Introduction
Hyperlipidemia is strongly associated with the
prevalence of type 2 diabetes mellitus (T2DM), and statins are the
first line of drugs for treating hyperlipidemia. However, a
previous study demonstrated that statins may increase the risk of
new-onset T2DM, and the underlying mechanism requires determination
(1). Notably, a series of studies
on patients with familial hypercholesterolemia (FH) have
investigated this (2,3). In these studies, a novel phenomenon
demonstrated that patients with FH were protected against diabetes
mellitus even when subjected to long-term statin therapy.
Considering that the majority of the patients in these studies had
a heterozygous mutation in the low-density lipoprotein receptor
(LDLR) gene, these findings suggest a noteworthy hypothesis that
LDLR-mediated cholesterol metabolism may be involved in the
diabetogenic effect of statins (4). Indeed, statins may enhance
cholesterol uptake in the liver and peripheral tissues via
upregulation of LDLR and thereby reduce plasma levels of
LDL-cholesterol (C) in the blood. Overaccumulation of cholesterol
in peripheral tissues may cause lipotoxicity, which promotes a
negative effect on the function of the pancreatic islets as
previously described (5).
Additionally, cholesterol impairs the function of pancreatic β
cells and causes cellular apoptosis, which have been demonstrated
in in vitro and in vivo studies (6).
Although LDLRs may provide a key association between
statin therapy and the risk of new-onset T2DM, whether LDLR
expression and its regulation in the pancreas and pancreatic islets
are altered when patients receive statins is unknown (2). A previous study conducted by the
present team demonstrated that 16 weeks of oral treatment with
atorvastatin did not affect glucose metabolism in rabbits (7). Therefore, it was hypothesized that
the short-term use of atorvastatin may disturb glucose homeostasis.
Accordingly, it was proposed that the short-term use of statins may
affect the function of pancreatic islets via regulation of LDLR
expression. Therefore, the present study aimed to elucidate glucose
homeostasis, LDLR expression and its possible regulation in the
pancreas following the short-term administration of
atorvastatin.
Materials and methods
Animals
The present study was conducted in accordance with
the Guidelines for Animal Experimentation of Xi'an Medical
University (Xi'an, China) and the Guide for the Care and Use of
Laboratory Animals published by the US National Institutes of
Health (NIH; Bethesda, MD, USA; NIH publication no. 85–23, revised
2011). The experimental protocols were approved by the Laboratory
Animal Administration Committee of Xi'an Medical University
(Institutional Animal Care and Use Committee; permit no.
XYJZS-1207011). Male C57BL/6j mice (12 weeks old; ~30 g) were
obtained from the laboratory animal center at The Fourth Military
Medical University (Xi'an, China), and male apolipoprotein E
(ApoE)-deficient mice (12 weeks old; ~30 g) were obtained from the
laboratory animal center at Xi'an Jiaotong University (Xi'an,
China). A total of 20 C57BL/6j mice were randomly divided into two
groups and received a normal chow diet. One group was treated with
atorvastatin via intragastric administration (10 mg/kg, each day;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 6 weeks; the
other group was given the same dose of double distilled water. A
total of 16 ApoE-deficient mice were additionally divided into two
groups and received a high-fat diet in addition to the treatment
described above. As previously suggested by Capel et al
(8), a 10 mg dose of atorvastatin
has been repeatedly demonstrated to be effective on lipid
metabolism in mice, and the relevant literature is listed in
Table I (8–17).
All mice were individually housed in plastic cages (30×20×13 cm)
throughout the extent of the present study and maintained on a
12/12 h light/dark cycle (light off at noon) at constant
temperature (22°C) with 10–15 hly cycles of fresh air and relative
humidity (60±10%). Food and water were available ad
libitum.
 | Table I.Summary of atorvastatin doses in mouse
models involving DM-associated research. |
Table I.
