Introduction
Type 1 diabetes mellitus (T1DM) is an autoimmune
disease caused by the destruction of pancreatic β cells (1). It affects more than 35 million people
worldwide. Reduced muscle mass and myofiber size as well as poor
metabolic control in T1DM can result from impaired muscle growth
and development (2–4). Diabetic muscle atrophy, a clinical
condition, means that the size and strength of skeletal muscles are
reduced (5,6), affecting normal daily activities.
Protein synthesis or protein degradation is one of the important
reasons for diabetic muscle atrophy. Some signaling pathways-such
as the activated protein kinase B (Akt)/ rapamycin (mTOR) and
Akt/forkhead box protein O1 (FoxO1) pathways-may be linked to
muscle loss in diabetic muscle atrophy (7,8).
Myostatin (MSTN), a member of the transforming growth factor β
family, is a negative regulator of skeletal muscle growth (9). The negative effect of MSTN may occur
via activation of the PI3K/Akt signaling pathway (10).
Growing evidence suggests that pulsed
electromagnetic fields (PEMFs) can serve as safe alternatives to
drug-based therapies for the treatment of some diseases. In recent
years, PEMFs have been widely used in the treatment of
osteoporosis, fracture, and other conditions and have produced good
therapeutic results. It has been reported, for example, that the
atrophy of type II fibers in denervated muscle was retarded by
magnetic stimulation (11).
However, whether PEMFs can alleviate streptozotocin (STZ)-induced
diabetic muscle atrophy has not been investigated.
Accordingly, we examined the effects of PEMFs on
STZ-induced diabetic muscle atrophy by evaluating muscle strength,
mass, and cross-sectional area of muscle fiber. Furthermore,
possible molecular mechanisms were explored through analyses of the
gene and protein expression of MSTN, Akt, activin type II receptor
(ActRIIB), mTOR, and FoxO1.
Materials and methods
Animals
This study was conducted with the approval of the
ethics committee of Shaanxi Normal University in Shaanxi, China,
and was performed in accordance with the Guide for the Care and Use
of Laboratory Animals published by the U.S. National Institutes of
Health (NIH; publication no. 85-23, revised 1996).
Healthy male Sprague-Dawley (SD) rats (200±20 g)
were obtained from the Laboratory Animal Breeding and Research
Center of Xi'an Jiaotong University (Xi'an, China). They were
housed in a temperature and humidity controlled room (22±2°C, 60±5%
humidity and 12-hour light/dark cycle). After 5 days of
acclimation, the rats were randomly divided into a normal control
group (NC; n=10) and a T1DM model group (n=50). Experimental T1DM
was induced via a peritoneal injection of STZ (Sigma, St. Louis,
MO, USA) and 60 mg/kg, 0.1 mol/l sodium citrate buffer, pH=4.5). An
equal volume of buffer was injected into the control rats at the
same time. The blood glucose levels in tail vein blood samples were
measured on the 1st, 3rd, 7th, and 10th days following injection.
The rats with blood glucose levels greater or equal to 16.7 mmol/l
(300 mg/dl) were considered diabetic. The diabetic rats were then
randomly assigned to the DM group (n=10), the diabetic
insulin-treated group (DT; n=10) as a positive control, and the
diabetic PEMFs therapy group (DP, n=10). The DT group was treated
with insulin (6–8 U/d twice a day for 6 weeks; Sigma), and the DP
group was exposed to PEMFs (15 Hz, 1.46 mT, 30 min/d for 6
weeks).
PEMFs treatment
The PEMFs exposure system was composed of coils and
a pulsed signal generator. There were three identical coils 800 mm
in diameter connected in series and placed coaxially 190 mm apart.
Each coil was made up of enameled coated copper wire 0.8 mm in
diameter. The number of turns on the central coil was 266, and the
number of turns on the two outside coils was 500. The pulsed signal
apparatus generated an open circuit waveform composed of a pulsed
burst (burst width, 5.16 ms; burst wait, 61.4 ms; pulse width,
0.171 ms; pulse wait, 0.171 ms) repeated at 15.08 Hz. The rats in
the DP group were put in the center of coils.
Oral glucose tolerance tests
Oral glucose load was administered at 2 g/kg of body
weight after overnight fasting. Glucose levels were measured from
tail bleeds by cutting off a small part of the tail at 0, 30, 60,
90, and 120 min after glucose administration.
Grip strength
During the final week, forelimb grip strength was
measured as maximum tensile force using a rat grip strength meter
(YLS-13A; Huaibei Zhenghua Bioinstrumentation Co., Ltd., Anhui,
China). Rats were tested 3 times in succession without rest and the
results of the 3 tests were averaged for each rat.
Weight and sample preparation
After 6 weeks of treatment, the rats were euthanized
with an overdose of diethyl ether and the final body weights were
recorded. Blood was collected and centrifuged in order to obtain
the serum fractions. Serum was stored at −80°C for further
analysis. After the animals were sacrificed, each quadriceps
femoris was harvested and weighed, then immediately stored in
liquid nitrogen at −80°C for reverse transcription-polymerase chain
reaction and western blot analysis.
Biochemical analysis
Blood glucose was measured using the eBsensor Blood
Glucose Monitor (Visgeneer Inc., Hsinchu, Taiwan). Serum insulin
levels were measured using a commercial enzyme linked immunosorbent
assay (ELISA; EMT Millipore, Billerica, MA, USA). The activity
levels of succinate dehydrogenase (SDH) and malate dehydrogenase
(MDH) in the quadriceps femoris were analyzed with standard
colorimetric tests using commercial kits (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China) according to the
protocols provided by the manufacturer.
Hematoxylin and eosin staining
The quadriceps femoris was cut into 1mmx1mmx1 mm
size and then placed in 2.5% glutaraldehyde to fix for 24 h at 4°C.
