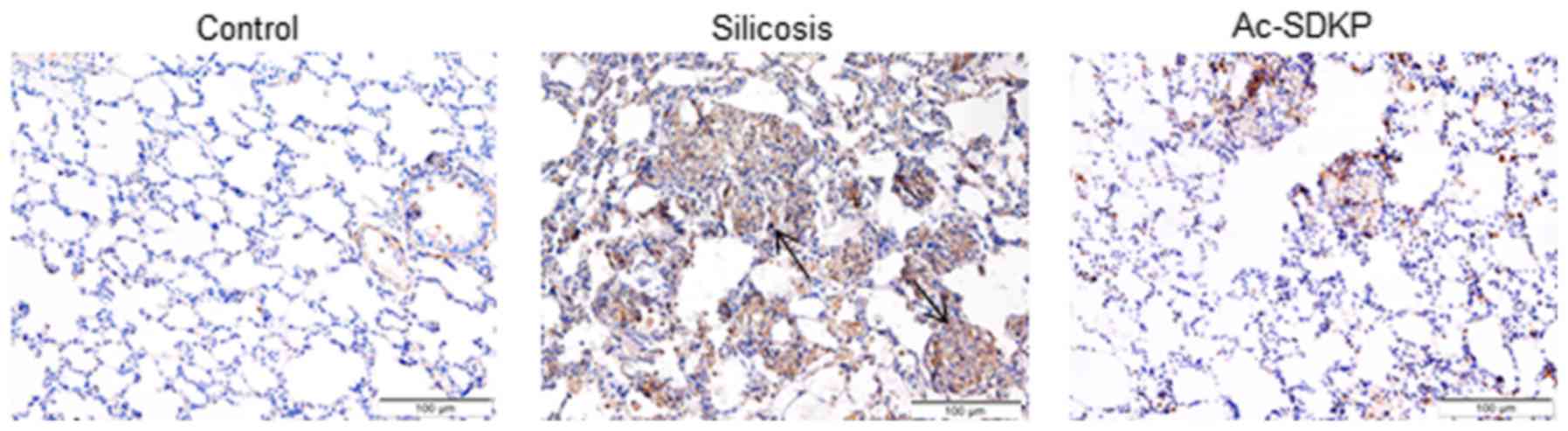
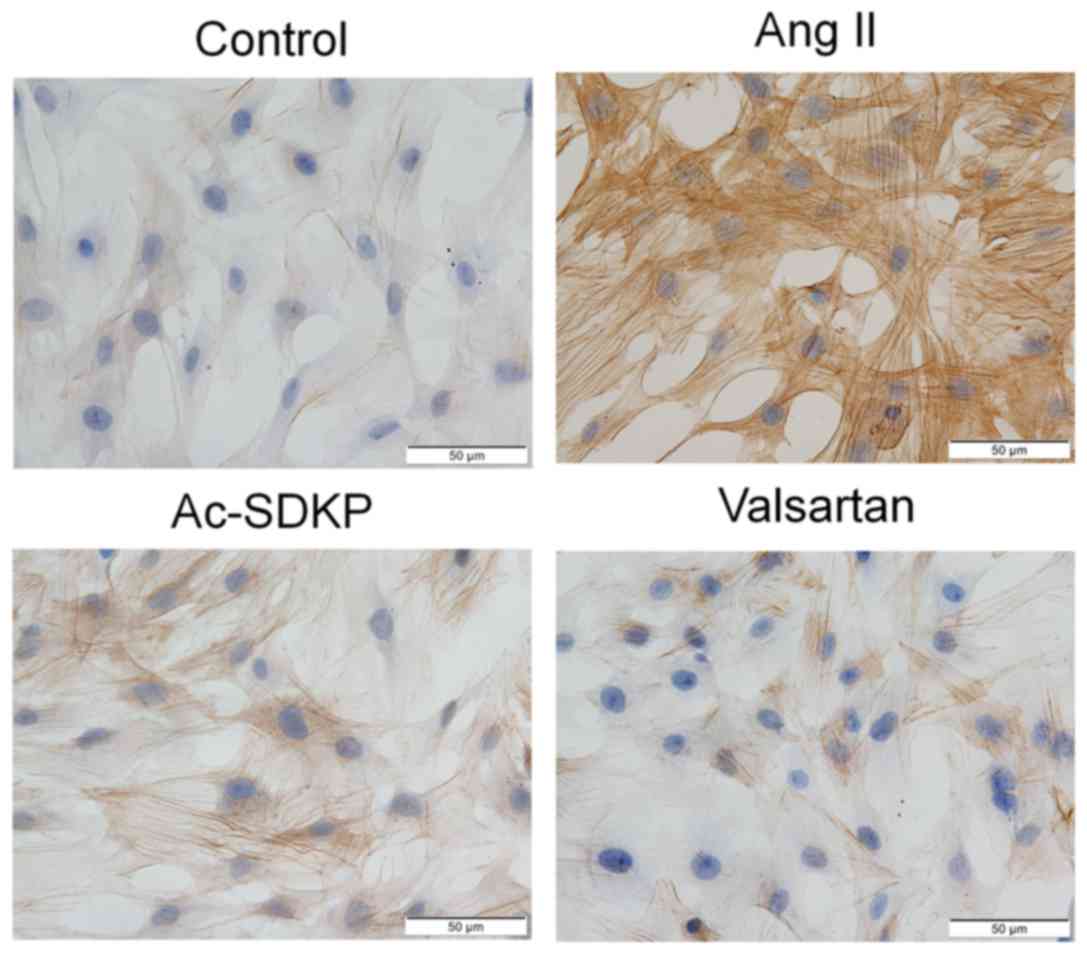
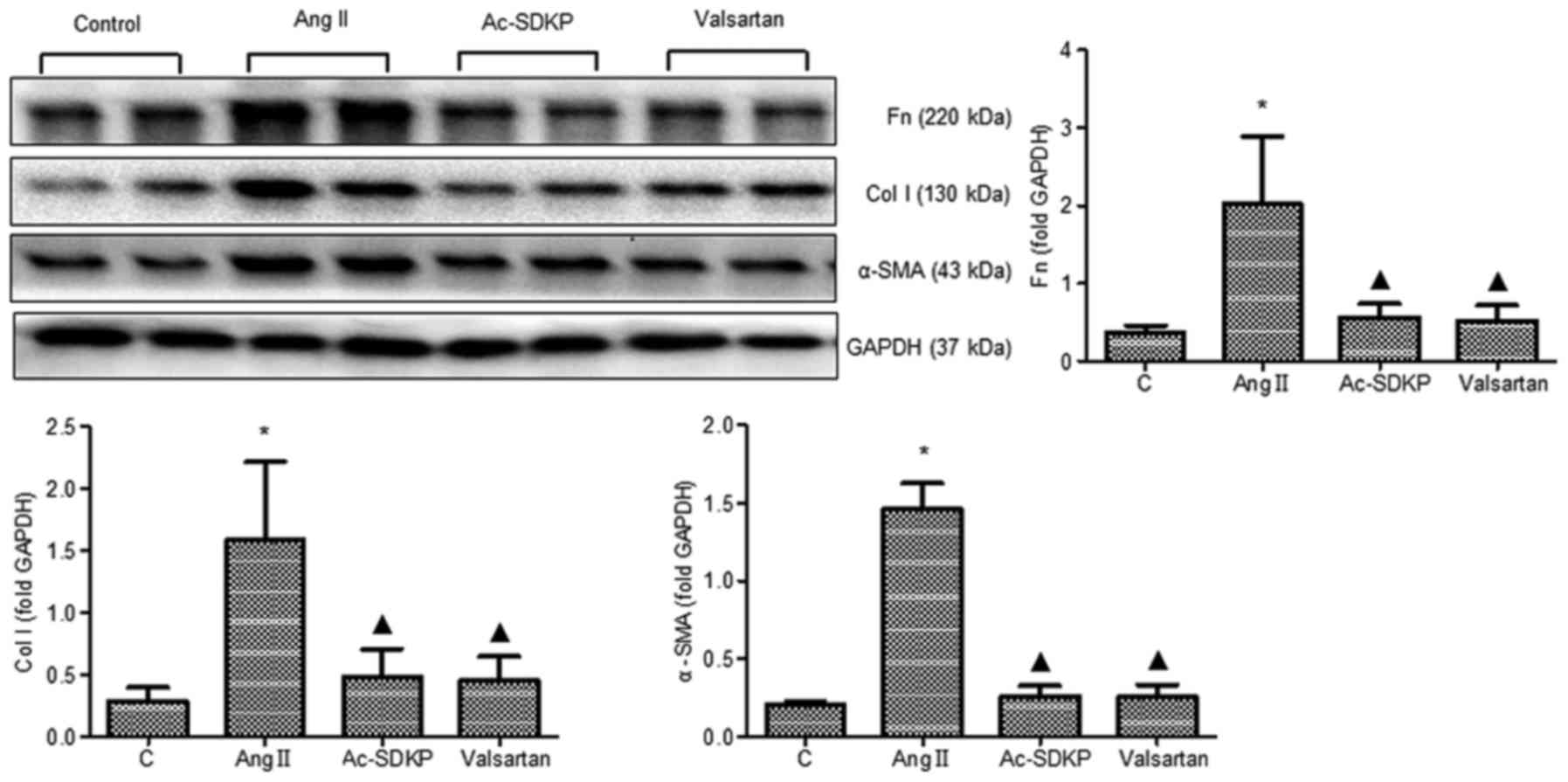
Ac-SDKP attenuates the expression of
α-SMA, Col I and Fn during silicosis and in Ang II-induced
fibroblasts
IHC staining revealed that α-SMA-positive
myofibroblasts were located in the nodules and distributed in the
interstitial fibrotic area in the 16-week silicosis group (Fig. 5). Ac-SDKP treatment reduced the
expression of α-SMA. Compared with the 16-week silicosis group,
α-SMA, Col I and Fn expression levels were significantly
downregulated in the Ac-SDKP post-treatment group (Fig. 6). Subsequently, α-SMA expression
was assessed using IHC staining. Compared with the control group,
α-SMA expression was demonstrated to be increased following Ang II
induction in cultured lung fibroblasts. Compared with the Ang II
group, the expression of α-SMA was decreased in the Ac-SDKP and
valsartan treatment groups (Fig.