Summary of atorvastatin doses in mouse
models involving DM-associated research.
| Author, year | PubMed-Indexed for
MEDLINE | Atorvastatin dose,
intervention time, mode of drug delivery | Mouse model | Purpose | (Refs.) |
|---|
| Capel et al,
2015 | 26228176 | 10 mg/kg/day, 3
weeks, gastric gavage | C57BL/6J mice | Diet-induced
obesity | (8) |
| Liang et al,
2017 | 28214881 | 10 mg/kg/day, 4
weeks, gastric gavage | Kunming mice | The treatment of
myocardial hypertrophy | (9) |
| Ren et al,
2016 | 27851811 | 10 mg/kg/day, 12
weeks, osmotic mini pumps | C57BL/6J mice | Experimental
diabetic cardiomyopathy | (10) |
| Wu et al,
2016 | 27428373 | 10 mg/kg/day, 8
weeks, gastric gavage | ApoE−/−mice | Inflammatory
stress | (11) |
| Lee et al,
2016 | 27565724 | 10 mg/kg/day, 7
weeks, gastric gavage | C57BL/6J mice | Immune dysfunction
in metabolic disorders | (12) |
| Roth et al,
2016 | 26826559 | 10 mg/kg/day, 15
weeks, gastric gavage | ApoE−/−mice | Cardiovascular
morbidity and mortality | (13) |
| Han et al,
2016 | 27574007 | 10 mg/kg/day, 15
weeks, gastric gavage | ApoE−/−mice | Atherogenesis and
plaque instability | (14) |
| Bruder-Nascimento
et al, 2015 | 25358739 | 10 mg/kg/day, 3
weeks, gastric gavage | db/db mice | Vascular injury in
DM | (15) |
| Park et al,
2015 | 26174316 | 10 mg/kg/day, 12
weeks, gastric gavage | ApoE−/−mice | Inflammatory and
vascular diseases in atherosclerosis | (16) |
| Van den Hoek et
al, 2014 | 24373179 | 10 mg/kg/day, 20
weeks, gastric gavage | E3L.CETP mice | The metabolic
syndrome | (17) |
Plasma lipid, glucose and insulin
metabolism
Following overnight fasting, blood samples were
collected via the retro-orbital sinus in tubes containing EDTA as
an anticoagulant. Blood samples were centrifuged (1,500 × g; 15
min; 4°C) to collect plasma. Plasma total cholesterol (TC), total
triglycerides (TG) and high-density lipoprotein (HDL-C) were
measured using a Varioskan Flash plate reader (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) with assay kits (BioSino
Bio-Technology and Science Inc., Beijing, China) during the final
week of the treatment as previously described (18). Intravenous glucose tolerance tests
(IGTT) and intravenous insulin tolerance tests (IITT) were
performed as previously described (7). The mice were injected with an
intravenous glucose solution following 12 h of fasting, and the
blood samples were subsequently drawn at 0, 15, 30, 60, 90, 120 and
150 min (1.5 g/kg body weight). For IITT, subsequent to the
intravenous injection of insulin (1 U/kg body weight; Wanbang
Biopharmaceuticals Co., Ltd., Xuzhou, China), blood samples were
collected to examine blood sugar levels at 0, 15, 30, 60, 90, 120
and 150 min.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA of the pancreas and liver were extracted
using RNAiso Plus (Takara Bio, Inc., Otsu, Japan). RT-qPCR was
performed and quantified using the 2−ΔΔCq method as
previously described (18–20). The sequences of the primers are
listed as follows: LDLR, 5′-TGACCTTCATCCCAGAGCCTTC-3′ and
5′-GGCATGAGCGGGTATCCATC-3′; proprotein convertase subtilisin/kexin
type 9 (PCSK9), 5′-TATCCCAGCATGGCACCAGA-3′ and
5′-ATGGTGACCCTGCCCTCAA-3′; sterol regulatory element-binding
protein 2 (SREBP-2), 5′-CCCTTGACTTCCTTGCTGCA-3′ and
5′-GCGTGAGTGTGGGCGAATC-3′; E3 ubiquitin-protein ligase MYLIP
(IDOL), 5′-AGGAGATCAACTCCACCTTCTG-3′ and 5′-ATCTGCAGACCGGACAGG-3;
GAPDH, 5′-ACTGAGGACCAGGTTGTC-3′ and 5′-TGCTGTAGCCGTATTCATTG-3′.
