Introduction
Thyroid cancer (TC) accounts for only 1% of all
malignancies and is a common endocrine cancer that derives from
follicular thyroid or parafollicular C cells (1). In the USA, TC incidence is rapidly
increasing with an estimated number of 56,870 individuals diagnosed
with TC and an annual total mortality of 2,010 cases in 2017
(2). TCs are typically classified
as papillary, follicular and anaplastic carcinomas (3). Anaplastic thyroid cancer (ATC)
accounts for 1–2% of all thyroid tumors and is characterized by
aggressive, local invasion and common distant metastases (3). Currently, available therapy for ATCs
includes chemotherapy, radiotherapy and surgery (3). However, no effective target
treatments are available. ATC is one of the most fatal cancer
types, with a mean survival of 6 months (3,4).
Therefore, an improved understanding of the molecular mechanisms
underlying carcinogenesis and progression in ATC may contribute to
the identification novel diagnostic markers and novel therapeutic
targets.
It is well known that >90% of the human genome is
actively transcribed, but only ~2% of the genome encodes proteins
(5). The rest of the genome
encodes non-coding RNAs (ncRNAs) including microRNAs (miRs) and
long non-coding RNAs (lncRNAs). lncRNAs are defined as transcripts
of >200 nucleotides in length with minimal to no protein-coding
function (6). lncRNAs have been
defined as a novel class of regulatory factors that modulate gene
expression in physiological and pathological states (7). Their biological roles have been
increasingly recognized; however, the functions of overexpressed
lncRNAs in malignant diseases remain to be elucidated. UCA1, a
sequence that consists of three exons with 1.4 kb in length, is a
lncRNA originally identified in bladder transitional cell carcinoma
(8) and promotes tumor progression
in certain carcinomas. However, the expression and function of UCA1
in ATC has not yet been elucidated.
The present study demonstrated that UCA1 was
significantly increased in ATC tissues and cell lines. Silencing of
UCA1 inhibited ATC cell viability, proliferation, migration and
invasion in vitro and inhibited the tumor growth in
vivo. In addition, it was identified that c-myc proto-oncogene
(c-myc) was a target gene of UCA1. UCA1 directly interacted with
miR-135a and decreased the binding of miR-135a to the c-myc 3′
untranslated region (UTR), which suppressed the degradation of
c-myc mRNA by miR-135a. The results revealed that the
UCA1/miR-135a/c-myc signaling pathway may be a potential novel
therapeutic target for patients with ATC.
Materials and methods
Cell culture and tissue
collection
The human Nthy-ori3-1 cell line and ATC cell lines,
SW1736 and KAT-18, were purchased from the American Type Culture
Collection (Manassas, VA, USA). The cell lines were authenticated
by short-tandem repeat profiling performing by BMR Genomics
(Padova, Italy). The cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; HyClone; GE Healthcare Life Sciences, Logan,
UT, USA) supplemented with 10% fetal bovine serum (FBS; HyClone; GE
Healthcare Life Sciences) in a 95% humidified atmosphere with 5%
CO2 at 37°C. Human ATC specimens and their adjacent
normal thyroid tissues (eight pairs) were collected from patients
(three males and five females; aged 43–67 years) who underwent
surgery, according to an approved human protocol at the Weifang
People's Hospital from February 2016 to December 2016. The present
study was approved by the Ethics Committee of Weifang People's
Hospital (Weifang, China). Written informed consent was obtained
from each patient.
Cell transfection
The LV3 (H1/GFP&Puro) vector was synthesized to
express the Lv-shRNA-UCA1 (Guangzhou Ribobio, Co., Ltd., Guangzhou,
China). A non-target scrambled oligonucleotide served as the
negative control (NC; Guangzhou RiboBio Co., Ltd., Guangzhou,
China). The short hairpin RNA (shRNA) sequences used in the present
study were as follows; shUCA1: 5′-GCCACCUACAUUAAAGCUAdTdT-3′ and
sh-control: 5′-CAGUACUUUUGUGUAGUACAA-3′. The SW1736 and KAT-18
cells (1×104 cells/well) were grown in 6-well plates
until the cells reached 50% confluency. The medium was replaced
with 1 ml fresh culture medium supplemented with 100 µl viral
supernatant (1×108 transducing units/ml) and 8 µg/ml
Polybrene (both Hanbio Biotechnology Co., Ltd., Shanghai, China)
for 24 h. The SW1736 and KAT-18 cells were further cultured in
medium containing 3 µg/ml puromycin for five passages and
subsequently used in further experiments. Individual
puromycin-resistant colonies were isolated during drug screening.