7). The Ang II-dependent increase in α-SMA, Col I and Fn
expression was also inhibited following Ac-SDKP and valsartan
treatment (Fig. 8).
Ac-SDKP regulates MMP-1, TIMP-1 and
the MMP-1/TIMP-1 ratio during silicosis and in fibroblasts induced
by Ang II
Western blotting demonstrated that the expression of
TIMP-1 was markedly increased in the silicosis group compared with
the control group, whereas MMP-1 expression and the MMP-1/TIMP-1
ratio was significantly decreased (Fig. 9A). Treatment with Ac-SDKP increased
the expression of MMP-1 and the MMP-1/TIMP-1 ratio. No significant
differences in TIMP-1 expression were observed between the Ac-SDKP
post-treatment and silicosis groups. In vitro experiments
revealed that MMP-1 expression and the MMP-1/TIMP-1 ratio was
reduced compared with the control and the expression of TIMP-1 was
markedly increased in fibroblasts induced by Ang II (Fig. 9B). Ac-SDKP or valsartan treatment
upregulated the expression of MMP-1 and enhanced the MMP-1/TIMP-1
ratio, whereas it had no significant effect on the expression of
TIMP-1.
Discussion
In the present study, a rat silicosis model was
established using HOPE MED8050 exposure control apparatus. This
system is a non-invasive instrument that allows animal inhalation
of silica particles and may be set to a certain dust concentration.
Compared with intratracheal administration of silica dust (20,21),
using the dynamic pollution control system facilitated the
establishment of an ideal silicosis model. Histopathological
observations demonstrated that, following inhalation of
SiO2 for 4 weeks, silicotic nodules containing
macrophages developed in the lung tissues of rats. Furthermore, the
number of silicotic nodules increased after 8 and 12 weeks of
inhalation. Fibrous and cellular silicotic nodules with diffuse
interstitial fibrosis were observed in rats at 16 weeks.