Protein extraction and western blot
analysis
Total protein was extracted from the pancreas and
liver as previously described (19). The primary antibodies were against
LDLR (1:500; cat. no. ab52818; Abcam, Cambridge, UK), ATP-binding
cassette transporter A1 (ABCA1; 1:1,000; cat. no. sc-58219),
SREBP-2 (1:1,000; cat. no. sc-271616; both Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), PCSK9 (1:1,000; cat. no.
ab181142; Abcam), IDOL (1:2,000; cat. no. SAB4501317;
Sigma-Aldrich; Merck KGaA) and GAPDH (1:2,000; cat. no. sc-32233;
Santa Cruz Biotechnology, Inc.) overnight at 4°C. Membranes were
subsequently incubated with horseradish peroxidase-conjugated
secondary antibodies (1:2,500; cat. nos. A0216 and A0208; Beyotime
Institute of Biotechnology, Haimen, China) for 3 h at room
temperature. A western blot analysis was performed as previously
described, and relative protein expression was quantified by ImageJ
(bundled with Java 1.8.0_172; NIH) (19).
Islet insolation and culture
Pancreatic islets in each mouse were isolated as
previously described (21).
Pancreatic islets were isolated from fasting mice with collagenase
type V (0.8 mg/ml; Sigma-Aldrich; Merck KGaA) digestion and,
subsequently, islets were purified using a continuous Histopaque
(Sigma-Aldrich; Merck KGaA) gradient. Purified islet fractions were
collected, and all islets (including islets with normal
architecture and islets more or less affected by fibrosis) were
selected under a stereomicroscope (magnification, ×10). Mouse
islets were cultured in RPMI 1640 medium (Invitrogen; Thermo Fisher
Scientific, Inc.) and seeded on a 24-well plate (20 islets/well)
coated with rat-tail collagen (Sigma-Aldrich; Merck KGaA). Islets
were left in wells for 24 h to adhere and were subsequently treated
with different concentrations of atorvastatin (0, 1, 10 and 100 nM;
Sigma-Aldrich; Merck KGaA) for 48 h, according to previously
described protocols (22).
Immunofluorescence
Following 48 h of incubation, islets were fixed for
1 h with 4% formaldehyde at room temperature and were blocked with
5% bovine serum albumin (cat. no. P0007; Beyotime Institute of
Biotechnology) in PBS for 1 h at room temperature. To avoid the
detection of internalized LDLR in the intracellular lysosomes,
islets were not permeabilized with Triton X-100 prior to
incubation. Pancreatic islets were incubated overnight at 4°C with
primary antibodies (1:500; cat. no. ab52818; Abcam) against LDLR,
and the islets were incubated with Alexa Fluor 488 (1:2,000; cat
no. Z25302; Thermo Fisher Scientific, Inc.) secondary antibodies
for 3 h at room temperature. Pictures were taken on the fluorescent
Nikon TE2000 Inverted Microscope (magnification, ×4; TE2000S; Nikon
Corporation, Tokyo, Japan) and analyzed with the ImageJ software as
previously described (23).
ELISA
Plasma PCSK9 and insulin levels were measured using
commercial ELISA kits (cat. no. MPC900; Mouse Quantikine ELISA for
PCSK9; R&D Systems, Inc., Minneapolis, MN, USA; cat. no. YK060;
Insulin ELISA; Yanaihara Institute Inc., Fujinomiya, Japan) during
the last week of intervention, as previously described (18).
Statistical analysis
All data are expressed as the mean ± standard error.
Comparisons between two groups were performed using the Student's
t-test. Comparisons between multiple groups were conducted using
one-way analysis of variance with the Bonferroni test. P<0.05
was considered to indicate a statistically significant difference.
The experiments were repeated three times and statistical
calculations were analyzed using the SPSS 19.0 software (IBM Corp.,
Armonk, NY, USA).
Results
Short-term use of atorvastatin affects
glucose homeostasis in mice with hyperlipidemia
To study the effect of acute atorvastatin
administration on glucose metabolism, IGTT and IITT were performed
following 3 and 6 weeks of treatment with atorvastatin in C57BL/6j
mice. As demonstrated in Fig. 1A,
a 3-week treatment with atorvastatin did not affect glucose
tolerance, and only the glucose levels at time 0 (fasting state)
were decreased in atorvastatin-treated mice. However, treatment
with atorvastatin for 3 weeks significantly affected insulin
tolerance following 50 min (P<0.05) and significantly increased
the AUC (P<0.05), indicating that atorvastatin decreased insulin
sensitivity (Fig. 1A). However,
when the AUCs were compared, it was observed that treatment with
atorvastatin for 6 weeks did not affect glucose tolerance or
insulin sensitivity (Fig. 1B).