The miR-135a mimic, mimic control (miR-NC), miR-135a inhibitor
(anti-miR-135a) and inhibitor control (anti-NC) were synthesized by
Shanghai GeneChem Co., Ltd. (Shanghai, China). The sequences were
as follows: miR-135a mimic:
5′-UAUGGCUUUUUAUUCCUAUGUGAAGCAUAGGAAUAAAAAGCCAUAUU-3′; mimic
control: 5′-CAGUACUUUUGUGUAGUACAA-3′; miR-135a inhibitor:
5′-UCACAUAGGAAUAAAAAGCCAUA-3′; and inhibitor control:
5′-CAGUACUUUUGUGUAGUACAA-3′. The SW1736 and KAT-18 Cells
(2×105) were seeded into 6-well plates 24 h prior to
transfection, and a final concentration of 50 nM miR-135a mimic,
miR-NC, anti-miR-135a and anti-NC was used for transfection.
Transfection was performed using Lipofectamine® 2000
reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) according to the manufacturer's protocol. Subsequently,
transfection efficiencies were determined following 48 h using the
quantitative polymerase chain reaction (qPCR) method.
Western blot analysis
The expression level of protein c-myc was analyzed
by western blotting. SW1736 and KAT-18 cells were lysed to extract
the total protein using radioimmunoprecipitation assay lysis buffer
(Promega Corporation, Madison, WI, USA) according to the
manufacturer's protocol. Protein concentration was determined using
a bicinchoninic acid Protein Assay kit (Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. The same amount of
protein (40 µg) from each cell line was separated by 12% SDS-PAGE
and transferred onto polyvinylidene difluoride membranes. The
membranes were blocked using 3% non-fat milk for 30 min at 4°C. The
membranes were subsequently respectively incubated with various
diluted primary antibodies against c-myc (1:500; cat. no. 5605;
Cell Signaling Technology, Inc., Danvers, MA, USA) and mouse
monoclonal GAPDH (1:500; cat. no. AG019; Beyotime Institute of
Biotechnology, Haimen, China). Following incubation overnight at
4°C, horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (1:2,000; cat. no. A0216; Beyotime Institute of
Biotechnology) was added and incubated at room temperature for 2 h.
Specific bands were visualized with an enhanced chemiluminescent
reagent (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China) on an
autoradiographic film. For quantitative assay, images were analyzed
using ImageJ software (version 1.48u; National Institutes of
Health, Bethesda, MD, USA).
Reverse transcription (RT)-qPCR
Total RNA from cells was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Complementary
(c)DNA was synthesized from 500 ng total RNA, and a reaction
mixture (20 µl) containing 1 µg total RNA was reverse transcribed
to cDNA using a PrimeScript RT Reagent kit with gDNA Eraser (Takara
Biotechnology Co., Ltd., Dalian, China) according to the
manufacturer's protocol. qPCR was performed using a SYBR Premix Ex
Taq (Takara Biotechnology Co., Ltd.). The following conditions were
applied for detecting mRNAs: 95°C for 30 sec; followed by 40 cycles
of 95°C for 30 sec; 60°C for 30 sec; and 72°C for 30 sec. GAPDH was
used as an internal control for the detection of c-myc and UCA1. U6
was used as an internal control for the detection of miR-135a. The
relative mRNA expression levels were calculated as the inverse log
of ΔΔCq and normalized to the reference (9). The primer sequences were as follows:
UCA1, forward 5′-TTTGCCAGCCTCAGCTTAAT-3′ and reverse
5′-TTGTCCCCATTTTCCATCAT-3′; miR-135a, forward
5′-GCGCGTATGGCTTTTTATTCCT-3′ and reverse 5′-CAGTGCAGGGTCCGAGGTC-3′;
U6, forward 5′-TGCGGGTGCTCGCTTCGCAGC-3′ and reverse
5′-CCAGTGCAGGGTCCGAGGT-3′; c-myc, forward
5′-TCAAGAGGCGAACACACAAC-3′ and reverse 5′-GGCCTTTTCATTGTTTTCCA-3′;
GAPDH, forward 5′-CTGACCTGCCGTCTAGAAA-3′ and reverse
5′-GTGGTGTGACTTAGAGGGG-3′.
