Introduction
Heart failure is the terminal stage of the
development of various cardiovascular diseases and it results in
high morbidity and mortality (1).
A variety of drug treatments used in recent years improve the
quality of life of patients with heart failure, but patients'
survival rate remains unaltered (2). Epigallocatechingallate (EGCG) is a
catechin monomer extracted from leaves or buds of Camellia
sinensis (3). It is an active
and water-soluble ingredient which extracted from tea tree and its
catechin content in was the highest among polyphenols (4). Previous studies demonstrated that
EGCG can effectively attenuatecardiac hypertrophy and remodeling
caused by pressure load, prevent cardiomyocyte apoptosis and
oxidative stress in cardiac hypertrophy, inhibit the abnormal
proliferation of cardiac fibroblasts and improve myocardial
remodeling (5,6). Sriram et al (7) demonstrated that EGCG attenuates
fibroblast proliferation and excessive collagen production by
interfering with transforming growth factor-β1 (TGF-β1) signaling.
Hsieh et al (8)
demonstrated that EGCG can inhibit TGF-β1-induced collagen
production by attenuating expression of early growth response
protein 1 in buccal mucosal fibroblasts. TGF-β1 is a pleiotropic
cytokine that regulates cell proliferation, differentiation and
apoptosis through autocrine and paracrine signaling pathways that
operate via cell surface receptors (9). It also serves a role in the
regulation of synthesis of extracellular matrix, tissue repair
following trauma and immune function (10–12).
TGF-β1 can bind to its receptors TGF-β receptor type-1 (TβRI) and
type-II (TβRII), regulate intracellular signal transduction and
exert numerous biological effects (13). Several studies have demonstrated
that TGF-β1 signaling pathway is a regulator of myocardial repair
and remodeling following myocardial infarction and it can
participate in the repair process of myocardial hypoxia through
Smad and non-Smad signaling pathways [including p38 protein, c-Jun
N-terminal kinase, TGF-β-activated kinase (TAK)] (14–16).
Smad proteins is are downstream regulators of the TGF-β1 signaling
pathway (17). Following binding
to its receptors TβRI and TβRII, TGF-β1 phosphorylates and
activates Smad proteins. Smad proteins exhibit specific DNA binding
activities and can function as transcription factors to regulate
gene expression of the associated cytokines [including
platelet-derived growth factor, fibroblast growth factor and tumor
necrosis factor (TNF)], leading to fibrosis. Downregulation or
inhibition of TGF-β1/Smad expression can reduce collagen synthesis
and improve myocardial fibrosis (18,19).
A previous study demonstrated that inhibition of activation of
TGF-β1/mothers against decapentaplegic homolog 3 (Smad3) or
TGF-β1/TAKl can attenuate cardiac hypertrophy and remodeling
(20).
In the present study, the protective effect of EGCG
in mouse models of heart failure and the underlying mechanism, were
investigated, providing a theoretical basis for clinical drug
therapy of this disease and development of novel drugs.
Materials and methods
Animal experimental protocols
A total of 40 (6 weeks old, male, weight 20–23 g)
C57/B6 mice were obtained from the Experimental Animal Center of
Liaoning University of Traditional Chinese Medicine (Shenyang,
China) [production license no. SCXK (Liao)2013-0004; application
license no. SYXK (Liao)-2013-0008]. The present study was approved
by the Liaoning University of Traditional Chinese Medicine
Laboratory Animal Welfare and Ethics Committee (approval no.
2016048). Mice were given a normal diet and water, and the
following housing conditions: Ambient temperature of 20–26°C,
40–70% relative humidity, 12-h light/dark cycle. TGF-β1 inhibitor
LY364947 (cat. no. ab141890) was supplied by Abcam (Cambridge, UK).
Mice were randomly divided into four groups with 8 mice in each
group, including sham, heart failure (HF), EGCG and LY
(EGCG+LY364947) groups. In the sham group, a skin incision was made
to bluntly separate the aortic arch and then the skin incision was
closed. In the HF group, a 27G blunt needle and the aortic arch
were ligated. Mouse models of heart failure were established, as
described below. In the EGCG group, following establishment of a
model of heart failure, 10 mg/kg/day EGCG was intraperitoneally
administered for 4 successive weeks. In the LY group, following
induction of heart failure, 10 mg/kg/day EGCG was intraperitoneally
administered and simultaneously TGF-β1 inhibitor LY364947 (1
µmol/l) was administered via the tail vein for 4 successive weeks.
