Introduction
Globally, lung cancer is the second most common
newly diagnosed cancer and the leading cause of cancer-associated
mortality. Non-small cell lung cancer (NSCLC) accounts for ~83% of
all newly diagnosed lung cancers (1). Numerous patients with NSCLC are
diagnosed at an advanced stage, thus surgery may not be suitable;
chemotherapy is a major option to treat such patients (2). First-generation epidermal growth
factor receptor-tyrosine kinase inhibitors (EGFR-TKIs), including
erlotinib and gefitinib have demonstrated notable response rates
and benefits in progression-free survival compared with first-line
conventional platinum-based chemotherapy (3). However, EGFR-TKIs have become
first-line therapy drugs for patients with NSCLC harboring
EGFR-activating mutations; the majority of patients with an initial
response to erlotinib or gefitinib will eventually develop
resistance due to the effects of drug-resistant mutations, with
T790M mutations accounting for ~60% of all resistance (4,5).
Additionally, patients with NSCLC and wild-type EGFR are primarily
resistant to erlotinib and gefitinib (6,7).
Thus, potential chemosensitizers are required to sensitize
wild-type EGFR NSCLC cells to EGFR-TKI treatment.
Shikonin, a natural naphthoquinone derivative
isolated from the traditional medical herb Lithospermum
erythrorhizon, has antitumor, anti-inflammatory, antimicrobial,
and antithrombotic properties (8).
Shikonin has been reported to exhibit antitumor properties via the
generation of reactive oxygen species (ROS), activating endoplasmic
reticulum (ER) stress, inducing caspase-dependent apoptosis and
inhibiting angiogenesis (9–12).
Furthermore, shikonin can sensitize cancer cells to numerous types
of treatment with cytotoxic drugs (11,13,14).
In addition, shikonin can also suppress EGFR signaling and enhance
the anti-glioblastoma efficacy of erlotinib via inhibiting EGFR
in vitro (15). Shikonin
inhibits gefitinib-resistant NSCLC cells by inhibiting thioredoxin
reductase and activating the EGFR proteasomal degradation pathway
in vitro (16); however,
whether shikonin can enhance the anticancer activity of erlotinib
or gefitinib in wild-type EGFR NSCLC cells requires further
investigation.
In the present study, the anticancer activity of
combining shikonin plus erlotinib/gefitinib was evaluated in
wild-type EGFR NSCLC cells in vitro and in vivo. In
addition, the findings of the resent study demonstrated that
shikonin could promote erlotinib/gefitinib-induced apoptosis in
wild-type EGFR NSCLC cells. ER stress and ROS were associated with
shikonin plus erlotinib/gefitinib-induced apoptosis. Thus, the
present study proposed that shikonin may be a potential sensitizer
to enhance the anticancer efficacy of erlotinib/gefitinib in
wild-type EGFR NSCLC cells primarily resistant to
erlotinib/gefitinib treatment.
Materials and methods
Materials
Shikonin (cat. no. S8279) and gefitinib (cat. no.
S1025) were obtained from Selleck Chemicals (Houston, TX, USA).
Erlotinib (cat. no. 183321-74-6) was obtained from LC Laboratories
(Woburn, MA, USA). N-acetylcysteine (NAC; cat. no. A0737) was
obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Cell culture
A549 (cat. no. TCHu150), NCI-H1299 (cat. no.
TCHu160), NCI-H460 (cat. no. TCHu205), 95-D (cat. no. TCHu 61) and
NCI-H1650 (cat. no. TCHu152) cells were obtained from the Shanghai
Institute of Biochemistry and Cell Biology (Chinese Academy of
Sciences, Shanghai, China). NCI-H1650, NCI-H1299, NCI-H460 and 95-D
cells were cultured in RPMI-1640 medium containing 10% fetal bovine
serum (FBS); A549 cells were maintained in Ham's F12 medium+10%
FBS.
Cell viability assay
NSCLC cells were treated with serial dilutions of
gefitinib, erlotinib and shikonin for 72 h. A sulforhodamine blue
(SRB) assay was used to test the proliferation of cancer cells as
described previously (17).
Determination of cell colony
formation
NSCLC cells were plated at ~700 cells per dish. The
next day, compounds (gefitinib, erlotinib, shikonin,
shikonin+erlotinib and shikonin+ gefitinib) were added into the
NSCLC cells; the medium containing 10% FBS plus the compounds was
replaced every 3 days. Dishes were stained with 0.5% crystal violet
after 14 days treatment. Finally, the colonies were scored and
photographed.