After washing, the specimens suffered the processes of dehydration
and transparency, then were embedded into paraffin and cut into 50
nm thickness. Subsequently, the paraffin sections undergo the
processes of deparaffinage and dehydration, then were stained with
hematoxylin for 8 min. After washing, sections were placed into the
eosin counterstain for 5 min. After resinene mount, sections were
visualized under the optical microscope (Olympus Corp., Tokyo,
Japan).
Western blot analysis
The removed quadriceps femoris muscles were
homogenized in ice-cold lysis buffer. Protein concentrations were
quantified with the BCA protein assay kit (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Protein concentrations were
determined and equal amounts of sample were uploaded for sodium
dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)
analysis. After electrophoresis and separation, samples were
transferred onto nitrocellulose membranes. The immunoblots were
incubated with primary antibodies overnight at 4°C, followed by
incubation with the corresponding secondary antibodies at room
temperature for 1 h. Blots were visualized with ECL-plus reagent,
and the results were quantified with Lab Image Version 2.7.1. The
primary antibodies used were as follows: the expression of MSTN
[EPR4567(2), ab124721], ActRIIB (EPR10739, ab180185) from Abcam
(Cambridge, UK); AKT (Rabbit Ab 9272S); phospho-AKT (S473 9271S);
mTOR (Rabbit 2972S), phospho-mTOR (S2448, 2971P), FoxO1 (C29H4,
Rabbit 2880P), and phospho-FoxO1 (S256, 9461P) from Cell Signaling
Technology, Inc. (Danvers, MA, USA).
Statistical analysis
Statistical analysis was performed using SPSS
version 20.0 software (IBM Corp., Armonk, NY, USA). One-way
analysis of variance was employed to evaluate the differences
between the three groups. Once a significant difference was
detected, Tukey's multiple comparisons test was used to determine
the significance between any two groups. P<0.05 was considered
to indicate a statistically significant difference. The results are
expressed as the mean ± standard deviation.
Results
Changes of blood glucose and oral
glucose tolerance tests before and after model establishment
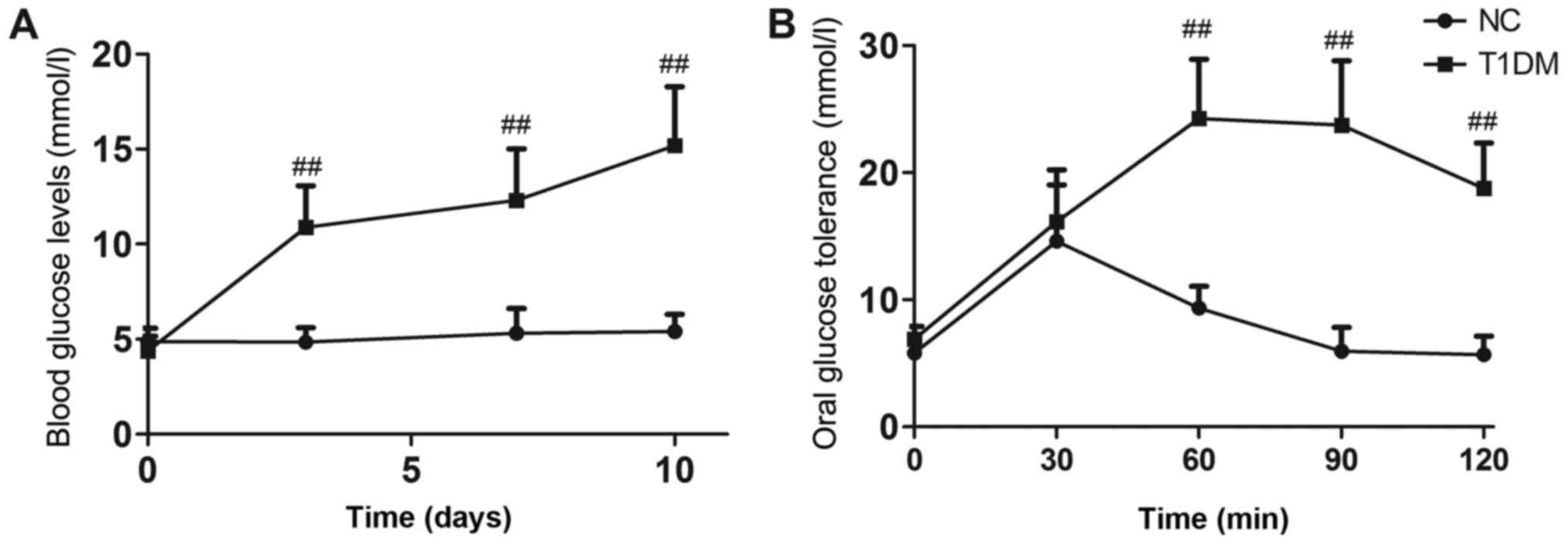
Blood glucose levels in diabetic mice remained
extremely high throughout the experiment (P<0.01, Fig. 1A). Oral glucose tolerance tests
were performed after 6 weeks of treatment. Rats in the DB group
showed impaired glucose tolerance compared with those in the NC
group (P<0.01, Fig. 1B). The
T1DM model was successfully established.