Myofibroblasts are α-SMA-expressing cells that originate from
epithelial-mesenchymal transition (22), endothelial-to-mesenchymal
transition (23), pericyte
transdifferentiation (24) and
pleural mesothelial cells (25) or
pulmonary interstitial fibroblasts (26). Myofibroblasts serve a pivotal role
in wound healing; however, the unusual persistent proliferative and
migratory properties of myofibroblasts leads to an excessive
accumulation of the ECM and lung fibrosis. In the present study,
IHC revealed that α-SMA-positive myofibroblasts surrounded
macrophages and were irregularly distributed in interstitial
fibrotic areas. Western blotting further demonstrated that α-SMA
protein expression in the 4-, 8-, 12- and 16-week silicosis groups
gradually increased, suggesting the accumulation of a large number
of activated myofibroblasts in the silicosis model. These findings
suggested that a silicosis model was successfully established using
a HOPE-MED8050 dynamic dust system.
RAS is a key mediator that is involved in the
pathogenesis of tissue remodeling, including the lungs (5). Studies have reported that the
expression of ACE in bronchoalveolar lavage fluid is higher
(27) and the angiotensinogen gene
is overexpressed in the lung tissues of patients with pulmonary
fibrosis (28). This contributes
to increase Ang II levels in pulmonary fibrosis. It has been
suggested that Ang II, acting via the AT1 receptor, modulates
pro-fibrotic downstream effects, including inflammatory cell
recruitment, cellular proliferation and the accumulation of ECM
(8). In the present study it was
demonstrated that the expression of ACE, Ang II and AT1 in the lung
tissue of rats increased with time, which was consistent with the
degree of silicosis. Differentiation of fibroblasts into
myofibroblasts is a critical event in fibrosis. Ang II serves a
major role in fibrosis by promoting myofibroblast differentiation
(29,30). The expression of α-SMA, Col I and
Fn in Ang II-stimulated lung fibroblasts was demonstrated to be
time-dependent in the present study, thereby confirming that
myofibroblast differentiation may be induced by Ang II from lung
fibroblasts and suggesting that the increased ECM deposition in the
rat silicosis model may be associated with Ang II elevation in
vivo. Silicosis in the rat model was mediated, at least
partially, by activation of the RAS signaling pathway.
Ac-SDKP is an ACE substrate that demonstrates
meaningful anti-fibrotic effects in various experimental models of
fibrotic disease (3). Ac-SDKP has
been reported to inhibit cell proliferation and myofibroblast
differentiation via weakening TGF-β-induced mothers against
decapentaplegic homolog 2 (Smad2)/3 signaling effects (31–33).
It has also been demonstrated that Ac-SDKP treatment promotes the
expression of Smad7, which antagonizes Smad2 activity (34). Both RAS and TGF-β have been
described as important regulators of the fibrotic disease (35,36).
Ang II and TGF-β1 do not appear to act independently from one
another, but rather as part of a signaling network in cardiac
remodeling and fibrosis (14,37).
Therefore, the interaction between Ac-SDKP and RAS in fibrosis
requires further investigation. In the present study, it was
observed that Ac-SDKP levels in lung tissues were increased in the
2-week silicosis group compared with the control group, whereas the
expression of Ac-SDKP decreased gradually at subsequent time
points. Decreased Ac-SDKP expression and increased ACE, Ang II and
AT1 expression may result in an imbalance between pro-fibrotic and
anti-fibrotic molecules, thereby facilitating the occurrence and
development of silicosis. In addition, the results of the present
study revealed that Ac-SDKP treatment did not significantly affect
ACE or Ang II levels, whereas Ac-SDKP did downregulate the
expression of AT1 in vivo and inhibited myofibroblast
differentiation and ECM synthesis. Ac-SDKP had the same effect as
valsatan, inhibiting Ang II-induced myofibroblast differentiation
and decreasing Col I and Fn accumulation in vitro. The
fibrogenic process is associated with an imbalance in MMPs/TIMPs,
which results in excessive expression of components of the ECM.
MMP-1 serves an important role in fibrosis by preferentially
degrading Col I (38). In
contrast, TIMP-1 moderately inhibits the activity of MMP1 (39). In the present study, Ac-SDKP
treatment increased the expression levels MMP-1 and the
MMP-1/TIMP-1 ratio in vivo and in vitro. The results
of the present study therefore suggest that Ac-SDKP may effectively
protect lung tissues against silicosis fibrosis and serve a vital
role in inhibiting the Ang II-induced myofibroblast differentiation
and accumulation of ECM in silicosis.