Additionally, it was identified that treatment with atorvastatin
for 6 weeks decreased insulin levels following 12 h of fasting
(Fig. 1B).
As it is known that hyperlipidemia increases the
risk of cardiovascular disease (CVD) and T2DM, it was hypothesized
that the short-term use of atorvastatin may affect glucose
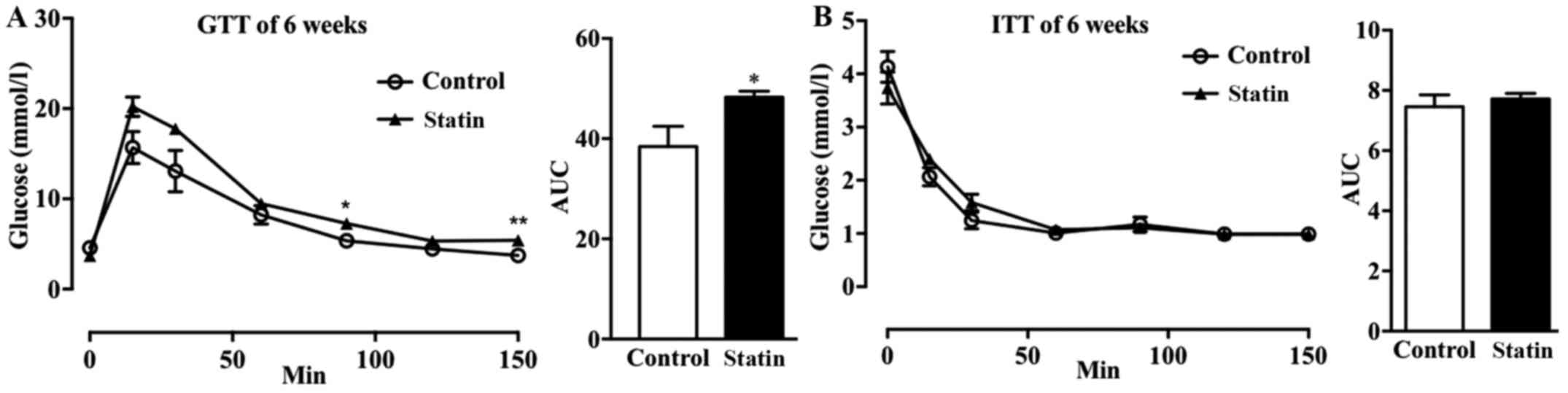
metabolism in mice with hyperlipidemia (24). It was observed that treatment with
atorvastatin for 6 weeks increased glucose tolerance in mice with
hyperlipidemia (Fig. 2A); whereas,
there was no marked effect on insulin sensitivity (Fig. 2B).
Short-term use of atorvastatin affects
blood lipid levels in normal mice
The lipid profile was examined in the two types of
mice following 6 weeks of treatment with atorvastatin. The present
results demonstrated that treatment with atorvastatin significantly
decreased the TC, TG and insulin levels in the C57BL/j mice
compared with the control plasma lipid levels (P<0.05); however,
there was no significant difference in plasma LDL-C, HDL-C or
glucose (Fig. 3A). In
hyperlipidemic mice, treatment with atorvastatin did not affect
plasma TC, LDL-C, HDL-C, TG or glucose (Fig. 3B). This result is consistent with a
previous study demonstrating that atorvastatin does not markedly
reduce the blood lipid levels in ApoE-deficient mice (25). Furthermore, it was identified that
treatment with atorvastatin for 6 weeks did not affect insulin
levels following 12 h of fasting (Fig.
3B).