Cell viability assay
Cell viability was measured using a Cell Counting
Kit-8 (CCK-8) assay (Beyotime Institute of Biotechnology). SW1736
and KAT-18 cells were plated onto 96-well plates at a density of
3,000 cells/well. Following culture at the indicated time points
(0, 24, 48 and 72 h), 10 µl CCK-8 solution was added into each well
at 37°C. After 3 h, the absorbance of each well was measured using
a Multiskan MK3 device (Thermo Fisher Scientific, Inc.) at a
wavelength of 450 nm.
Colony formation assay
The cells (at a density of 400 cells/well)
transfected with shUCA1 and sh-control were placed into 6-well
plates. After 1 week of culture, the colonies were fixed with 10%
methanol for 15 min at room temperature and stained with 0.1%
crystal violet (Beyotime Institute of Biotechnology) for 20 min at
room temperature. Colonies with >50 cells were counted using the
CKX41 light microscope (magnification, ×100; Olympus Corporation,
Tokyo, Japan).
Cell migration and invasion assay
Cell migration and invasion were assessed by
performing a Transwell assay (Corning Incorporated, Corning, NY,
USA). For the invasion assay, the upper chambers were coated with
50 µl Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). Cells
were harvested at 48 h post-transfection. A total of
5×104 cells in 200 µl DMEM (HyClone; GE Healthcare Life
Sciences) were seeded in the upper chamber. The lower chamber was
filled with DMEM supplemented with 5% FBS. Following incubation at
37°C with 5% CO2 for 8 h (migration) or 12 h (invasion),
the cells on the lower chamber were fixed with 10% methanol for 15
min at room temperature and stained with crystal violet for 20 min
at room temperature. The number of the colonies that had migrated
through the pores was quantified by randomly counting 10
independent visual fields using the CKX41 light microscope
(magnification, ×200; Olympus Corporation, Tokyo, Japan).
Bioinformatics analysis
Targetscan software (http://www.targetscan.org/vert72/) was used to predict
the possible targets of miR-135a by searching miR-135a. The
putative miRNA binding sites on UCA1 sequences were predicted by an
RNAhybrid software program (http://bibiserv2.cebitec.uni-bielefeld.de/rnahybrid)
with the minimum free energy cutoff set at 22 kcal/mol.
Luciferase reporter assay
The binding site of miR-135a in the 3′-UTR of the
target mRNA was cloned into the pmirGLO Dual-Luciferase miRNA
Target Expression Vector (Promega Corporation), according to the
manufacturer's protocol. SW1736 and KAT-18 cells (4×105)
cultured in 24-well plates were co-transfected with luciferase
reporter plasmids [wild type (wt)-c-myc, mutant (mut)-c-myc
containing miR-135a binding site, wt-UCA1 and mut-UCA1 containing
miR-135a binding site] and miR-135a mimics or miR-135a inhibitor
(Shanghai GeneChem Co., Ltd., Shanghai, China) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). The sequences were as follows; miR-135a mimic:
5′-UAUGGCUUUUUAUUCCUAUGUGAAGCAUAGGAAUAAAAAGCCAUAUU-3′ and miR-135a
inhibitor: 5′-UCACAUAGGAAUAAAAAGCCAUA-3′. After 48 h
co-transfection, the luciferase activity was measured using a
dual-luciferase reporter assay system (Promega Corporation,
Madison, WI, USA) according to the manufacturer's protocol. The
data was normalized to Renilla luciferase activity.
Animal studies
All experiments involving animals were approved by
the Animal Care and Welfare Committee of Weifang People Hospital.
SW1736 cells (1×106) transfected with sh-control or
sh-UCA1 were subcutaneously injected into the right flanks of
four-week-old BALB⁄c athymic nude mice (n=12; female; weight range,
20–22 g; Yangzhou University, Yangzhou, China) and maintained in a
specific pathogen free environment with constant humidity (45–50%)
and constant temperature (25–27°C) under a 12 h light/dark cycle
with free access to food and water. Tumor volume (mm3)
was calculated every 3 days for 3 weeks using the formula V=0.5×
length × width2. Tumors were collected and images were
captured 3 weeks after inoculation.