PEMFs increased body weight, muscle
weight and muscle strength
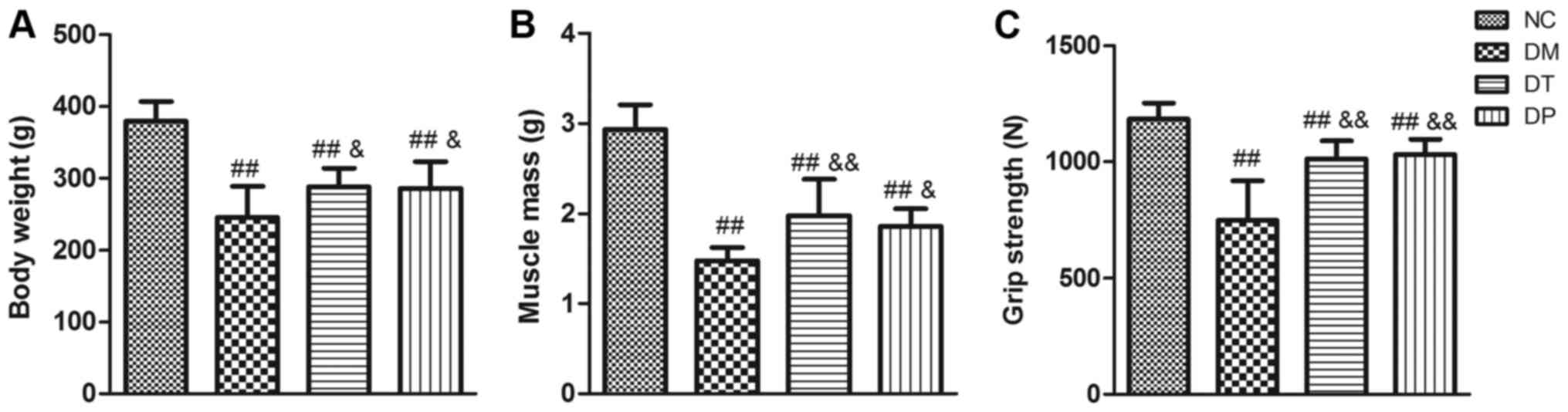
After 6 weeks of PEMFs treatment, the body weight,
quadriceps wet weight, and grip strength of the rats were measured
(as shown in Fig. 2). Compared
with the NC group, the body weight, quadriceps wet weight, and grip
strength of the DM group were significantly decreased (P<0.01,
P<0.01, and P<0.01, respectively). Although the body weight,
muscle weight, and grip strength in the DT and DF groups were
significantly lower than those in the NC group (P<0.01,
P<0.01, and P<0.01, respectively), insulin treatment
statistically significantly affected the loss of body weight,
muscle weight, and grip strength (P<0.01, P<0.01, and
P<0.01, respectively) in this group compared with the DM group.
Furthermore, the body weight, muscle weight, and grip strength in
the DF group were significantly increased (P<0.05, P<0.05,
and P<0.01, respectively) compared with the DM group.
PEMFs decreased blood glucose level
and increased insulin level
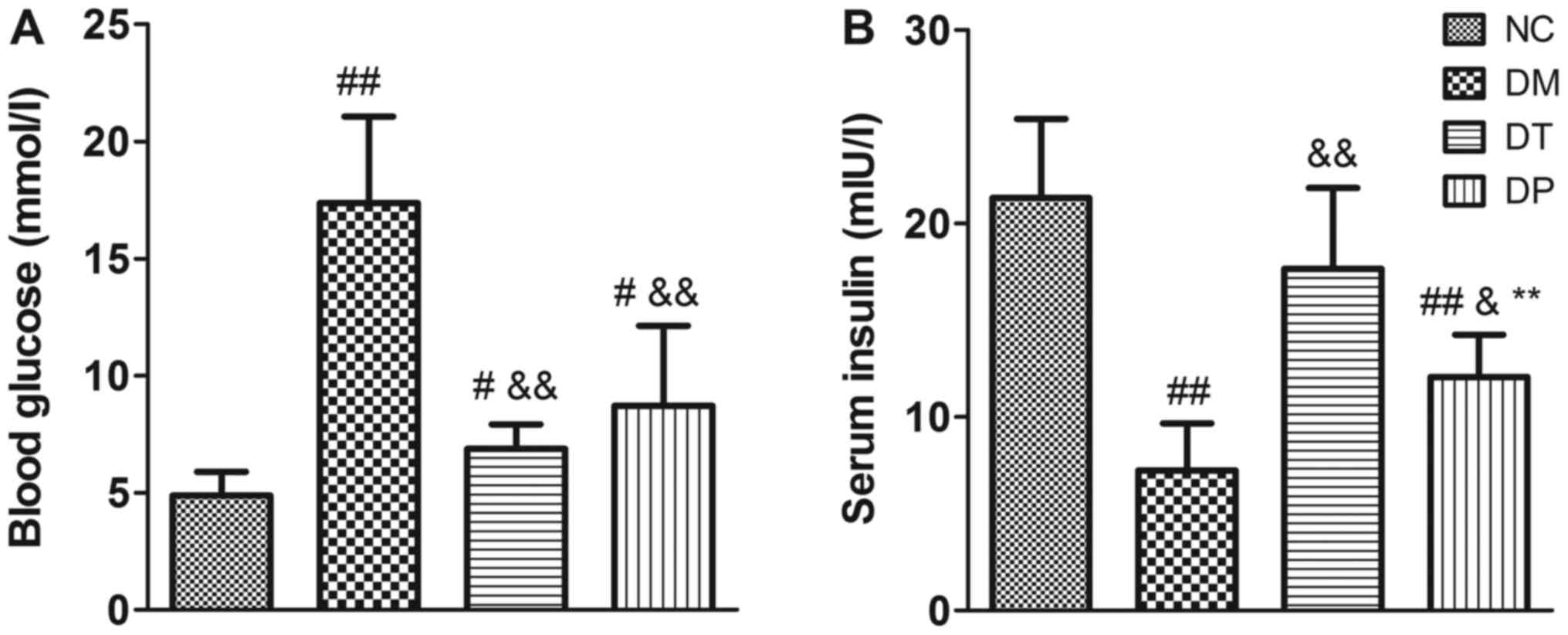
Before the animals were sacrificed, the blood
glucose levels were detected. The blood glucose levels of the DM
group were significantly higher than those of the NC group
(P<0.01); insulin and PEMFs treatment caused a decrease in blood
glucose levels compared with the DM group (P<0.01) (Fig. 3A). Moreover, insulin levels were
significantly decreased in the DM group (P<0.01). Insulin and
PEMFs treatment caused a significant decrease in blood glucose
(P<0.01 and P<0.01, respectively) and a significant increase
in serum insulin compared with the DM group (P<0.01 and
P<0.05, respectively) (Fig.
3B).