In summary, the results of the present study suggest
that an imbalance between Ac-SDKP and ACE/Ang II/AT1 molecules
promotes the development of silicosis. Ac-SDKP blocked fibroblast
differentiation induced by Ang II and inhibited ECM production,
thereby preventing the development of silicotic fibrosis. As
Ac-SDKP is inhibited by ACE in vivo, the authors of the
present study intend to use an ACE inhibitor to determine if
similar anti-fibrosis results can be observed. The present study
indicates a possibility of use of Ac-SDKP as an inhibitor of RAS
for the treatment of silicosis.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81472953), the
Natural Science Foundation of Hebei Province (grant no.
H2016209170), the National College Students Innovation and
Entrepreneurship Training Program (grant no. 201710081010) and
Doctoral Fund of North China University of Science and
Technology.
Glossary
Abbreviations
Abbreviations:
|
Ac-SDKP
|
N-acetyl-seryl-aspartyl-lysyl-proline
|
|
ECM
|
extracellular matrix
|
|
ACE
|
angiotensin-converting enzyme
|
|
Ang II
|
angiotensin II
|
|
RAS
|
renin-angiotensin system
|
|
α-SMA
|
α-smooth muscle actin
|
|
TGF-β
|
transforming growth factor-β
|
|
H&E
|
hematoxylin and eosin
|
|
IHC
|
immunohistochemistry
|
|
MMP-1
|
matrix metalloproteinase-1
|
|
TIMP-1
|
tissue inhibitor of
metalloproteinases-1
|
References
|
1
|
Rimal B, Greenberg AK and Rom WN: Basic
pathogenetic mechanisms in silicosis: Current understanding. Curr
Opin Pulm Med. 11:169–173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cavasin MA, Rhaleb NE, Yang XP and
Carretero OA: Prolyl oligopeptidase is involved in release of the
antifibrotic peptide Ac-SDKP. Hypertension. 43:1140–1145. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mnguni AT, Engel ME, Borkum MS and Mayosi
BM: The effects of angiotensin converting enzyme inhibitors (ACE-I)
on human N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) levels: A
systematic review and meta-analysis. PLoS One. 10:e01433382015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kumar N, Nakagawa P, Janic B, Romero CA,
Worou ME, Monu SR, Peterson EL, Shaw J, Valeriote F, Ongeri EM, et
al: The anti-inflammatory peptide Ac-SDKP is released from
thymosin-β4 by renal meprin-α and prolyl oligopeptidase. Am J
Physiol Renal Physiol. 310:F1026–F1034. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meng Y, Yu CH, Li W, Li T, Luo W, Huang S,
Wu PS, Cai SX and Li X: Angiotensin-converting enzyme
2/angiotensin-(1–7)/Mas axis protects against lung fibrosis by
inhibiting the MAPK/NF-κB pathway. Am J Respir Cell Mol Biol.
50:723–736. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ntsekhe M, Matthews K, Wolske J, Badri M,
Wilkinson KA, Wilkinson RJ, Sturrock ED and Mayosi BM: Scientific
letter: Ac-SDKP (N-acetyl-seryl-aspartyl-lysyl-proline) and
Galectin-3 levels in tuberculous pericardial effusion: Implications
for pathogenesis and prevention of pericardial constriction. Heart.
98:1326–1328. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang J, Chen L, Chen B, Meliton A, Liu SQ,
Shi Y, Liu T, Deb DK, Solway J and Li YC: Chronic activation of the
renin-angiotensin system induces lung fibrosis. Sci Rep.
5:155612015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Murphy AM, Wong AL and Bezuhly M:
Modulation of angiotensin II signaling in the prevention of
fibrosis. Fibrogenesis Tissue Repair. 8:72015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wei L, Alhenc-Gelas F, Corvol P and
Clauser E: The two homologous domains of human angiotensin
I-converting enzyme are both catalytically active. J Biol Chem.