In the sham group, mouse were injected intraperitoneally with the
same amount of saline for 4 weeks.
Sample collection
Folllowing the 4 weeks of treatment, all mice were
sacrificed by cervical dislocation under anesthesia. Mouse venous
blood was collected and serum was separated by centrifugation at
500 × g at 4°C for 10 min, and used for ELISA. Mouse heart tissue
was harvested, as previously described (21). One portion of heart tissue was
preserved in liquid nitrogen for subsequent western blot analysis
and reverse transcription-quantitative polymerase chain reaction
(RT-qPCR). The remaining tissue was fixed in formaldehyde at room
temperature for 48 h for hematoxylin and eosin (H&E) and Masson
staining.
Establishment of mouse models of heart
failure
Heart failure model was performed as previously
reported with minor modifications (21). Mice were anesthetized by
intraperitoneal administration of 2% pentobarbital sodium. A 0.5 cm
long incision was made on the sternum to expose the aortic arch. A
27G blunt needle and the aortic arch were ligated. The needle was
removed and the incision was closed.
Color Doppler ultrasonography
A total of 4 weeks following surgery, mice were
weighted. Following anesthesia, mice were placed in the dorsal
position. Echocardiography was performed using a Philips CX50
ultrasound system (probe pattern S-12-4, frequency 4–12 MHz) to
measure the ejection fraction (EF), left ventricular internal
diastolic diameter (LVIDd) and left ventricular internal systolic
diameter (LVIDs). The mean value for each index across three
cardiac cycles was calculated.
H&E staining
A total of 48 h following fixation in formaldehyde,
heart tissue was dehydrated, cleared, embedded, 5 µm sliced,
dewaxed, hydrated, stained with hematoxylin for 20 min at room
temperature, differentiated with hydrochloric acid and ethanol for
3–5 sec, stained with eosin for 1 min at room temperature,
dehydrated in alcohol gradients, cleared and mounted. Pathological
alteration of myocardial tissue was observed under a light
microscope.
Masson staining
Formaldehyde sections were dewaxed, hydrated,
stained with Regaud's hematoxylin for 5–10 min at room temperature,
washed, stained with Masson ponceau acid fuchsin solution for 5–10
min at room temperature, soaked in 2% acetic acid aqueous solution
for 3–5 sec, differentiated with 1% phosphomolybdic acid aqueous
solution for 3–5 min at room temperature, stained with aniline blue
for 5 min at room temperature, immersed in 0.2% acetic acid aqueous
solution for 3–5 sec, treated with 95% ethanol and absolute
ethanol, cleared with xylene, and mounted with neutral gum.
ELISA
Mouse peripheral blood was collected. Serum levels
of brain natriuretic peptide (BNP; cat. no. JM-E10001485; Tsz
Biosciences, San Francisco, CA, USA), N-terminal proBNP (NT-proBNP;
cat. no. JM-E10001510; Tsz Biosciences), IL-1β (cat. no. SEA563Mu;
Cloud-Clone Corp, Katy, TX, USA), IL-6 (cat. no. SEA079Mu;
Cloud-Clone Corp), TNF-α (cat. no. SEA133Mu; Cloud-Clone Corp),
malondialdehyde (MDA; cat. no. CEA597Ge; Cloud-Clone Corp),
superoxide dismutase (SOD; cat. no. SES134Mu; Cloud-Clone Corp),
glutathione peroxidase (GSH-Px; cat. no. CEA294Ge; CCC) were
measured according to manufacturer's instructions of the kits.
Optical density was measured at a wavelength of 450 nm with a
microplate reader. A standard curve was generated by plotting
optical density value vs. the standard concentration. The curve
equation and r value were calculated and used to determine
concentrations of samples using Excel (2010; Microsoft Corporation,
Redmond, WA, USA).