PEMFs altered the activities of
metabolic enzymes
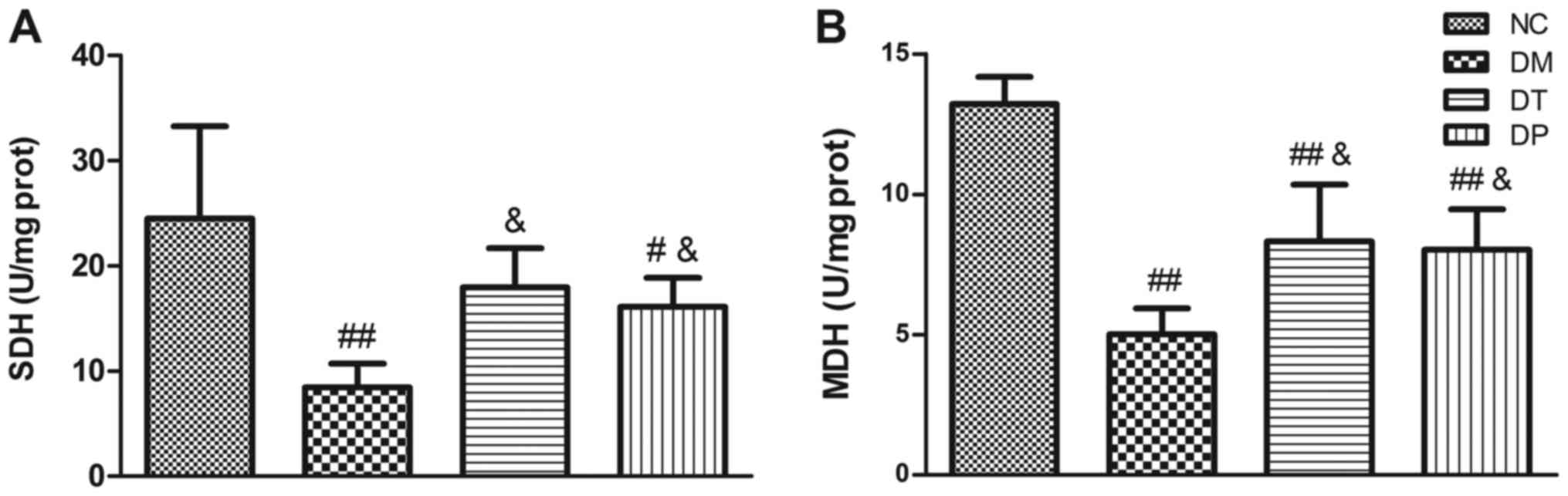
Fig. 4 shows the
effect of PEMFs on activity levels of key muscle metabolism enzymes
in the several experimental groups. In the diabetic rats, activity
levels of SDH (Fig. 4A) and MDH
(Fig. 4B) were significantly
decreased (P<0.01 and P<0.01, respectively) as compared with
the normal, nondiabetic rats. Insulin treatment statistically
significantly increased the SDH and MDH levels (P<0.05 and
P<0.05, respectively). PEMFs treatment resulted in significantly
higher SDH and MDH activity levels (P<0.05 and P<0.05,
respectively) as compared with the untreated diabetic rats.
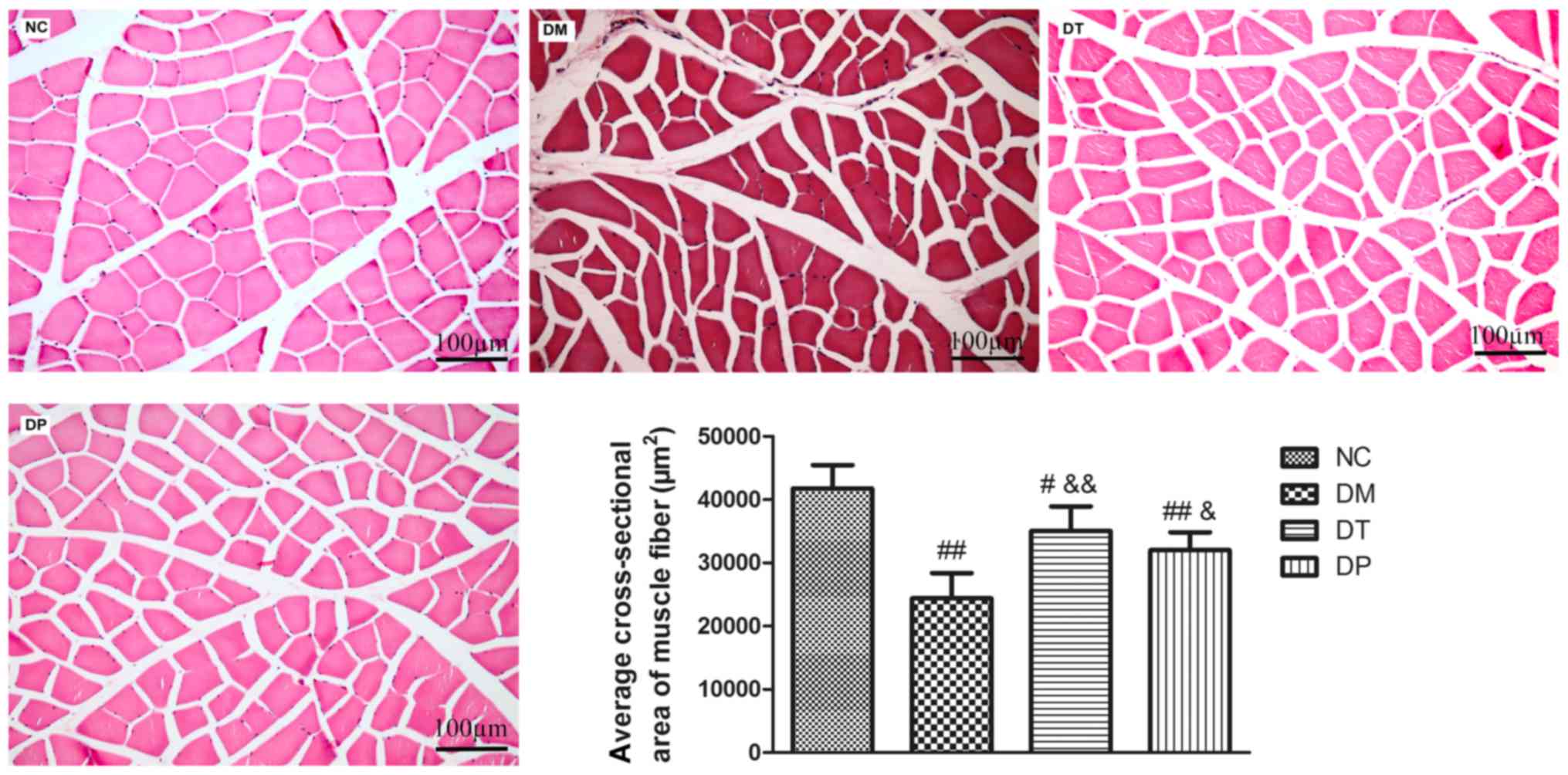
Effect of PEMFs on cross-sectional
area of muscle fiber
As shown in Fig. 5,
the cross-sectional area of muscle fiber in DM group was
significantly decreased (P<0.01) as compared with the normal.