Treatment with atorvastatin suppresses
LDLR expression in the pancreas of C57BL/j mice
To study whether treatment with atorvastatin may
affect LDLR expression in the pancreas and liver, the protein and
mRNA levels of LDLR were measured by western blotting and PCR. It
was observed that 6 weeks of treatment with atorvastatin
significantly reduced LDLR protein in the pancreas (P<0.05), and
this result was additionally confirmed with PCR, which demonstrated
that LDLR mRNA expression was altered significantly (Fig. 4A; P<0.05). As it is known that
ABCA1 is a key receptor that mediates cellular cholesterol efflux,
ABCA1 expression was additionally examined; however, no alterations
in ABCA1 expression were observed in the pancreas with acute
atorvastatin administration (Fig.
4A) (26). Contrary to the
findings in the pancreas, it was identified that 6 weeks of
treatment with atorvastatin did not affect expression of LDLR and
ABCA1 at the protein and mRNA levels in the liver (Fig. 4B).
To test whether the effect of atorvastatin on LDLR
expression may be affected by hyperlipidemia, the LDLR expression
was measured in the pancreas and liver of ApoE-deficient mice. It
was observed that 6 weeks of treatment with atorvastatin did not
affect the protein and mRNA expression levels of LDLR in the
pancreas (Fig. 5A). Furthermore,
it was identified that treatment with atorvastatin did not affect
the protein and mRNA expression of LDLR in the liver (Fig. 5B).
Treatment with atorvastatin suppresses
LDLR expression in pancreatic islets
To test whether atorvastatin may affect LDLR
expression in pancreatic islets, increasing concentrations of
atorvastatin were used to treat adhered islets from C57BL/6j mice
for 48 h. It was identified that incubation with atorvastatin
affected LDLR expression in islets in a concentration-dependent
manner (Fig. 6). Subsequent to
analyzing the fluorescence intensity by using ImageJ software, it
was observed that islets incubated with a high dose of atorvastatin
(100 nM) exhibited a significantly decreased fluorescence intensity
compared with the 0 nm group (Fig.
6B; P<0.05).
Short-term use of atorvastatin induces
an increase in PCSK9 in the pancreas
To investigate the possible regulation of LDLR by
atorvastatin, the protein expression of SREBP-2, PCSK9 and IDOL was
measured in the pancreas of C57BL/6j mice. It was observed that 6
weeks of treatment with atorvastatin did not affect SREBP-2 or IDOL
expression; whereas, treatment with atorvastatin significantly
increased PCSK9 expression in the pancreas compared with the
control (Fig. 7A; P<0.05). To
confirm these results, RT-qPCR was used to measure the mRNA
expression of SREBP-2, PCSK9 and IDOL in the pancreas. It was
observed that the mRNA expression of PCSK9 was upregulated by
treatment with atorvastatin; however, SREBP-2 and IDOL were not
affected (Fig. 7B). Furthermore,
given that plasma PCSK9 may affect LDLR expression in peripheral
tissues, a commercial ELISA kit was used to detect plasma PCSK9
expression levels. It was identified that 6 weeks of treatment with
atorvastatin did not affect plasma PCSK9 expression levels
(Fig. 7C). A schematic diagram
represents the regulation of hepatic LDLR by statins: Statins
upregulate LDLR via SREBP-2; SREBP-2 additionally increases
transcription of PCSK9 to degrade LDLR; and cellular cholesterol
accumulation may activate LXR, which upregulates IDOL to degrade
LDLR (Fig. 7D).
Discussion
Statins are 3-hydroxy-3-methyl-glutaryl coenzyme-A
reductase inhibitors, which are used to decrease blood LDL-C levels
by inhibiting cellular cholesterol synthesis and upregulating LDLR
expression levels in the liver and peripheral tissues (27). Although previous research suggests
that statins increase the risk of new-onset T2DM, it is noteworthy
that statins are highly effective for the prevention of CVD in
individuals with or without DM (28). A recent study additionally
concluded that the net absolute benefit observed with statin
therapy in such individuals is >50 times greater compared with
any putative effect on DM (29).
As statins have an important role in the primary and secondary
prevention of CVD, a mechanistic understanding of the diabetogenic
effect of statins requires investigation. However, there are
inconsistent findings on the diabetogenic effect of statins in
animal experiments (28). A
previous study conducted by the present team demonstrated that
long-term treatment with atorvastatin does not affect glucose
homeostasis in rabbits with normal blood lipid levels (7). Given that adaptation may occur in
islets when experimental animals are subjected to long-term
treatment with atorvastatin, it was hypothesized that acute
administration of atorvastatin may affect glucose hemostasis.