Propidium iodide (PI) staining and
determination of mitochondrial membrane depolarization
Cancer cells (3×105/well) were exposed to
the drugs, harvested and washed with PBS. Then, PI staining was
used to detect apoptosis, and the mitochondrial membrane
depolarization was determined by
5,5′,6,6′tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine
iodide staining as described previously (17).
Western blot analysis
Western blotting was conducted as described
previously (17). The primary
antibodies below were used in the present study: Induced myeloid
leukemia cell differentiation protein 1 (Mcl-1; cat. no. sc-819),
poly (ADP-ribose) polymerase (PARP; cat. no. sc-7150) and caspase-3
(cat. no. sc-7148) were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Antibodies for cleaved-caspase-3 (cat. no.
9661), activating transcription factor 4 (ATF-4; cat. no. 11815),
phosphorylated (p)-eukaryotic initiation factor 2α (eIf2α; cat. no.
3398) and eIf2α (cat. no. 5324) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). β-actin (cat. no. BD-612656)
was obtained from BD Biosciences (Franklin Lakes, NJ, USA).
Animals and antitumor activity in
vivo
Human lung cancer A549 ×enografts were established
as described previously (18). The
nude mice were randomized to 6 groups and then treated with
vehicle, shikonin [20 mg/kg, intraperitoneal (i.p.) administration]
once per week, erlotinib [50 mg/kg, intragastric (i.g.)
administration] twice per week, gefitinib (50 mg/kg, i.g.
administration) twice per week, shikonin (20 mg/kg, i.p., once per
week) + erlotinib (50 mg/kg, i.g., twice per week) and shikonin (20
mg/kg, i.p., once per week) + gefitinib (50 mg/kg, i.g., twice per
week) for 35 days. All animal protocols were performed in
accordance with the National Institutes of Health Guide for the
Care and Use of Laboratory Animals, and were approved by the
Zhejiang University City College Animal Care and Use committee.
This article does not contain any studies with human participants
performed by any of the authors.
Statistical analysis
One-way analysis of variance followed with the
Tukey's post hoc test was used to examine the significance of
differences among groups, and two-tailed student's t tests was used
to examine the significance of differences two groups. Data points
in graphs were presented as the mean ± standard deviation.
*P<0.05 was considered to indicate a statistically significant
difference, **P<0.01 and ***P<0.001 were considered to
indicate a highly statistically significant difference. For the SRB
assay, combination index (CI) values were calculated using Calcusyn
(http://www.biosoft.com/w/calcusyn.htm) and the mean CI
values were chosen for presentation (19). CI< 0.9 indicated synergism; 0.9
to 1.10, additive and >1.10, antagonism.
Results
Shikonin sensitizes the
antiproliferative effects of erlotinib/gefitinib in wild-type EGFR
NSCLC cells
Firstly, antiproliferative effects were investigated
by combining shikonin with erlotinib/gefitinib in vitro. As
expected, shikonin could potentiate erlotinib/gefitinib-induced
cytotoxicity in NSCLC cells (Fig.
1). CI values were calculated with Calcusyn software; treatment
with shikonin plus erlotinib/gefitinib revealed synergistic
cytotoxic effects in NSCLC cells, with the mean CI values below
0.9. In addition, the combined treatment of shikonin plus
erlotinib/gefitinib was markedly more effective in attenuating
colony formation than either agent alone (Fig. 2). Thus, shikonin plus
erlotinib/gefitinib was effective in limiting colony formation and
cell growth in wild-type EGFR NSCLC cells.
Shikonin promotes
erlotinib/gefitinib-induced apoptosis
The present study also investigated whether shikonin
could promote erlotinib/gefitinib-induced apoptosis in wild-type
EGFR NSCLC cells; PI staining was used to detect apoptosis.
Consistent with the cytotoxicity data, it was demonstrated that
shikonin could promote erlotinib-induced apoptosis in NSCLC cells
(Fig. 3A, upper panel and 3B). In
addition, shikonin plus erlotinib resulted in greater mitochondrial
membrane depolarization than either drug alone in wild-type EGFR
NSCLC cells (Fig. 3A, lower panel
and C). Furthermore, shikonin was revealed to potentiate
mitochondrial-mediated apoptosis induced by gefitinib in wild-type
EGFR NSCLC cells (Fig. 4).