The cross-sectional area of muscle fiber in DT and DP groups were
significantly increased (P<0.01 and P<0.05, respectively) as
compared with the DM group.
Effect of PEMFs on MSTN/Akt/mTOR/FoxO
signaling pathways
To investigate the molecular mechanism of the effect
of PEMFs on reversal of muscle atrophy in diabetic rats, we
explored the MSTN, Akt/mTOR, and FoxO3 signaling pathways, which
are closely related to muscle protein synthesis and subject to
degradation by Western blotting. The results showed that STZ
markedly decreased p-Akt (P<0.01) (Fig. 6D), mTOR (P<0.01) (Fig. 6E), and p-mTOR (P<0.01) (Fig. 6F) while it increased MSTN
(P<0.01) (Fig. 6A), ActRIIB
(P<0.01) (Fig. 6B), FoxO1
(P<0.01) (Fig. 6G), and p-FoxO1
(P<0.01) (Fig. 6H) as compared
with the nondiabetic rats. Insulin and PEMF increased p-Akt
(P<0.01 and P<0.05, respectively) (Fig. 6D), mTOR (P<0.01 and P<0.01,
respectively) (Fig. 6E), p-mTOR
(P<.01 and P<0.01, respectively) (Fig. 6F) whereas it reduced MSTN
(P<0.01) (Fig. 6A), ActRIIB
(P<0.01 and P<0.01, respectively) (Fig. 6B), FoxO1 (P<0.01 and P<0.05,
respectively) (Fig. 6G), and
p-FoxO1 (P<0.01 and P<0.01, respectively) (Fig. 6H) as compared with the DM group.
Akt expression did not change significantly among the groups
(Fig. 6C). These results suggest
that PEMFs can not only increase muscle protein synthesis by
activating Akt/mTOR but also inhibit protein degradation by
inactivating MSTN and FoxO1 protein.
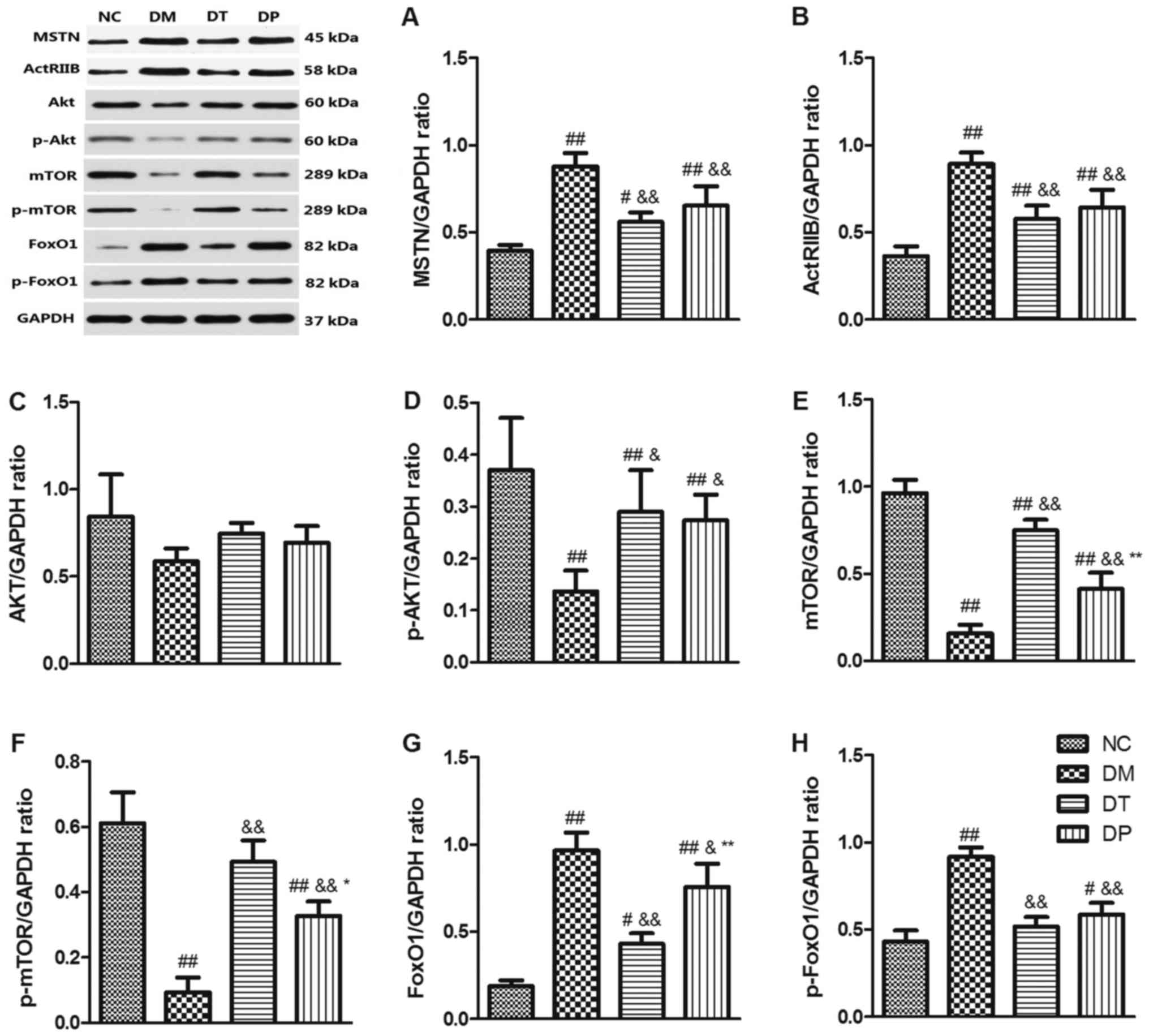 | Figure 6.Effects of pulsed electromagnetic
fields on the protein expressions of (A) MSTN, (B) ActRIIB, (C)
Akt, (D) p-Akt, (E) mTOR, (F) p-mTOR, (G) FoxO1 and (H) p-FoxO1 in
quadriceps. Data are expressed as the mean ± standard deviation.