Notably, the present study supports the hypothesis that short-term
administration of atorvastatin may slightly affect glucose
homeostasis in normal and hyperlipidemic mice. In clinical studies,
treatment with statins has been associated with the risk of
new-onset T2DM; there is additional evidence to suggest that
statins may induce the onset of T2DM (29,30).
Although diabetic rodent models are useful for the majority of
diabetes studies, these animals usually have impaired pancreatic
islets and present dysfunction of β cells, which may not be
suitable for observation of early changes in the diabetogenic
process or a slight effect of statins on normal islets (31). It was hypothesized that the
diabetogenic effect of statins may contribute to LDLR-mediated
LDL-C uptake in β cells, and hyperlipidemia may be involved in the
diabetogenic effect of statins. Therefore, normal C57BL/J and
hyperlipidemic ApoE-deficient mice were examined in the present
study.
On the basis of previous studies, statins may
increase the transcription of LDLRs in the liver by upregulating
SREBP-2. As a transcription factor, SREBP-2 additionally
upregulates PCSK9 to induce the degradation of LDLR (27). Statins may promote an increase in
IDOL, which serves as a ubiquitin ligase to degrade LDLR and may
reduce LDLR expression (32). As
statins may dually upregulate and downregulate LDLR, the abundance
of the LDLR protein is not markedly increased in the liver
(33). In contrast to the liver,
the pancreas and islets may not be fully subjected to such dual
regulation. In the present study, it was observed that atorvastatin
suppressed LDLR expression in the pancreas and islets, whereas this
phenomenon was not observed in the liver. Furthermore, it was not
observed that atorvastatin affected LDLR expression in the pancreas
and liver in hyperlipidemic mice. As mentioned previously, LDLR
expression is primarily regulated by SREBP-2, PCSK9 and IDOL.
Therefore, these genes were investigated in the pancreas.
Interestingly, compared with no difference in plasma PCSK9,
treatment with atorvastatin may promote an increase in PCSK9 in the
pancreas, indicating that decreased LDLR may be attributed to
upregulated PCSK9 in situ.
As previously mentioned, upregulation of LDLR may
enhance LDLR-mediated LDL-C (or modified LDL-C) uptake and causes
dysfunction or cytotoxicity in these cells (2). LDLR therefore provides an association
between treatment with statins and new-onset T2DM. Although the
present data demonstrated that atorvastatin decreases LDLR in the
pancreas, the pathological role of LDLR in T2DM requires further
investigation. Firstly, it is unclear whether treatment with
atorvastatin may induce a similar effect on LDLR expression in the
pancreas and islets of humans, and this effect may be enhanced or
weakened in patients with hyperlipidemia or T2DM. Secondly,
ApoE-deficient mice present hypercholesterolemia due to
accumulation of remnant lipoproteins compared with LDLs, which is
not completely consistent with the physiopathological process of
patients with hyperlipidemia or FH. Therefore, a clinical study may
provide novel insight into the pathological role of LDLR in the
diabetogenic effect of statins. Notably, it was additionally
identified that atorvastatin did not promote an increase in ABCA1
in the liver and pancreas. Given that ABCA1 is a pivotal receptor
for mediating cholesterol efflux, whether atorvastatin may enhance
the ABCA1-mediated cholesterol efflux in mice requires
determination (34).
In conclusion, the present study presents novel
findings on the effect of acute administration of atorvastatin on
disturbing glucose homeostasis. It was additionally demonstrated
that acute administration of atorvastatin downregulates LDLR
expression in the pancreas of normal mice; however, this reduction
in LDLR was not observed in hyperlipidemic mice. Furthermore, the
present data demonstrated that LDLR expression in the pancreas is
more affected by atorvastatin compared with in the liver of mice
with normal blood lipid levels, suggesting that the role of LDLR in
the diabetogenic effect of statins requires further
investigation.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science of China (grant nos. 81400328 and 81773795), China
Postdoctoral Science Foundation (grant nos. 2015M582800 and
2016T90972), Natural Science Basic Research Plan in Shaanxi
Province of China (grant nos. 2016JM8025 and 2016SF-107) and
Scientific Research Fund of Shannxi Provincial Education Department
(grant no. 17JS116).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
QY, CX and SW contributed to the experimental
design, discussion of result and critical revision of the
manuscript. QY contributed to the drafting of the manuscript. QY,
FW, XM, YG and YW conducted the experiments. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
The experimental protocols were approved by the
Laboratory Animal Administration Committee of Xi'an Medical
University (Institutional Animal Care and Use Committee; permit no.