Additionally, shikonin plus erlotinib/gefitinib markedly induced
cleavage of caspase-3 and PARP, and inhibition of Mcl-1 in NSCLC
cells (Fig. 5A). Collectively, the
results of the present study suggested that shikonin could promote
erlotinib/gefitinib-induced apoptosis via the mitochondrial
apoptotic pathway in wild-type EGFR NSCLC cells.
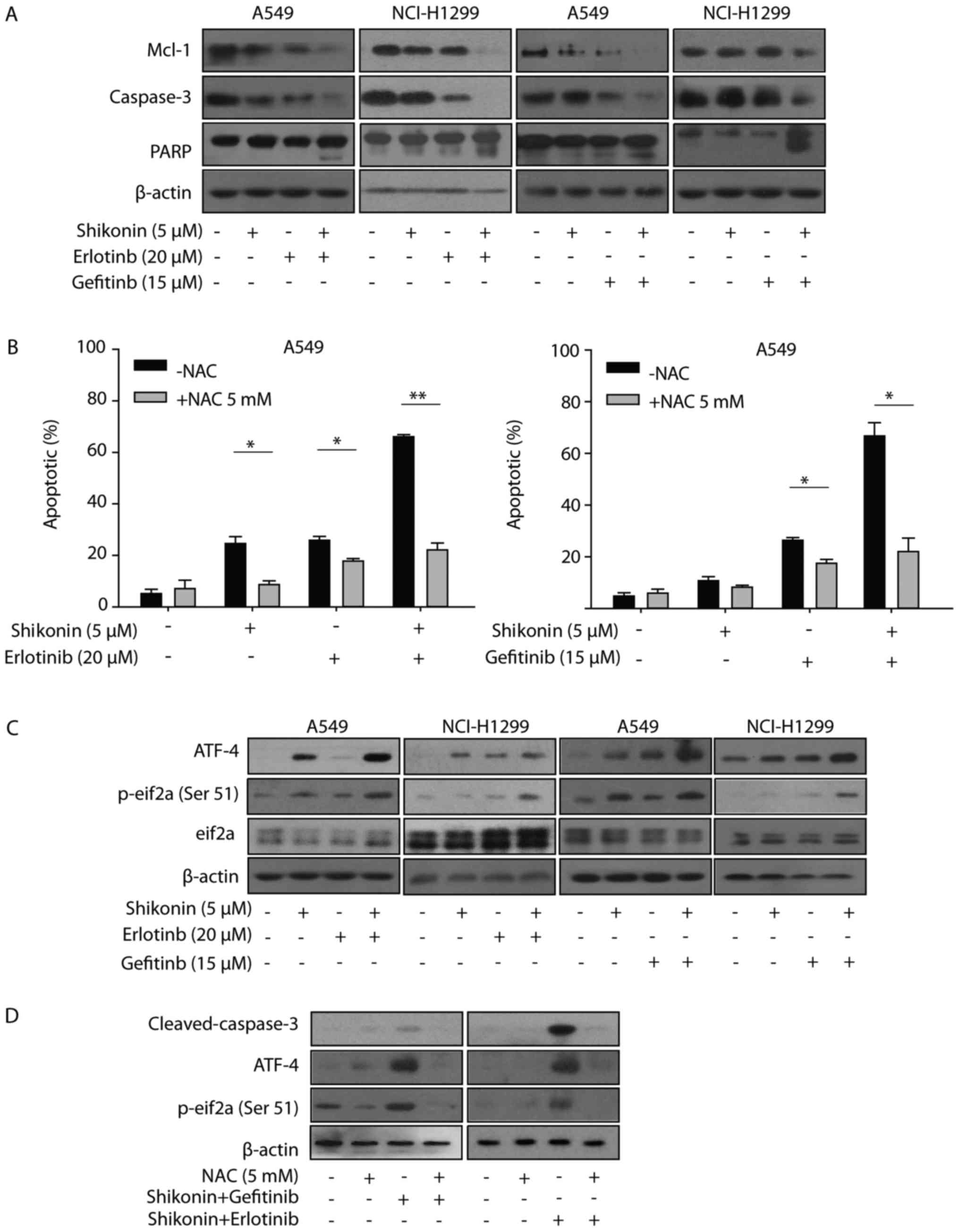 | Figure 5.ER stress and ROS are involved in
shikonin plus erlotinib/gefitinib treatment-induced apoptosis. (A)
A549/NCI-H1299 cells were treated with compounds at indicated
concentrations for 48 h, after which the expression of proteins
were detected by western blotting. (B) A549 cells were pre-treated
with NAC (5 mM) for 1 h, and then treated with 5 µM shikonin and/or
20 µM erlotinib (left panel), 5 µM shikonin and/or 15 µM gefitinib
(right panel) for 48 h. PI staining was used to detect apoptosis.