266:9002–9008. 1991.PubMed/NCBI
|
|
10
|
Masuyer G, Schwager SL, Sturrock ED, Isaac
RE and Acharya KR: Molecular recognition and regulation of human
angiotensin-I converting enzyme (ACE) activity by natural
inhibitory peptides. Sci Rep. 2:7172012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stawski L, Han R, Bujor AM and Trojanowska
M: Angiotensin II induces skin fibrosis: A novel mouse model of
dermal fibrosis. Arthritis Res Ther. 14:R1942012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cavin S, Maric D and Diviani D: A-kinase
anchoring protein-Lbc promotes pro-fibrotic signaling in cardiac
fibroblasts. Biochim Biophys Acta. 1843:335–345. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wong MH, Chapin OC and Johnson MD:
LPS-stimulated cytokine production in type I cells is modulated by
the renin-angiotensin system. Am J Respir Cell Mol Biol.
46:641–650. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Uhal BD, Kim JK, Li X and Molina-Molina M:
Angiotensin-TGF-beta 1 crosstalk in human idiopathic pulmonary
fibrosis: Autocrine mechanisms in myofibroblasts and macrophages.
Curr Pharm Des. 13:1247–1256. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Masuyer G, Douglas RG, Sturrock ED and
Acharya KR: Structural basis of Ac-SDKP hydrolysis by Angiotensin-I
converting enzyme. Sci Rep. 5:137422015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Y, Xu H, Geng Y, Xu D, Zhang L, Yang
Y, Wei Z, Zhang B, Li S, Gao X, et al: Dibutyryl-cAMP attenuates
pulmonary fibrosis by blocking myofibroblast differentiation via
PKA/CREB/CBP signaling in rats with silicosis. Respir Res.
18:382017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu H, Yang F, Sun Y, Yuan Y, Cheng H, Wei
Z, Li S, Cheng T, Brann D and Wang R: A new antifibrotic target of
Ac-SDKP: Inhibition of myofibroblast differentiation in rat lung
with silicosis. PLoS One. 7:e403012012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li S, Gao X, Xu D, Wang X, Liu Y, Zhang L,
Deng H, Wei Z, Tian J, Xu H and Yang F: Inhibition effect of
N-acetyl-seryl-aspartyl-lysyl-proline on myofibroblast
differentiation by regulating acetylated tubulin α in silicotic rat
model. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 33:816–821.
2015.(In Chinese). PubMed/NCBI
|
|
19
|
Meng Y, Li T, Zhou GS, Chen Y, Yu CH, Pang
MX, Li W, Li Y, Zhang WY and Li X: The angiotensin-converting
enzyme 2/angiotensin (1–7)/Mas axis protects against lung
fibroblast migration and lung fibrosis by inhibiting the
NOX4-derived ROS-mediated RhoA/Rho kinase pathway. Antioxid Redox
Signal. 22:241–258. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Morimoto Y, Nagatomo H, Hirohashi M, Oyabu
T, Ogami A, Yamato H, Kuroda K, Obata Y, Higashi T and Tanaka I:
Expression of clara cell secretory protein in the lungs of rats
exposed to crystalline silica in vivo. J Occup Health. 47:504–509.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dong H, Jing W, Yabo Y, Xiaokang Y, Wan W,
Min M, Wenyang W, Zhaoquan C, Yingru X and Rongbo Z: Establishment
of rat model of silicotuberculosis and its pathological
characteristic. Pathog Glob Health. 108:312–316. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou B, Liu Y, Kahn M, Ann DK, Han A, Wang
H, Nguyen C, Flodby P, Zhong Q, Krishnaveni MS, et al: Interactions
between β-catenin and transforming growth factor-β signaling
pathways mediate epithelial-mesenchymal transition and are
dependent on the transcriptional co-activator cAMP-response
element-binding protein (CREB)-binding protein (CBP). J Biol Chem.