Statistical analysis
All experiments were performed in triplicate. Unless
otherwise indicated, the data were presented as the mean ± standard
deviation. Statistical significance was determined using an
unpaired Student's t-test or one-way analysis of variance with
Tukey's test as the post hoc test using SPSS software version 13.0
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
UCA1 is upregulated in ATC tissues and
cell lines
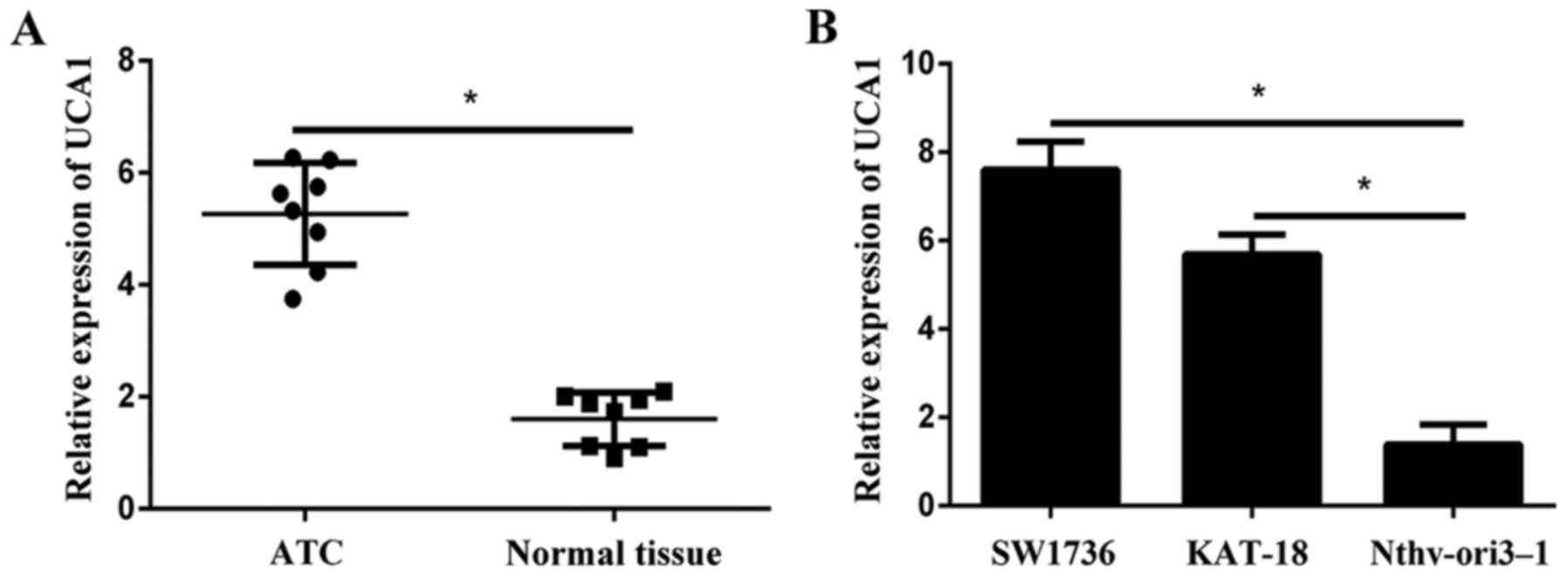
Initially the expression levels of UCA1 in eight
paired samples (ATC specimens and corresponding adjacent normal
tissues) were examined using RT-qPCR. Results revealed that UCA1
expression was significantly higher in tumor tissues compared with
adjacent normal tissues (Fig. 1A).
In addition, the expression of UCA1 in ATC cell lines (SW1736 and
KAT-18) was significantly increased compared with the normal human
follicular thyroid cell Nthy-ori3-1 (Fig. 1B).
UCA1 silencing suppresses cell
viability, proliferation, migration and invasion in vitro
Partial UCA1 knockdown was achieved by transfection
with shUCA1 in SW1736 and KAT-18, as verified using RT-qPCR
(Fig. 2A and B). As demonstrated
by CCK-8 assays, the cell viability of SW1736 and KAT-18 cells was
significantly inhibited after UCA1 knockdown (Fig. 2C and D). In addition, the colony
formation assay indicated that the proliferation of UCA1 knockdown
cells was inhibited (Fig. 2E and
F). The Transwell assay indicated that UCA1 knockdown
significantly inhibited cell migration and invasion (Fig. 2G and H).