(C) Cells were treated with compounds at indicated concentrations
for 24 h, after which the expression of proteins were detected by
western blotting. (D) A549 cells were pre-treated with NAC (5 mM)
for 1 h and then treated with shikonin+erlotinib or
shikonin+gefitinib for 24 h, after which the expression of proteins
were detected by western blotting. *P<0.05 and **P<0.01.
Mcl-1, myeloid leukemia cell differentiation protein 1; PARP, poly
(ADP-ribose) polymerase; ER, endoplasmic reticulum; ROS, reactive
oxygen species; NAC, N-acetylcysteine; ATF-4, activating
transcription factor 4; PI, propidium iodide; p-eIf2α,
phosphorylated-eukaryotic initiation factor 2α. |
ER stress and ROS are associated with
shikonin plus erlotinib/gefitinib-induced apoptosis in vitro
Shikonin induced ROS generation, activated ER stress
and induced mitochondrial-associated apoptosis in human prostate
cancer cells (12). Therefore, the
present study first examined whether shikonin could induce
ROS-mediated ER stress in wild-type EGFR NSCLC cells. As presented
in Fig. 5B, NAC was observed to
reverse the apoptosis induced by shikonin treatment. Furthermore,
NAC was demonstrated to also protect cells from apoptosis induced
by shikonin plus erlotinib/gefitinib. These data indicated that ROS
may be associated with the enhanced apoptosis induced by shikonin
plus erlotinib/gefitinib. The present study also detected the
expression levels of ER stress marker proteins (eIF2a and ATF4),
which may be activated during ER stress. Shikonin increased the
expression levels of p-eIF2a and ATF4 in the absence of
erlotinib/gefitinib, and this effect was markedly enhanced in the
presence of erlotinib/gefitinib (Fig.
5C). In addition, NAC pretreatment inhibited the activation of
ATF-4 and eIF2a induced by shikonin plus erlotinib/gefitinib.
Furthermore, the activation of caspase-3 induced by shikonin plus
erlotinib/gefitinib was completely inhibited by NAC pretreatment
(Fig. 5D). Thus, the data of the
present study suggested that ROS-mediated ER stress was involved in
shikonin plus erlotinib/gefitinib-induced apoptosis in wild-type
EGFR NSCLC cells.
Shikonin sensitizes the anti-NSCLC
effects of erlotinib/gefitinib in vivo
Finally, the in vivo activity of shikonin
plus erlotinib/gefitinib was detected on A549 ×enograft model. The
dose of erlotinib/gefitinib selected in the present study was not
expected to exhibit significant anticancer efficacy. As presented
in Fig. 6, the low dose of
erlotinib/gefitinib did not result in significant tumor growth
inhibition; however, combining shikonin and erlotinib/gefitinib
significantly inhibited tumor growth [mean Relative Tumor Volume
(RTV)shikonin plus erlotinib group=3.40 vs. mean
RTVcontrol group=11.58, P<0.01; mean RTV
shikonin plus gefitinib group=4.88 vs. mean
RTVcontrol group=11.58, P<0.05] (Fig. 6). Thus, shikonin sensitized the
anti-NSCLC effects of erlotinib/gefitinib on A549 ×enografts in
vivo.
Discussion
The first generation EGFR-TKIs, including erlotinib
and gefitinib, are used as the first-line treatment of metastatic
NSCLC with EGFR exon 19 deletions or exon 21 (L858R) (20,21);
however, patients with NSCLC and wild-type EGFR are primarily
resistant to erlotinib and gefitinib (6). The T790M mutation in EGFR kinase is
the most common (~50%) acquired resistance mechanism to first- and
second-generation EGFR-TKIs (22).
The third generation EGFR-TKIs (rociletinib, osimertinib,
HM61713/BI1482694, ASP8273 and EGF816) can overcome EGFR-TKIs
resistance mediated by EGFR T790M (23). However, there are numerous
mechanisms underlying non-T790M resistance against EGFR-TKIs,
including mesenchymal-epithelial transition factor receptor
tyrosine kinase (MET) gene amplification, erb B2 receptor tyrosine
kinase gene amplification, reactivation of mitogen-activated
protein kinase kinase (MEK-extracellular signal-regulated kinase
(ERK) or phosphoinositide 3-kinase (PI3K)-protein kinase B (AKT)
signaling pathways (24).