Western blot analysis
Mouse myocardial tissue was lysed in a RIPA lysate
containing protease inhibitor (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) and homogenized on ice. Supernatant was collected
following centrifugation at 4,000 × g at 4°C for 10 min to
determine protein concentration using the Bicinchoninic acid
Protein Assay kit. Protein samples (30 µg) were subjected to 10%
SDS-PAGE and transferred to PVDF membranes. Following addition of
TGF-β1 (1:1,000; cat. no. ab92486), Smad3 (1:1,000; cat. no.
ab40854), phosphorylated (p)-Smad3 (1:1,000; cat. no. ab52903),
collagen I (1:1,000; cat. no. ab34710) and collagen III (1:1,000;
cat. no. ab7778), Bcl-2 (1:1,000; cat. no. ab59348), apoptosis
regulator BAX (Bax; 1:1,000; cat. no. ab32503), caspase-3 (1:1,000;
cat. no. ab13847) and GAPDH (1:2,000; cat no. ab8245) antibodies
(all Abcam), protein samples were incubated at 4°C overnight.
Following addition of goat anti-rabbit immunoglobulin G/horseradish
peroxidase conjugated antibody (1:2,000; cat. no. bs-0295G-HRP;
BIOSS, Beijing, China), protein samples were incubated at room
temperature for 2 hand western blots were developed using ECL-Plus
chemiluminescence reagent kit (Thermo Fisher Scientific, Inc.) and
visualized by UVP Bio-Imaging Systems. Thereafter, the optical
density analysis was performed (Image J 1.8.0 software; National
Institutes of Health, Bethesda, MD, USA).
RT-qPCR
Primers were designed according to the sequences of
collagen I and III reported in GenBank (Table I), and were synthesized in Sangon
Biotech Co., Ltd. (Shanghai, China). Total RNA was isolated using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and
reverse transcribed (42°C for 1 h; 70°C for 5 min) to cDNA using
High-Capacity RNA-to-cDNA kit (Invitrogen; Thermo Fisher
Scientific, Inc.). Fluorescent qPCR kit (SYBR-Green Master Mix:
SYBR Premix Ex Taq II, TliRNase H Plus; Takara Biotechnology Co.,
Ltd., Dalian, China) was used for the detection. The following
thermocycling conditions were used for qPCR: Initial denaturation
at 95°C for 30 sec; 40 cycles of 95°C for 5 sec, 60°C for 30 sec.
The relative gene expression data were analyzed using the
2−ΔΔCt method (21,22).
Primers used for RT-qPCR are listed in Table I.
 | Table I.Primers used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers used for reverse
transcription-quantitative polymerase chain reaction.
|
|
| Primer sequence
(5′→3′) |
|---|
|
|
|
|
|---|
| Gene | GenBankID | Forward | Reverse |
|---|
| Collagen I | 12842 |
TGCTCATAGCAGCCCCCTCCG |
TCCTTGACTTGGGAAGGTTT |
| Collagen III | 12825 |
GCAACAGGCATACTAACT |
GGTCCCCGAGGAAACAA |
| GAPDH | 14433 |
AACTTTGGCATTGTGGAAGG |
CACATTGGGGGTAGGAACAC |
Immunofluorescence staining
Mouse myocardial tissue was dewaxed, dehydrated,
treated with 0.1 M sodium citrate buffer (OriGene Technologies,
Inc., Beijing, China) for antigen retrieval, washed three times
with PBS and incubated at 4°C overnight following addition of
TGF-β1 (1:100), p-Smad3 (1:200) antibodies (the same antibodies as
used for western blotting). Subsequently, samples were washed three
times and incubated with fluorescent secondary antibodies [goat
anti-rabbit IgG H&L (Alexa Fluor® 488), 1:500, cat.
no. ab150077; goat anti-rabbit IgG H&L (Alexa Fluor®
594), 1:250, cat. no. ab150080, all Abcam] at room temperature for
2 h in the dark. Samples were washed with PBS, stained with DAPI
for 10 min at room temperature, mounted with glycerine, and finally
observed under fluorescence microscope.