miR-135a negatively regulates c-myc
expression
To further investigate the underlying mechanism by
which UCA1 regulates ATC progression, Targetscan 7.2 (http://www.targetscan.org/vert72/) was used. A
previous study has illustrated that c-myc is involved in the
progression of various tumors (10). c-myc silencing was demonstrated to
decrease the tumor viability, proliferation and migration in
vitro and in vivo (10). c-myc was predicted to be a target
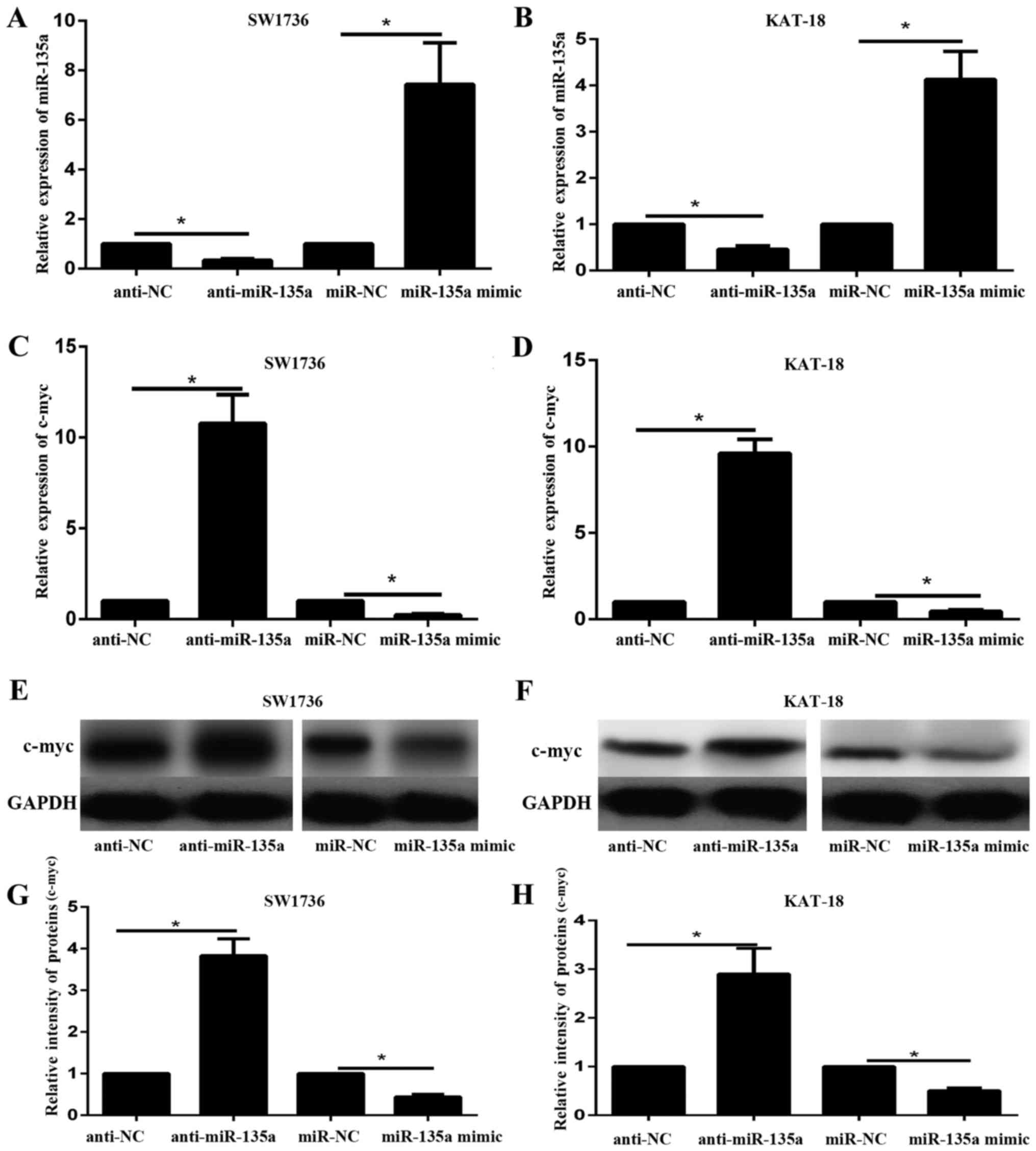
of miR-135a. SW1736 and KAT-18 cells were transfected with miR-135a
mimic, miR-NC, anti-miR-135a or anti-NC. RT-qPCR results
demonstrated the expression level of miR-135a in SW1736 and KAT-18
cell lines transfected with with miR-135a mimic, miR-NC,
anti-miR-135a and anti-NC (Fig. 3A and
B). miR-135a mimic significantly increased the expression of
miR-135a in SW1736 and KAT-18 cells; in contrast, anti-miR-135a
significantly decreased the expression of miR-135a in the two cell
lines (P<0.05). The mRNA and protein expression levels of c-myc
in SW1736 and KAT-18 cell lines in response to miR-135a
overexpression or inhibition were subsequently examined via RT-qPCR
and western blotting. The results indicated that the mRNA and
protein expression of c-myc decreased following miR-135a
overexpression and increased by miR-135a inhibition (Fig. 3C-H). These data suggested that
miR-135a may negatively regulate c-myc expression in ATC cell
lines.