287:7026–7038. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hashimoto N, Phan SH, Imaizumi K, Matsuo
M, Nakashima H, Kawabe T, Shimokata K and Hasegawa Y:
Endothelial-mesenchymal transition in bleomycin-induced pulmonary
fibrosis. Am J Respir Cell Mol Biol. 43:161–172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marriott S, Baskir RS, Gaskill C, Menon S,
Carrier EJ, Williams J, Talati M, Helm K, Alford CE, Kropski JA, et
al: ABCG2pos lung mesenchymal stem cells are a novel pericyte
subpopulation that contributes to fibrotic remodeling. Am J Physiol
Cell Physiol. 307:C684–C698. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zolak JS, Jagirdar R, Surolia R, Karki S,
Oliva O, Hock T, Guroji P, Ding Q, Liu RM, Bolisetty S, et al:
Pleural mesothelial cell differentiation and invasion in fibrogenic
lung injury. Am J Pathol. 182:1239–1247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tumelty KE, Smith BD, Nugent MA and Layne
MD: Aortic carboxypeptidase-like protein (ACLP) enhances lung
myofibroblast differentiation through transforming growth factor β
receptor-dependent and -independent pathways. J Biol Chem.
289:2526–2536. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Specks U, Martin WJ II and Rohrbach MS:
Bronchoalveolar lavage fluid angiotensin-converting enzyme in
interstitial lung diseases. Am Rev Respir Dis. 141:117–123. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li X, Molina-Molina M, Abdul-Hafez A,
Ramirez J, Serrano-Mollar A, Xaubet A and Uhal BD: Extravascular
sources of lung angiotensin peptide synthesis in idiopathic
pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol.
291:L887–L895. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bai J, Zhang N, Hua Y, Wang B, Ling L,
Ferro A and Xu B: Metformin inhibits angiotensin II-induced
differentiation of cardiac fibroblasts into myofibroblasts. PLoS
One. 8:e721202013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo X, Yan F, Li J, Zhang C and Bu P:
SIRT3 attenuates AngII-induced cardiac fibrosis by inhibiting
myofibroblasts transdifferentiation via STAT3-NFATc2 pathway. Am J
Transl Res. 9:3258–3269. 2017.PubMed/NCBI
|
|
31
|
Peng H, Carretero OA, Peterson EL and
Rhaleb NE: Ac-SDKP inhibits transforming growth
factor-beta1-induced differentiation of human cardiac fibroblasts
into myofibroblasts. Am J Physiol Heart Circ Physiol.
298:H1357–H1364. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pokharel S, Rasoul S, Roks AJ, van Leeuwen
RE, van Luyn MJ, Deelman LE, Smits JF, Carretero O, van Gilst WH
and Pinto YM: N-acetyl-Ser-Asp-Lys-Pro inhibits phosphorylation of
Smad2 in cardiac fibroblasts. Hypertension. 40:155–161. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kanasaki K, Koya D, Sugimoto T, Isono M,
Kashiwagi A and Haneda M: N-Acetyl-seryl-aspartyl-lysyl-proline
inhibits TGF-beta-mediated plasminogen activator inhibitor-1
expression via inhibition of Smad pathway in human mesangial cells.
J Am Soc Nephrol. 14:863–872. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin CX, Rhaleb NE, Yang XP, Liao TD,
D'Ambrosio MA and Carretero OA: Prevention of aortic fibrosis by
N-acetyl-seryl-aspartyl-lysyl-proline in angiotensin II-induced
hypertension. Am J Physiol Heart Circ Physiol. 295:H1253–H1261.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yamazaki KG, Gonzalez E and Zambon AC:
Crosstalk between the renin-angiotensin system and the advance
glycation end product axis in the heart: Role of the cardiac
fibroblast. J Cardiovasc Transl Res. 5:805–813. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Samarakoon R, Overstreet JM and Higgins
PJ: TGF-β signaling in tissue fibrosis: Redox controls, target
genes and therapeutic opportunities. Cell Signal. 25:264–268. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rosenkranz S: TGF-beta1 and angiotensin
networking in cardiac remodeling. Cardiovasc Res. 63:423–432. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Craig VJ, Zhang L, Hagood JS and Owen CA:
Matrix metalloproteinases as therapeutic targets for idiopathic
pulmonary fibrosis. Am J Respir Cell Mol Biol. 53:585–600. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yoshiji H, Kuriyama S, Yoshii J, Ikenaka
Y, Noguchi R, Nakatani T, Tsujinoue H, Yanase K, Namisaki T, Imazu
H and Fukui H: Tissue inhibitor of metalloproteinases-1 attenuates
spontaneous liver fibrosis resolution in the transgenic mouse.
Hepatology. 36:850–860. 2002. View Article : Google Scholar : PubMed/NCBI
|