Statistical analysis
All data were statistically analyzed using SPSS 19.0
software (IBM Corp., Armonk, NY, USA). Multiple comparisons were
made using one-way analysis of variance with Student-Newman-Keuls
test. Each experiment was repeated three times. All data are
presented as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
EGCG improves heart failure in
mice
Following ligation of the aortic arch, morphological
alterations of mouse myocardial tissue were observed by H&E
staining. In the sham group, cardiomyocytes were well stained and
swollen and necrotic cells were observed. In the heart failure
group, widening of myocardial cell gap and myocardial fiber rupture
were observed. Following EGCG treatment, myocardial fiber rupture
attenuated, the gap slightly widened. In the LY group, injury to
cardiomyocytes was not alleviated (Fig. 1A). Color Doppler ultrasonography
revealed that EF significantly decreased, and LVIDs and LVIDd
significantly increased, in the heart failure group compared with
the sham group (all P<0.05). EF significantly increased, and
LVIDs and LVIDd significantly decreased in the EGCG group compared
with the HF group (all P<0.05). There were no significant
differences in EF, LVIDs and LVIDd between LY and HF group
(Fig. 1B). To further confirm the
role of EGCG in mouse with heart failure, serum levels of BNP and
NT-proBNP were determined. Serum levels of BNP and NT-proBNP
significantly increased in the HF group compared with the sham
group (P<0.05). Serum levels of BNP and NT-proBNP significantly
decreased in the EGCG group compared with the HF group (P<0.05)
and there was no significant difference between EGCG and LY groups
(Fig. 1C). These results suggest
that EGCG markedly improved heart failure in mice and that the
observed effects may be associated with TGF-β1 signaling
pathway.
 | Figure 1.The model of heart failure in mice
was established and verified in all treatment groups. (A) The
pathological alterations of myocardium were observed by hematoxylin
and eosin staining (arrows represents widened myocardial cell gap
and myocardial fiber rupture). (B) Alterations of LVIDs, LVIDd and
EF in mice were observed by echocardiography in order to determine
whether the model of heart failure was successfully established.
(C) The peripheral blood of mice was collected, and serum was
isolated, ELISA was used to detect BNP and NT-proBNP levels.
*P<0.05 vs. the sham group; #P<0.05 vs. the HF
group. HF, heart failure; LVIDs, left ventricular internal systolic
diameter; LVIDd, left ventricular internal diastolic diameter; BNP,
brain natriuretic peptide; NT-proBNP, N-terminal-proBNP; EGCG,
epigallocatechingallate; LY group, LY364947 inhibitor and EGCG
treatment group. |
EGCG attenuates myocardial fibrosis in
mice with heart failure
Masson staining was performed to investigate the
extent of myocardial fibrosis. Myocardial fibrosis was visibly
aggravated in the heart failure group compared with the sham group.
Following EGCG treatment, the size of collagen fibers were
significantly reduced and myocardial fibrosis was alleviated.
Following inhibition of TGF-β1 receptor, the expression of
cardiomyocyte collagen fibers increased (Fig. 2A). Expression levels of collagen I
and III were determined in myocardial tissue. The expression levels
of collagen I and III significantly decreased in the HF group
compared with the sham group (P<0.05), but significantly
decreased in the EGCG group compared with the HF group (P<0.05).
The expression levels of collagen I and III were not significantly
different in the LY group compared with the heart failure group
(Fig. 2B). RT-qPCR confirmed these
results (Fig. 2C). The above
results suggest that EGCG attenuated myocardial fibrosis in mice
with heart failure and suggest that EGCG alleviated heart failure
and myocardial fibrosis.
EGCG attenuates oxidative stress in
mice with heart failure
ELISA was used to detect the level of SOD, MDA and
GSH-Px in serum. Results demonstrated that the level of MDA was
significantly increased in the HF group compared with the sham
group (P<0.05; Fig. 3). The
levels of SOD and GSH-Px were significantly decreased in the HF
group compared with the sham group (P<0.05). Following EGCG
treatment, the level of MDA significantly decreased, SOD and GSH-Px
levels were significantly elevated compared with the HF group
(P<0.05). Following treatment with TGF-β1 receptor inhibitor,
the effect of EGCG was reduced. Those results suggested that
following treatment with EGCG, antioxidative effect of
cardiomyocytes increased and oxidative stress injury in heart
failure was attenuated.