UCA1 competes with c-myc for miR-135a
binding
The present study demonstrated that miR-135a
negatively regulated c-myc expression. The association between UCA1
and miR-135a was subsequently determined. The expression level of
UCA1 in response to miR-135a inhibition and overexpression in
SW1736 and KAT-18 cells was determined via RT-qPCR. The results
illustrated that UCA1 decreased following miR-135a overexpression
and increased by miR-135a inhibition (Fig. 4A and B). Furthermore, RT-qPCR
results indicated that miR-135a expression was significantly
increased following UCA1 silencing (Fig. 4C and D). These data suggested a
dual regulation between UCA1 and miR-135a. According to online
tools, UCA1 shared the same binding site in miR-135a with c-myc
(Fig. 4E). Luciferase assays were
performed to determine the association between miR-135a and UCA1.
Luciferase reporter gene vectors were constructed and
co-transfected into SW1736 and KAT-18 cells with miR-135a mimic or
anti-miR-135a. The luciferase activity was monitored using dual
luciferase assays. The results indicated that the luciferase
activity of wt-UCA1 and wt-c-myc vectors was significantly
decreased by miR-135a mimics and increased by the miR-135a
inhibitor. Following mutation in the predicted binding sites of
miR-135a, the alterations in the luciferase activity were abolished
(Fig. 4F and G). These data
suggested that UCA1 and c-myc may bind to miR-135a, and UCA1 may
compete with c-myc for miR-135a binding.
 | Figure 4.UCA1 competes with c-myc for miR-135a
binding. UCA1 expression in response to miR-135a overexpression and
inhibition in (A) SW1736 and (B) KAT-18 cells was examined using
RT-qPCR. *P<0.05. miR-135a expression in response to UCA1
knockdown in (C) SW1736 and (D) KAT-18 cells was determined by
RT-qPCR. *P<0.05 vs. the sh-control group. (E) Wt and mut UCA1
containing 5 bp mutation in the predicted binding sites of
miR-135a, or c-myc 3′UTR (wt-c-myc 3′UTR and mut-c-myc 3′UTR
containing a 5 bp mutation in the predicted binding sites of
miR-135a) luciferase reporter gene vectors were constructed. After
culturing overnight, (F) SW1736 and (G) KAT-18 cells were
co-transfected with the indicated vectors and miR-135a mimics or
anti-miR-135a. Luciferase assays were performed 48 h after
transfection using the Dual Luciferase Reporter Assay system to
determine the luciferase activity. *P<0.05. Wt, wild type; mut,
mutant; UCA1, urothelial carcinoma-associated 1; miR, microRNA;
3′UTR, 3′ untranslated region; c-myc, c-myc proto-oncogene;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; anti-miR-135a, miR-135a inhibitor. |
UCA1 regulates c-myc expression via
miR-135a
The present study measured the protein expression
levels of c-myc in SW1736 and KAT-18 cells, in response to UCA1
silencing. The RT-qPCR and western blotting results indicated that
UCA1 knockdown significantly decreased the expression of c-myc at
the mRNA and protein level, respectively (Fig. 5). These results suggested that UCA1
may regulate tumor progression through c-myc. To validate whether
UCA1 regulated c-myc expression through miR-135a, the present study
co-transfected SW1736 and KAT-18 cells with the miR-135a inhibitor
and shUCA1, and examined the expression of c-myc by qPCR and
western blot. The results demonstrated that the mRNA and protein
expression levels of c-myc were increased by miR-135a inhibition
and reduced by shUCA1 (Fig. 5A-D).
The promotive effect of the miR-135a inhibitor on the expression of
c-myc may be partially abolished by shUCA1 (Fig. 5A-D). These data suggested that UCA1
regulated c-myc expression through miR-135a.