#P<0.05 and ##P<0.01 vs. NC;
&P<0.05 and &&P<0.01 vs.
DM; *P<0.05 and **P<0.01 vs. DT. NC, normal control; DM,
diabetic mellitus group; DT, diabetic insulin-treated group; DP,
diabetic pulsed electromagnetic fields-therapy group; MSTN,
myostatin; ActRIIB, activin type II receptor; Akt, protein kinase
B; mTOR, mammalian target of rapamycin; p-, phosphorylated; FoxO1,
forkhead box protein O1. |
Discussion
Because of the absence of insulin in T1DM, blood
glucose levels rise dramatically when glucose cannot be taken up
into the major insulin-sensitive tissue-skeletal muscle.
Individuals with T1DM are at a high risk of muscle
atrophy. This means that many of these individuals' lives are lost,
despite insulin therapies (12).
PEMFs are dynamic and able to penetrate all the way through the
body, thus having many effects. It has been proved that PEMFs can
promote the proliferation and differentiation of C2C12 myoblasts
(13,14) and facilitate tendon healing
(15). Therefore, we sought to
investigate the effects and potential mechanisms of PEMFs on
STZ-induced diabetic muscle atrophy. Our findings indicate that
PEMFs alleviate diabetic myopathy by increasing protein synthesis
and decreasing protein degradation in MSTN-associated signaling
pathways.
Increase in blood glucose, appetite, urination, and
thirst during the experimental period confirmed the induction of
T1DM. The insufficient insulin therapy could cause the development
of muscle function in individuals with T1DM. Furthermore, glycemic
control is directly related to muscle metabolism and could be an
important determinant of muscle force and power in T1DM (16). In accordance with earlier studies
(17–19), our experimental rats with T1DM were
characterized by increased blood glucose, appetite, urination, and
thirst and by decreased blood insulin, weight mass, muscle mass,
and grip strength compared with the NC group rats. Furthermore,
compared with DM, PEMFs significantly increased strength and mass
of quadriceps muscle, and cross-sectional area of quadriceps muscle
fiber. These results indicate that PEMFs can improve the muscle
atrophy induced by STZ.
It is well known that SDH and MDH are marker enzymes
in the metabolism of muscle (20,21).
Decreased SDH and MDH activity in patients with diabetes has been
reported (22–25). In the current study, SDH and MDH
activity was decreased in the muscles of diabetic rats. These
results are in line with previous studies. The observed increase in
the activity of SDH and MDH in the quadriceps femoris muscles of
the diabetic rats was significantly enhanced by PEMFs therapy,
indicating that PEMFs contributed to increasing the metabolic
capacity of skeletal muscle by alleviating STZ-induced diabetic
muscle atrophy.
MSTN is a potent negative regulator of skeletal
muscle mass as demonstrated by the hypermuscularity caused by its
inactivation (26). MSTN is
expressed in skeletal muscle predominantly, and the muscle mass
increases significantly if its gene is disrupted (9). It has been demonstrated that MSTN can
activate the TGFβ2/activin type II receptor (ActRIIB) and then
activate the downstream signaling pathway (27,28).
Animals with STZ-induced T1DM that were treated with follistatin
(an inhibitor of MSTN) demonstrated improvement in the regenerative
capacity of skeletal muscle (29),
and elevations in MSTN expression have been observed in STZ-induced
T1DM (7,30). In accordance with earlier studies,
our results also pointed to a significant increase in MSTN
expression in the animals with STZ-induced T1DM. PEMFs
significantly inhibited mRNA and protein expression of MSTN and
ActRIIB compared with DM. These results indicate that inhibition of
MSTN may play a role in PEMFs promotion of STZ-induced diabetic
muscle atrophy.
Muscle mass depends on a homeostatic balance between
protein synthesis and degradation. It is believed that the Akt-mTOR
pathway is the principal signaling protein cascade regulating
protein synthesis (31,32). Akt is a key regulator of several
signaling pathways associated with skeletal muscle homeostasis, and
its major direct target downstream is the mammalian target of mTOR
kinase (33). The mTOR blocker can
inhibit muscle hypertrophy (34).
Furthermore, inhibition of the Akt/mTOR pathway can lead to muscle
atrophy (35). Rodriguez et
al (36) have reported that
MSTN negatively regulates the activity of the Akt pathway, which
promotes protein synthesis.
Moreover, muscle-specific FoxO1 overexpression in
mice has been linked to muscle atrophy (37), and Akt can control the activation
of FoxO transcription factors (38). In the present study, STZ enhanced
the activity of Akt and mTOR and inhibited FoxO1 activity and then
induced skeletal muscle hypertrophy (7,39).