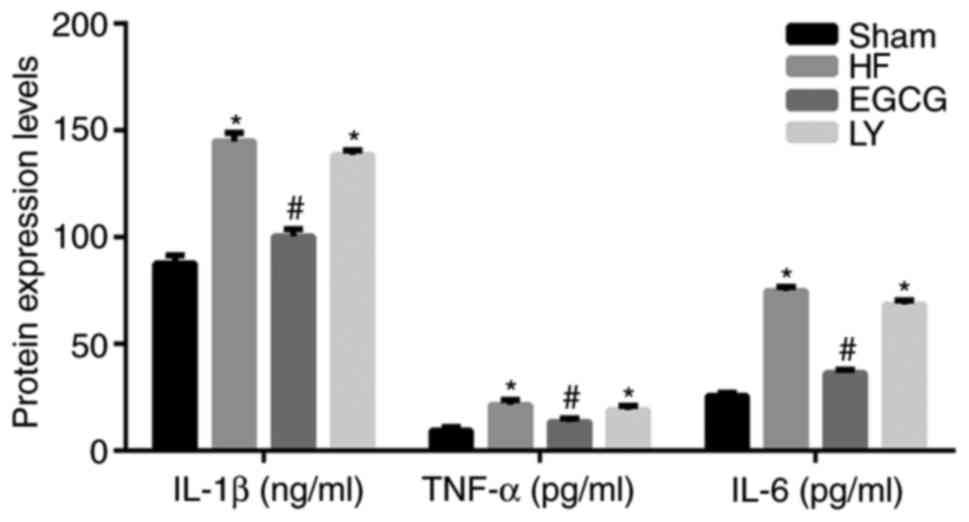
EGCG attenuates inflammation in mice
with heart failure
ELISA was used to detect inflammatory factors in
serum. Data demonstrated that the levels of IL-1β, IL-6 and TNF-α
significantly increased in the HF group compared with the sham
group (all P<0.05; Fig. 4).
Following treatment with EGCG, the levels of IL-1β, IL-6 and TNF-α
significantly decreased compared with the HF group (all P<0.05).
These results suggest that EGCG attenuated inflammation in mice
with heart failure.
EGCG attenuates cardiomyocyte
apoptosis in mice with heart failure
Western blot analysis was used to detect the
expression of Bax, apoptosis regulator Bcl2 (Bcl2) and caspase-3.
In the HF group, Bax and caspase-3 expression significantly
increased compared with the sham group (both P<0.05; Fig. 5). Bcl2 expression significantly
decreased in the HF group, compared with the sham group
(P<0.05). Following EGCG treatment, expression of Bax and
caspase-3 significantly decreased and expression of Bcl2
significantly increased compared with the HF group (all P<0.05).
Following treatment with TGF-β1 inhibitor, the effect of EGCG was
reduced. Those results indicate that EGCG attenuates cardiomyocyte
apoptosis in mice with heart failure.
EGCG attenuates myocardial injury in
mice with heart failure through TGF-β1/Smad3 signaling pathway
To identify the mechanism by which EGCG attenuates
myocardial fibrosis in mice with heart failure, the expression
levels of TGF-β1/Smad3 signaling pathway-associated proteins were
detected. Expression levels of TGF-β1 and p-Smad3 significantly
increased in the HF group compared with the sham group (both
P<0.05). Expression levels of these proteins significantly
decreased in the EGCG group compared with the heart failure group
(P<0.05). Expression levels of these proteins were significantly
increased in the LY group compared with the sham group (P<0.05;
Fig. 6). Immunofluorescence
staining (Fig. 6) confirmed these
results. All above results suggest that EGCG attenuated myocardial
injury in mice with heart failure through TGF-β1/Smad3 signaling
pathway.
Discussion
EGCG is a catechin monomer extracted from leaves or
buds of Camellia sinensis that exhibits antioxidative,
anti-inflammatory and anticancer effects (23). In the present study, mouse models
of heart failure were established and it was determined that EGCG
alleviated heart failure. It was also determined that EGCG
decreased expression if collagen I and III and decreased TGF-β1 and
p-Smad3 expression. These results suggest that EGCG exhibits
protective effects against myocardial injury in mice with heart
failure, which is inhibited through TGF-β1/Smad3 signaling
pathway.
Ventricular remodeling serves a role in the
occurrence and development of chronic heart failure (24). Myocardial fibrosis is a common
pathological alteration during the development of various heart
diseases (25). It is the primary
manifestation of cardiac remodeling (26). The main pathological alterations
include deposition of excessive extracellular matrix, fibrin
hyperplasia and disproportion of various collagens, which lead to
increased cardiac stiffness and decreased cardiac diastolic and
systolic functions, and may result in chronic heart failure
(27). The myocardial collagen
network is primarily composed of collagen I and collagen III, which
provide supportive framework for cardiomyocytes, and determine
ventricular compliance (28).