Knockdown of UCA1 inhibits tumor
growth in vivo
To further illustrate the effects of UCA1 on ATC
growth in vivo, the present study used a xenograft model in
which SW1736 cells transfected with shUCA1 or sh-NC were
subcutaneously injected into the flanks of athymic mice and were
allowed to develop measurable tumors. The results demonstrated that
tumors formed by transfected shUCA1 grew more slowly compared with
those formed by transfected sh-NC (Fig. 6A-C). Furthermore, RT-qPCR indicated
that the c-myc expression levels in the shUCA1 tumor xenografts
were lower compared with those in the control xenografts (Fig. 6D). In addition, the RT-qPCR results
indicated that the expression level of miR-135a was significantly
downregulated in shUCA1-transfected tumors compared with
sh-NC-transfected tumors (Fig.
6E).
Discussion
The present study demonstrated the oncogenic role of
UCA1 in the progression of ATC. It was revealed that expression
levels of UCA1 significantly increased in ATC cell lines and
tissues. The present study also indicated that UCA1 knockdown
significantly inhibited cell viability, proliferation, migration
and invasion in ATC in vitro and in vivo. Finally, it
was predicted that UCA1 binds to miR-135a and modulates c-myc
expression.
LncRNAs are transcribed RNA molecules that are
>200 nucleotides in length and lack significant protein-coding
potential, however, they may regulate protein-coding genes at
epigenetic, transcriptional and post-transcriptional levels, and
serve a critical role in physiological processes (11). Previous studies have demonstrated
that a variety of lncRNAs are frequently aberrantly expressed in
cancer, and the differential expression of lncRNAs is closely
associated with tumor progression, serving roles of oncogenes or
tumor suppressor genes (12). To
date, only a number of lncRNAs, including lncRNA LINC00312, have
been demonstrated to be involved in TC development and progression
(13). UCA1 is a recently
identified lncRNA (8). Previous
studies have identified that UCA1 promoted tumor progression in a
wide range of tumor types, including bladder cancer, breast cancer,
hepatocellular carcinoma, colorectal cancer and gastric cancer
(14–18). However, the role and mechanism of
UCA1 in ATC has not been illustrated. The present study initially
demonstrated that UCA1 expression was significantly increased in
ATC tissues and cell lines. The data suggested that UCA1 served as
an oncogene in ATC, consistent with previous studies in the other
tumor types (8,14–18).
A previous study illustrated that UCA1 regulated the growth and
metastasis of pancreatic cancer by binding to miR-135a (19). The present study illustrated the
dual interaction of UCA1 and miR-135a in ATC, possibly for the
first time. UCA1 and miR-135a may regulate each other in a negative
way. To further demonstrate the underlying molecular mechanisms of
the UCA1/miR-135a axis-induced cell proliferation in SW1736 and
KAT-18 cells, the present study particularly focused on c-myc for
further experiments.
The c-myc protein encodes a basic helix-loop-helix
leucine zipper transcription factor and acts as an important
regulator of several cellular processes, including protein
synthesis, cell growth and proliferation in various cell types
(20). Furthermore, the c-myc
oncogene was overexpressed in a number of types of human cancer,
including anaplastic thyroid cancer, and targeting of the c-myc
protein may be a potential treatment modality for human ATC
(10,21). A previous study demonstrated that
miR-135a inhibited cancer cell proliferation by targeting the c-myc
oncogene in renal cell carcinoma (22). The present study measured the
protein and mRNA expression levels of c-myc and UCA1. UCA1
knockdown reduced c-myc expression at the protein and mRNA level,
indicating the involvement of c-myc in the regulation of UCA1 cell
proliferation and invasion. Furthermore, by dual luciferase assays,
the present study illustrated that UCA1 may bind to miR-135a, and
miR-135a may directly bind to the 3′UTR of c-myc. The results
suggested that UCA1 may compete with c-myc for miR-135a binding,
and inhibit miR-135a and promote c-myc expression, to affect ATC
progression.
In conclusion, the UCA1/miR-135a/c-myc axis may
serve a role in ATC cell proliferation and invasion, and may
provide a potential therapeutic application in patients with
ATC.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Foundation of
Shandong People and Family Planning Commission (grant no.
2015WSA07008).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YW and DL conceived and designed the experiments. YW
wrote and revised the manuscript. ZH conducted all experiments. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of Weifang People's Hospital (Weifang, China). All
patients provided written informed consent.