XYJZS-1207011).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Brault M, Ray J, Gomez YH, Mantzoros CS
and Daskalopoulou SS: Statin treatment and new-onset diabetes: A
review of proposed mechanisms. Metabolism. 63:735–745. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yu Q, Chen Y and Xu CB: Statins and
new-onset diabetes mellitus: LDL receptor may provide a key link.
Front Pharmacol. 8:3722017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Besseling J, Kastelein JJ, Defesche JC,
Hutten BA and Hovingh GK: Association between familial
hypercholesterolemia and prevalence of type 2 diabetes mellitus.
JAMA. 313:1029–1036. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu Q, Su X and Liu E: Could familial
hypercholesterolemia oppose the diabetogenic effect of statin?
Comments on a new SAFEHEART study. Int J Cardiol. 202:954–955.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ishikawa M, Iwasaki Y, Yatoh S, Kato T,
Kumadaki S, Inoue N, Yamamoto T, Matsuzaka T, Nakagawa Y, Yahagi N,
et al: Cholesterol accumulation and diabetes in pancreatic
beta-cell-specific SREBP-2 transgenic mice: A new model for
lipotoxicity. J Lipid Res. 49:2524–2534. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brunham LR, Kruit JK, Verchere CB and
Hayden MR: Cholesterol in islet dysfunction and type 2 diabetes. J
Clin Invest. 118:403–408. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheng D, Wang Y, Gao S, Wang X, Sun W, Bai
L, Cheng G, Chu Y, Zhao S and Liu E: Atorvastatin delays the
glucose clearance rate in hypercholesterolemic rabbits. Biomed
Pharmacother. 72:24–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Capel F, Chabrier G, Pitois E, Rigaudière
JP, Le Plenier S, Durand C, Jouve C, de Bandt JP, Cynober L and
Moinard C: Combining citrulline with atorvastatin preserves glucose
homeostasis in a murine model of diet-induced obesity. Br J
Pharmacol. 172:4996–5008. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liang J, Yin K, Cao X, Han Z, Huang Q,
Zhang L, Ma W, Ding F, Bi C, Feng D, et al: Attenuation of low
ambient temperature-induced myocardial hypertrophy by atorvastatin
via promoting Bcl-2 expression. Cell Physiol Biochem. 41:286–295.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ren XM, Zuo GF, Wu W, Luo J, Ye P, Chen SL
and Hu ZY: Atorvastatin alleviates experimental diabetic
cardiomyopathy by regulating the GSK-3β-PP2Ac-NF-κB signaling axis.
PLoS One. 11:e01667402016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu W, Zhao L, Yang P, Zhou W, Li B,
Moorhead JF, Varghese Z, Ruan XZ and Chen Y: Inflammatory stress
sensitizes the liver to atorvastatin-induced injury in ApoE−/−mice.
PLoS One. 11:e01595122016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee S, Lee Y, Kim J, An J and Kim K, Lee
H, Kong H, Song Y and Kim K: Atorvastatin and rosuvastatin improve
physiological parameters and alleviate immune dysfunction in
metabolic disorders. Biochem Biophys Res Commun. 478:1242–1247.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Roth L, Rombouts M, Schrijvers DM,
Martinet W and De Meyer GR: Cholesterol-independent effects of
atorvastatin prevent cardiovascular morbidity and mortality in a
mouse model of atherosclerotic plaque rupture. Vascul Pharmacol.