Under normal condition, interstitial collagen depends on a dynamic
balance between synthesis and degradation. Under pathological
condition, the balance between collagen synthesis and degradation
is disturbed, collagen gradually accumulates and the proportion of
collagen I and collagen III is altered, resulting in collagen
remodeling which leads to increased wall hardness, decreased
compliance, impaired ventricular systolic and diastolic function.
Therefore, delaying or reversing myocardial fibrosis is the key to
prevention and treatment of heart failure (29). Zhou et al (30) demonstrated that EGCG attenuates
angiotensin II-induced oxidative stress and apoptosis in human
umbilical vein endothelial cells through the activation of
Nrf2/caspase-3 signaling. In the present study, mouse models of
heart failure were established by ligating the aortic arch.
Following heart failure, Masson staining revealed occurrence of
fibrous myocardial tissue. ELISA assay revealed that EGCG reduced
the expression of MDA, increased the expression of SOD and GSH-Px
and enhanced the anti-oxidative stress in cardiomyocyte. Western
blot assay revealed that expression levels of collagen I and III in
myocardial tissue were markedly increased. These results suggested
that following heart failure, cardiac collagen remodeling
occurred.
EGCG is an active ingredient in the leaves or buds
of Camellia sinensis. Oyama et al (5) demonstrated that in
H/M-SOD2−/− mouse models of heart failure, EGCG markedly
increased the survival rate of mice and alleviated cardiac
contraction and myocardial dilatation. Feng et al (31) demonstrated that EGCG alleviated
heart failure by inducing alteration of Ca2+ content in
the sarcoplasmic reticulum, inhibition of
Na+-Ca2+ exchange, regulation of
Ca2+ load in the sarcoplasmic reticulum and regulation
of the contraction of cardiomyocytes.
In the present study, following EGCG treatment, EF
was markedly increased, and LVIDs and LVIDd were markedly decreased
compared with the HF group. Serum levels of BNP and NT-proBNP were
markedly decreased compared with the HF group. The above results
suggest that EGCG can effectively treat heart failure. Masson
staining demonstrated that following EGCG treatment, myocardial
fibrosis was markedly decreased and the expression levels of
collagen I and III in the myocardial tissue were markedly
decreased. These results were confirmed by RT-qPCR. The results of
the present study suggest that EGCG can effectively inhibit
myocardial fibrosis and collagen remodeling following heart
failure.
TGF-β1, as a pro-fibrogenic factor, exhibits
anti-fibrotic, anti-proliferative, and anti-inflammatory effects
(32,33). It participates in the
proliferation, transformation, migration, and apoptosis of
fibroblasts and the synthesis of extracellular matrix, mainly of
collagen (34). Simultaneously, it
induces cardiac fibroblasts to differentiate into myofibroblasts
with stronger connection function (35). Smads are downstream signaling
molecules of TGF-β1 and are the only substrates that can bind to
TGF-β1 (36). One study
demonstrated that EGCG attenuates fibroblast proliferation and
excessive collagen production by interfering with TGF-β1 signaling
(37). EGCG inhibits the
expression of tumor necrosis factor receptor associated factor 6,
inhibits the activation of TGF-b1 and regulates rheumatoid
arthritis (38). The present study
demonstrated that following EGCG treatment, TGF-β1 p-Smad3
expression levels were significantly increased in mouse models of
heart failure. The authors of the present study hypothesized that
TGF-β1 receptor is likely to be targeted by treatment with EGCG.
Therefore, LY364947, a competitive TGF-β receptor inhibitor was
used in the present study and it was determined that the
therapeutic effects of EGCG on myocardial fibrosis in mice with
heart failure were markedly decreased. This suggests that EGCG
inhibits the myocardial fibrosis through TGF-β1/Smad3 signaling
pathway.
Taken together, EGCG can inhibit the progression of
heart failure through inhibiting myocardial fibrosis and reducing
ventricular collagen remodeling. The regulatory mechanism of EGCG
may be associated with TGF-β1/smad3 signaling pathway.
Acknowledgements
The present study was supported by the Liaoning
Natural Fund Project (grant no. 2015020382).
References
|
1
|
Kharchenko EP: Heart failure in
cardiorenal syndromes. Ter Arkh. 85:85–91. 2013.(In Russian).