Patient consent for publication
All patients provided written informed consent for
the publication of any associated data and accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xing M: Molecular pathogenesis and
mechanisms of thyroid cancer. Nat Rev Cancer. 13:184–199. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nagaiah G, Hossain A, Mooney CJ,
Parmentier J and Remick SC: Anaplastic thyroid cancer: A review of
epidemiology, pathogenesis, and treatment. J Oncol.
2011:5423582011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Neill JP and Shaha AR: Anaplastic
thyroid cancer. Oral Oncol. 49:702–706. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ezkurdia I, Juan D, Rodriguez JM, Frankish
A, Diekhans M, Harrow J, Vazquez J, Valencia A and Tress ML:
Multiple evidence strands suggest that there may be as few as
19,000 human protein-coding genes. Hum Mol Genet. 23:5866–5878.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Geisler S and Coller J: RNA in unexpected
places: Long non-coding RNA functions in diverse cellular contexts.
Nat Rev Mol Cell Biol. 14:699–712. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee JT and Bartolomei MS: X-inactivation,
imprinting, and long noncoding RNAs in health and disease. Cell.
152:1308–1323. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang F, Li X, Xie X, Zhao L and Chen W:
UCA1, a non-protein-coding RNA up-regulated in bladder carcinoma
and embryo, influencing cell growth and promoting invasion. FEBS
Lett. 582:1919–1927. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Enomoto K, Zhu X, Park S, Zhao L, Zhu YJ,
Willingham MC, Qi J, Copland JA, Meltzer P and Cheng SY: Targeting
MYC as a therapeutic intervention for anaplastic thyroid cancer. J
Clin Endocrinol Metab. 102:2268–2280. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mercer TR and Mattick JS: Structure and
function of long noncoding RNAs in epigenetic regulation. Nat
Struct Mol Biol. 20:300–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin CY and Xu HM: Novel perspectives of
long non-coding RNAs in esophageal carcinoma. Carcinogenesis.
36:1255–1262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu K, Huang W, Yan DQ, Luo Q and Min X:
Overexpression of long intergenic noncoding RNA LINC00312 inhibits
the invasion and migration of thyroid cancer cells by
down-regulating microRNA-197-3p. Biosci Rep. 37:pii: BSR20170109.
2017. View Article : Google Scholar
|
|
14
|
Zhang S, Dong X, Ji T, Chen G and Shan L:
Long non-coding RNA UCA1 promotes cell progression by acting as a
competing endogenous RNA of ATF2 in prostate cancer. Am J Transl
Res. 9:366–375. 2017.PubMed/NCBI
|
|
15
|
Wang X, Yang B and Ma B: The
UCA1/miR-204/Sirt1 axis modulates docetaxel sensitivity of prostate
cancer cells. Cancer Chemother Pharmacol. 78:1025–1031. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiao C, Song Z, Chen J, Zhong J, Cai W,
Tian S, Chen S, Yi Y and Xiao Y: lncRNA-UCA1 enhances cell
proliferation through functioning as a ceRNA of Sox4 in esophageal
cancer. Oncol Rep. 36:2960–2966. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang ZQ, Cai Q, Hu L, He CY, Li JF, Quan
ZW, Liu BY, Li C and Zhu ZG: Long noncoding RNA UCA1 induced by SP1
promotes cell proliferation via recruiting EZH2 and activating AKT
pathway in gastric cancer. Cell Death Dis. 8:e28392017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang X, Gao Z, Liao J, Shang M, Li X, Yin
L, Pu Y and Liu R: lncRNA UCA1 inhibits esophageal squamous-cell
carcinoma growth by regulating the Wnt signaling pathway. J Toxicol
Environ Health A. 79:407–418. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang X, Gao F, Zhou L, Wang H, Shi G and
Tan X: UCA1 regulates the growth and metastasis of pancreatic
cancer by sponging MiR-135a. Oncol Res. 25:1529–1541. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eilers M and Eisenman RN: Myc's broad
reach. Genes Dev. 22:2755–2766. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pelengaris S, Khan M and Evan G: c-MYC:
More than just a matter of life and death. Nat Rev Cancer.
2:764–776. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yamada Y, Hidaka H, Seki N, Yoshino H,
Yamasaki T, Itesako T, Nakagawa M and Enokida H: Tumor-suppressive
microRNA-135a inhibits cancer cell proliferation by targeting the
c-MYC oncogene in renal cell carcinoma. Cancer Sci. 104:304–312.
2013. View Article : Google Scholar : PubMed/NCBI
|