Some findings suggest that PEMFs exposure might function in a
manner analogous to soluble growth factors by activating a unique
set of signaling pathways, including the Akt/mTOR pathway (40). We found that PEMFs significantly
activated Akt and mTOR and inhibited the activity of MSTN, ActRIIB,
and FoxO1 compared with DM. This means that both the Akt/mTOR and
Akt/FoxO1 signaling pathways may be involved in the alleviation of
STZ-induced diabetic muscle atrophy by PEMFs.
In terms of broadness, future studies should
investigate the mechanistic links between the proteins identified
in our study and muscle atrophy in various diabetic models and
investigate methods of inhibiting or knocking down elements of the
MSTN pathway. In terms of depth, changes in vascular lesions and
nerve evoked potentials as a more in-depth discussion of
electromagnetic field effects on diabetic muscle atrophy, which is
the focus of our next study. We will be ready to cooperate with
electrophysiology laboratories to research this issue.
Our results show that PEMFs stimulation can
alleviate diabetic muscle atrophy in an STZ model in association
with the alteration of multiple signaling pathways, wherein MSTN
may be an important factor. MSTN-associated signaling pathways may
provide therapeutic targets for the attenuation of severe diabetic
muscle wasting.
Acknowledgements
The authors would like to thank the graduate
students of the Institute of Sports Biology, Shaanxi Normal
University (Shaanxi, China) for their cooperation, and the College
of Life Sciences, Shaanxi Normal University, and Department of
Physical Education, Xi'an University of Post and Telecommunications
(Shaanxi, China).
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 11774213, 11727813
and 11502134).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LT and SA conceived the study. JY, BY, XF, LS and YK
performed the experiments, and collected and analyzed the data. JY
and LS prepared the manuscript. XF revised the manuscript and all
authors edited the manuscript. All authors contributed to the
writing of the manuscript.
Ethics approval and consent to
participate
The present study was conducted with the approval of
the Ethics Committee of Shaanxi Normal University (Shaanxi,
China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Atkinson MA and Eisenbarth GS: Type 1
diabetes: New perspectives on disease pathogenesis and treatment.
Lancet. 358:221–229. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Andersen H, Gjerstad MD and Jakobsen J:
Atrophy of foot muscles: A measure of diabetic neuropathy. Diabetes
Care. 27:2382–2385. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fritzsche K, Blüher M, Schering S,
Buchwalow IB, Kern M, Linke A, Oberbach A, Adams V and Punkt K:
Metabolic profile and nitric oxide synthase expression of skeletal
muscle fibers are altered in patients with type 1 diabetes. Exp
Clin Endocrinol Diabetes. 116:606–613. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krause MP, Riddell MC, Gordon CS, Imam SA,
Cafarelli E and Hawke TJ: Diabetic myopathy differs between
Ins2Akita+/- and streptozotocin-induced Type 1 diabetic models. J
Appl Physiol (1985). 106:1650–1659. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jakobsen J and Reske-Nielsen E: Diffuse
muscle fiber atrophy in newly diagnosed diabetes. Clin Neuropathol.
5:73–77. 1986.PubMed/NCBI
|
|
6
|
Riddell MC and Iscoe KE: Physical
activity, sport, and pediatric diabetes. Pediatr Diabetes. 7:60–70.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hulmi JJ, Silvennoinen M, Lehti M, Kivelä
R and Kainulainen H: Altered REDD1, myostatin, and
Akt/mTOR/FoxO/MAPK signaling in streptozotocin-induced diabetic
muscle atrophy. Am J Physiol Endocrinol Metab. 302:E307–E315. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang L, Liu CT, Wang XD, Luo K, Zhang DD,
Chi AP, Zhang J and Sun LJ: A prepared anti-MSTN polyclonal
antibody reverses insulin resistance of diet-induced obese rats via
regulation of PI3K/Akt/mTOR&FoxO1 signal pathways. Biotechnol
Lett. 36:2417–2423. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
McPherron AC, Lawler AM and Lee SJ:
Regulation of skeletal muscle mass in mice by a new TGF-beta
superfamily member. Nature. 387:83–90. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Glass DJ: PI3 kinase regulation of
skeletal muscle hypertrophy and atrophy. Curr Top Microbiol
Immunol. 346:267–278. 2010.PubMed/NCBI
|
|
11
|
Chang CW and Lien IN: Tardy effect of
neurogenic muscular atrophy by magnetic stimulation. Am J Phys Med
Rehabil. 73:275–279. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livingstone SJ, Levin D, Looker HC,
Lindsay RS, Wild SH, Joss N, Leese G, Leslie P, McCrimmon RJ,
Metcalfe W, et al: Estimated life expectancy in a Scottish cohort
with type 1 diabetes, 2008–2010. JAMA. 313:37–44. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu H, Zhang J, Lei Y, Han Z, Rong D, Yu Q,
Zhao M and Tian J: Low frequency pulsed electromagnetic field
promotes C2C12 myoblasts proliferation via activation of MAPK/ERK
pathway. Biochem Biophys Res Commun. 479:97–102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu M, Lee C, Laron D, Zhang N, Waldorff
EI, Ryaby JT, Feeley B and Liu X: Role of pulsed electromagnetic
fields (PEMF) on tenocytes and myoblasts-potential application for
treating rotator cuff tears. J Orthop Res. 35:956–964. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tucker JJ, Cirone JM, Morris TR, Nuss CA,
Huegel J, Waldorff EI, Zhang N, Ryaby JT and Soslowsky LJ: Pulsed
electromagnetic field therapy improves tendon-to-bone healing in a
rat rotator cuff repair model. J Orthop Res. 35:902–909. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fricke O, Seewi O, Semler O, Tutlewski B,
Stabrey A and Schoenau E: The influence of auxology and long-term
glycemic control on muscle function in children and adolescents
with type 1 diabetes mellitus. J Musculoskelet Neuronal Interact.