PubMed/NCBI
|
|
2
|
Greenberg B: Gene therapy for heart
failure. Trends Cardiovasc Med. 27:216–222. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schramm L: Going green: The role of the
green tea component EGCG in chemoprevention. J Carcinog Mutagen.
4:10001422013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Suzuki T, Pervin M, Goto S, Isemura M and
Nakamura Y: Beneficial effects of tea and the green tea catechin
epigallocatechin-3-gallate on obesity. Molecules. 21:pii: E1305.
2016. View Article : Google Scholar
|
|
5
|
Oyama JI, Shiraki A, Nishikido T, Maeda T,
Komoda H, Shimizu T, Makino N and Node K: EGCG, a green tea
catechin, attenuates the progression of heart failure induced by
the heart/muscle-specific deletion of MnSOD in mice. J Cardiol.
69:417–427. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hao J, Kim CH, Ha TS and Ahn HY:
Epigallocatechin-3 gallate prevents cardiac hypertrophy induced by
pressure overload in rats. J Vet Sci. 8:121–129. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sriram N, Kalayarasan S, Manikandan R,
Arumugam M and Sudhandiran G: Epigallocatechin gallate attenuates
fibroblast proliferation and excessive collagen production by
effectively intervening TGF-β1 signalling. Clin Exp Pharmacol
Physiol. 42:849–859. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hsieh YP, Chen HM, Lin HY, Yang H and
Chang JZ: Epigallocatechin-3-gallate inhibits
transforming-growth-factor-β1-induced collagen synthesis by
suppressing early growth response-1 in human buccal mucosal
fibroblasts. J Formos Med Assoc. 116:107–113. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moses HL, Roberts AB and Derynck R: The
discovery and early days of TGF-β: A historical perspective. Cold
Spring Harb Perspect Biol. 8:pii: a021865. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Travis MA and Sheppard D: TGF-β activation
and function in immunity. Annu Rev Immunol. 32:51–82. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim YJ, Carvalho FC, Souza JA, Gonçalves
PC, Nogueira AV, Spolidório LC, Roque-Barreira MC and Cirelli JA:
Topical application of the lectin Artin M accelerates wound healing
in rat oral mucosa by enhancing TGF-β and VEGF production. Wound
Repair Regen. 21:456–463. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sugiyama D, Kulkeaw K and Mizuochi C:
TGF-beta-1 up-regulates extra-cellular matrix production in mouse
hepatoblasts. Mech Dev. 130:195–206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mira YE, Muhuyati, Lu WH, He PY, Liu ZQ
and Yang YC: TGF-β1 signal pathway in the regulation of
inflammation in patients with atrial fibrillation. Asian Pac J Trop
Med. 6:999–1003. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu CP, Dandapat A, Liu Y, Hermonat PL and
Mehta JL: Blockade of hypoxia-reoxygenation-mediated collagen type
I expression and MMP activity by overexpression of TGF-beta1
delivered by AAV in mouse cardiomyocytes. Am J Physiol Heart Circ
Physiol. 293:H1833–H1838. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang H, Cui YC, Li K, Yang BQ, Liu XP,
Zhang D, Li H, Wu AL and Tang Y: Glutamine protects cardiomyocytes
from hypoxia/reoxygenation injury under high glucose conditions
through inhibition of the transforming growth factor-β1-Smad3
pathway. Arch Biochem Biophys. 596:43–50. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cheng CI, Lee YH, Chen PH, Lin YC, Chou MH
and Kao YH: Cobalt chloride induces RhoA/ROCK activation and
remodeling effect in H9c2 cardiomyoblasts: Involvement of PI3K/Akt
and MAPK pathways. Cell Signal. 36:25–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lan HY and Chung AC: TGF-β/Smad signaling
in kidney disease. Semin Nephrol. 32:236–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou P, Shi L, Li Q and Lu D:
Overexpression of RACK1 inhibits collagen synthesis in keloid
fibroblasts via inhibition of transforming growth factor-β1/Smad
signaling pathway. Int J Clin Exp Med. 8:15262–15268.