80:50–58. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han H, Chen Y, Zhu J, Ni J, Sun J and
Zhang R: Atorvastatin attenuates p-cresyl sulfate-induced
atherogenesis and plaque instability in ApoE knockout mice. Mol Med
Rep. 14:3122–3128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bruder-Nascimento T, Callera GE, Montezano
AC, He Y, Antunes TT, Cat Nguyen Dinh A, Tostes RC and Touyz RM:
Vascular injury in diabetic db/db mice is ameliorated by
atorvastatin: Role of Rac1/2-sensitive Nox-dependent pathways. Clin
Sci (Lond). 128:411–423. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park SH, Sung YY, Nho KJ and Kim HK:
Protective activity ethanol extract of the fruits of Illicium verum
against atherogenesis in apolipoprotein E knockout mice. BMC
Complement Altern Med. 15:2322015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van den Hoek AM, van der Hoorn JW, Maas
AC, van den Hoogen RM, van Nieuwkoop A, Droog S, Offerman EH,
Pieterman EJ, Havekes LM and Princen HM: APOE*3Leiden. CETP
transgenic mice as model for pharmaceutical treatment of the
metabolic syndrome. Diabetes Obes Metab. 16:537–544. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu Q, Liu Z, Waqar AB, Ning B, Yang X,
Shiomi M, Graham MJ, Crooke RM, Liu E, Dong S and Fan J: Effects of
antisense oligonucleotides against C-reactive protein on the
development of atherosclerosis in WHHL rabbits. Mediators Inflamm.
2014:9791322014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu Q, Wang Y, Yu Y, Li Y, Zhao S, Chen Y,
Waqar AB, Fan J and Liu E: Expression of TRPV1 in rabbits and
consuming hot pepper affects its body weight. Mol Biol Rep.
39:7583–7589. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li DS, Yuan YH, Tu HJ, Liang QL and Dai
LJ: A protocol for islet isolation from mouse pancreas. Nat Protoc.
4:1649–1652. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao W and Zhao SP: Different effects of
statins on induction of diabetes mellitus: An experimental study.
Drug Des Devel Ther. 9:6211–6223. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jensen EC: Quantitative analysis of
histological staining and fluorescence using ImageJ. Anat Rec
(Hoboken). 296:378–381. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ansar S, Koska J and Reaven PD:
Postprandial hyperlipidemia, endothelial dysfunction and
cardiovascular risk: Focus on incretins. Cardiovasc Diabetol.
10:612011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bot I, Jukema JW, Lankhuizen IM, van
Berkel TJ and Biessen EA: Atorvastatin inhibits plaque development
and adventitial neovascularization in ApoE deficient mice
independent of plasma cholesterol levels. Atherosclerosis.
214:295–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Du XM, Kim MJ, Hou L, Le Goff W, Chapman
MJ, Van Eck M, Curtiss LK, Burnett JR, Cartland SP, Quinn CM, et
al: HDL particle size is a critical determinant of ABCA1-mediated
macrophage cellular cholesterol export. Circ Res. 116:1133–1142.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goldstein JL and Brown MS: A century of
cholesterol and coronaries: From plaques to genes to statins. Cell.
161:161–172. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Axsom K, Berger JS and Schwartzbard AZ:
Statins and diabetes: The good, the bad and the unknown. Curr
Atheroscler Rep. 15:2992013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hennekens CH, Teng B and Pfeffer MA:
Statins and diabetes: Current perspectives and implications for
clinicians. Am J Med. 130:504–506. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sattar N and Taskinen MR: Statins are
diabetogenic-myth or reality? Atheroscler Suppl. 13:1–10. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Furman BL: Streptozotocin-induced diabetic
models in mice and rats. Curr Protoc Pharmacol. 70:5.47.1–20. 2015.
View Article : Google Scholar
|
|
32
|
Hong C, Marshall SM, McDaniel AL, Graham
M, Layne JD, Cai L, Scotti E, Boyadjian R, Kim J, Chamberlain BT,
et al: The LXR-Idol axis differentially regulates plasma LDL levels
in primates and mice. Cell Metab. 20:910–918. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Attie AD and Seidah NG: Dual regulation of
the LDL receptor-some clarity and new questions. Cell Metab.
1:290–292. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kruit JK, Kremer PH, Dai L, Tang R, Ruddle
P, de Haan W, Brunham LR, Verchere CB and Hayden MR: Cholesterol
efflux via ATP-binding cassette transporter A1 (ABCA1) and
cholesterol uptake via the LDL receptor influences
cholesterol-induced impairment of beta cell function in mice.
Diabetologia. 53:1110–1119. 2010. View Article : Google Scholar : PubMed/NCBI
|