8:188–195. 2008.PubMed/NCBI
|
|
17
|
Junod A, Lambert AE, Stauffacher W and
Renold AE: Diabetogenic action of streptozotocin: Relationship of
dose to metabolic response. J Clin Invest. 48:2129–2139. 1969.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li RJ, Qiu SD, Tian H and Zhou SW:
Diabetes induced by multiple low doses of STZ can be spontaneously
recovered in adult mice. Dongwuxue Yanjiu. 34:238–243. 2013.(In
Chinese). PubMed/NCBI
|
|
19
|
Tsai CC, Chan P, Chen LJ, Chang CK, Liu Z
and Lin JW: Merit of ginseng in the treatment of heart failure in
type 1-like diabetic rats. Biomed Res Int. 2014:4841612014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lewis MI, Fournier M, Wang H, Storer TW,
Casaburi R and Kopple JD: Effect of endurance and/or strength
training on muscle fiber size, oxidative capacity, and capillarity
in hemodialysis patients. J Appl Physiol (1985). 119:865–871. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eprintsev AT, Falaleeva MI, Lyashchenko
MS, Gataullinaa MO and Kompantseva EI: Isoformes of malate
dehydrogenase from rhodovulum steppense A-20s grown
chemotrophically under aerobic condtions. Prikl Biokhim Mikrobiol.
52:168–173. 2016.PubMed/NCBI
|
|
22
|
Chen V and Ianuzzo CD: Metabolic
alterations in skeletal muscle of chronically
streptozotocin-diabetic rats. Arch Biochem Biophys. 217:131–138.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ianuzzo CD and Armstrong RB:
Phosphofructokinase and succinate dehydrogenase activities of
normal and diabetic rat skeletal muscle. Horm Metab Res. 8:244–245.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cai F: Studies of enzyme histochemistry
and ultrastructure of the myocardium in rats with
streptozotocin-induced diabetes. Zhonghua Yi Xue Za Zhi. 69276–278.
(20)1989.(In Chinese). PubMed/NCBI
|
|
25
|
Jia Q, Ma S, Liu X, Li S, Wang Y, Gao Q
and Yang R: Effects of hydrogen sulfide on contraction capacity of
diaphragm from type 1 diabetic rats. Zhong Nan Da Xue Xue Bao Yi
Xue Ban. 41:496–501. 2016.(In Chinese). PubMed/NCBI
|
|
26
|
Lee SJ: Sprinting without myostatin: A
genetic determinant of athletic prowess. Trends Genet. 23:475–477.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rebbapragada A, Benchabane H, Wrana JL,
Celeste AJ and Attisano L: Myostatin signals through a transforming
growth factor beta-like signaling pathway to block adipogenesis.
Mol Cell Biol. 23:7230–7242. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee SJ: Regulation of muscle mass by
myostatin. Annu Rev Cell Dev Biol. 20:61–86. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jeong J, Conboy MJ and Conboy IM:
Pharmacological inhibition of myostatin/TGF-β receptor/pSmad3
signaling rescues muscle regenerative responses in mouse model of
type 1 diabetes. Acta Pharmacol Sin. 34:1052–1060. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sriram S, Subramanian S, Juvvuna PK,
McFarlane C, Salerno MS, Kambadur R and Sharma M: Myostatin induces
DNA damage in skeletal muscle of streptozotocin-induced type 1
diabetic mice. J Biol Chem. 289:5784–5798. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Glass DJ: Skeletal muscle hypertrophy and
atrophy signaling pathways. Int J Biochem Cell Biol. 37:1974–1984.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Leger B, Cartoni R, Praz M, Lamon S,
Dériaz O, Crettenand A, Gobelet C, Rohmer P, Konzelmann M, Luthi F
and Russell AP: Akt signalling through GSK-3beta, mTOR and Foxo1 is
involved in human skeletal muscle hypertrophy and atrophy. J
Physiol. 576:923–933. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dang K, Li YZ, Gong LC, Xue W, Wang HP,
Goswami N and Gao YF: Stable atrogin-1 (Fbxo32) and MuRF1 (Trim63)
gene expression is involved in the protective mechanism in soleus
muscle of hibernating Daurian ground squirrels (Spermophilus
dauricus). Biol Open. 5:62–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bodine SC, Stitt TN, Gonzalez M, Kline WO,
Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC,
Glass DJ and Yancopoulos GD: Akt/mTOR pathway is a crucial
regulator of skeletal muscle hypertrophy and can prevent muscle
atrophy in vivo. Nat Cell Biol. 3:1014–1019. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hoffman EP and Nader GA: Balancing muscle
hypertrophy and atrophy. Nat Med. 10:584–585. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rodriguez J, Vernus B, Chelh I,
Cassar-Malek I, Gabillard JC, Sassi Hadj A, Seiliez I, Picard B and
Bonnieu A: Myostatin and the skeletal muscle atrophy and
hypertrophy signaling pathways. Cell Mol Life Sci. 71:4361–4371.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kamei Y, Miura S, Suzuki M, Kai Y,
Mizukami J, Taniguchi T, Mochida K, Hata T, Matsuda J, Aburatani H,
et al: Skeletal muscle FOXO1 (FKHR) transgenic mice have less
skeletal muscle mass, down-regulated Type I (slow twitch/red
muscle) fiber genes, and impaired glycemic control. J Biol Chem.
279:41114–41123. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bonaldo P and Sandri M: Cellular and
molecular mechanisms of muscle atrophy. Dis Model Mech. 6:25–39.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang J, Zhuang P, Wang Y, Song L, Zhang
M, Lu Z, Zhang L, Wang J, Alemu PN, Zhang Y, et al: Reversal of
muscle atrophy by Zhimu-Huangbai herb-pair via Akt/mTOR/FoxO3
signal pathway in streptozotocin-induced diabetic mice. PLoS One.
9:e1009182014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Patterson TE, Sakai Y, Grabiner MD,
Ibiwoye M, Midura RJ, Zborowski M and Wolfman A: Exposure of murine
cells to pulsed electromagnetic fields rapidly activates the mTOR
signaling pathway. Bioelectromagnetics. 27:535–544. 2006.
View Article : Google Scholar : PubMed/NCBI
|