2015.PubMed/NCBI
|
|
19
|
Zhao M, Zheng S, Yang J, Wu Y, Ren Y, Kong
X, Li W and Xuan J: Suppression of TGF-β1/Smad signaling pathway by
sesamin contributes to the attenuation of myocardial fibrosis in
spontaneously hypertensive rats. PLoS One. 10:e01213122015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yan L, Wei X, Tang QZ, Feng J, Zhang Y,
Liu C, Bian ZY, Zhang LF, Chen M, Bai X, et al: Cardiac-specific
mindin overexpression attenuates cardiac hypertrophy via blocking
AKT/GSK3β and TGF-β1-Smad signalling. Cardiovasc Res. 92:85–94.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang H, Kwak D, Fassett J, Hou L, Xu X,
Burbach BJ, Thenappan T, Xu Y, Ge JB, Shimizu Y, et al: CD28/B7
deficiency attenuates systolic overload-induced congestive heart
failure, myocardial and pulmonary inflammation, and activated T
cell accumulation in the heart and lungs. Hypertension. 68:688–696.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feng WY: Metabolism of green tea
catechins: An overview. Curr Drug Metab. 7:755–809. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Braunwald E: Heart failure. JACC Heart
Fail. 1:1–20. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Neubauer S and Bull S: Myocardial fibrosis
in aortic stenosis. JACC Cardiovasc Imaging. 10:1334–1336. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac fibrosis: The fibroblast awakens. Circ Res.
118:1021–1040. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kong P, Christia P and Frangogiannis NG:
The pathogenesis of cardiac fibrosis. Cell Mol Life Sci.
71:549–574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Weber KT: Cardiac interstitium in health
and disease: The fibrillar collagen network. J Am Coll Cardiol.
13:1637–1652. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Edgley AJ, Krum H and Kelly DJ: Targeting
fibrosis for the treatment of heart failure: A role for
transforming growth factor-β. Cardiovasc Ther. 30:e30–e40. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou X, Liang L, Zhao Y and Zhang H:
Epigallocatechin-3-gallate ameliorates angiotensin II-induced
oxidative stress and apoptosis in human umbilical vein endothelial
cells through the activation of Nrf2/caspase-3 signaling. J Vasc
Res. 54:299–308. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Feng W, Hwang HS, Kryshtal DO, Yang T,
Padilla IT, Tiwary AK, Puschner B, Pessah IN and Knollmann BC:
Coordinated regulation of murine cardiomyocyte contractility by
nanomolar (−)-epigallocatechin-3-gallate, the major green tea
catechin. Mol Pharmacol. 82:993–1000. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
TGF-β: The master regulator of fibrosis. Nat Rev Nephrol.
12:325–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tian X, Zhang J, Tan TK, Lyons JG, Zhao H,
Niu B, Lee SR, Tsatralis T, Zhao Y, Wang Y, et al: Association of
β-catenin with P-Smad3 but not LEF-1 dissociates in vitro
profibrotic from anti-inflammatory effects of TGF-β1. J Cell Sci.
126:67–76. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Park B, Hwang E, Seo SA, Cho JG, Yang JE
and Yi TH: Eucalyptus globulus extract protects against UVB-induced
photoaging by enhancing collagen synthesis via regulation of
TGF-β/Smad signals and attenuation of AP-1. Arch Biochem Biophys.
637:31–39. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lijnen P, Petrov V, Rumilla K and Fagard
R: Transforming growth factor-beta 1 promotes contraction of
collagen gel by cardiac fibroblasts through their differentiation
into myofibroblasts. Methods Find Exp Clin Pharmacol. 25:79–86.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee YS, Kim JH, Kim ST, Kwon JY, Hong S,
Kim SJ and Park SH: Smad7 and Smad6 bind to discrete regions of
Pellino-1 via their MH2 domains to mediate TGF-beta1-induced
negative regulation of IL-1R/TLR signaling. Biochem Biophys Res
Commun. 393:836–843. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Singh AK, Umar S, Riegsecker S, Chourasia
M and Ahmed S: Regulation of transforming growth factor β-activated
kinase activation by epigallocatechin-3-gallate in rheumatoid
arthritis synovial fibroblasts: Suppression of K(63)-linked
autoubiquitination of tumor necrosis factor receptor-associated
factor 6. Arthritis Rheumatol. 68:347–358. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chang JZ, Hsieh YP, Lin WH, Chen HM and
Kuo MY: Activation of transforming growth factor-β1 by thrombin via
integrins αvβ1, αvβ3, and αvβ5 in buccal fibroblasts: Suppression
by epigallocatechin-3-gallate. Head Neck. 39:1436–1445. 2017.
View Article : Google Scholar : PubMed/NCBI
|