Investigating novel combinations of treatment regimens is one of
main therapeutic strategies to overcome EGFR-TKI-associated
resistance, including primary and acquired resistance, and prolong
survival durations for patients with NSCLC. Shikonin may inhibit
numerous molecular markers associated with EGFR-TKI resistance,
including those involved in the MET-ERK and PI3K-AKT signaling
pathways (25,26). In addition, shikonin may be a
sensitizer for erlotinib/gefitinib in vitro (15,16).
In the present study, shikonin was reported to potentiate the
anticancer effects of erlotinib/gefitinib on wild-type EGFR NSCLC
cells resistant to erlotinib and gefitinib both in vitro and
in vivo; however, whether shikonin may sensitize other
EGFR-TKIs requires further investigation. Additionally, the dual
targeting of EGFR with cetuximab and EGFR-TKIs may be an effective
strategy to overcome T790M-mediated drug resistance (27). As shikonin can induce EGFR
proteasomal degradation, further investigation is needed to
evaluate the anticancer efficiency of shikonin plus EGFR-TKIs on
NSCLC cells with EGFR T790M.
Heterogeneous nuclear ribonucleoprotein A1
(hnRNPA1), an abundant ubiquitous nuclear protein, has numerous
functions associated with RNA metabolism (28). Shikonin can induce cell death via
direct binding-interference with hnRNPA1 (29). HnRNPA1 binds to the 5′untranslated
region of the mRNA encoding the RON receptor tyrosine kinase and
regulates its expression; RON is a member of the c-MET receptor
tyrosine kinase family, which has been reported to correlate with
EGFR-TKI-associated resistance and poor clinical outcome (30). In addition, co-targeting EGFR and
c-MET has been reported to efficiently overcome EGFR-TKI resistance
in NSCLC cells (31). Thus, the
present study hypothesized that shikonin may suppress MET via
hnRNPA1 inhibition and sensitive the anticancer activity of
EGFR-TKI, however, further research is required to test this
hypothesis.
ER stress induces the unfolded protein response
(UPR), which is initiated by the activation of three main stress
sensors, including inositol-requiring protein-1α, PERK protein
kinase RNA-like ER kinase, and ATF6 (32). ER stress serves a dual role in
determining cell survival or cell death. The UPR is primarily a
pro-survival process; sustained and/or prolonged stress may result
in the induction of cell death (33). ER stress can promote the survival
of cancer cells that are tolerant to EGFR-TKIs and has been
associated with the pro-survival process (34). In addition, increased levels of
intracellular ROS can also induce ER stress and UPR activation, and
ROS-mediated ER stress can induce mitochondria-mediated apoptosis
via PERK (35–37). Shikonin induces the apoptosis of
prostate cancer cells via the induction of ROS production and ER
stress (12). In the present
study, it was reported that shikonin may induce apoptosis via
ROS-mediated ER stress in wild-type EGFR NSCLC cells. Furthermore,
erlotinib/gefitinib can also induce ER stress; shikonin plus
erlotinib/gefitinib was more effective in activating ER stress than
either agent alone. These findings indicated that ROS-mediated ER
stress may be associated with the enhanced mitochondrial apoptosis
induced by shikonin plus erlotinib/gefitinib, and that shikonin may
promote the transition of cytoprotective ER stress-inducing
EGFR-TKI tolerance to apoptotic-promoting ER stress. However,
further investigation is required to determine the molecular
mechanism underlying the transition between cytoprotective ER
stress and apoptosis-promoting ER stress induced by shikonin plus
erlotinib/gefitinib.
In summary, the findings of the present study
demonstrated that shikonin could sensitize the anticancer activity
of erlotinib/gefitinib in wild-type EGFR NSCLC cells both in
vitro and in vivo. Furthermore, ROS-mediated ER stress
may be associated with the enhanced anti-NSCLC effects induced by
shikonin plus erlotinib/gefitinib. The present study suggested that
combining shikonin with EGFR-TKIs may be a novel theray for
wild-type EGFR NSCLC and may also provide insight into the
application of shikonin in other clinical research fields.
Acknowledgements
Not applicable.
Funding
The present study was funded by Zhejiang Province
Medical Key Discipline Construction (grant no. 2018-2-03), National
natural science foundation of china (grant no. 81702887), Hangzhou
major science and technology project (grant no. 20172016A01), Fund
of Hangzhou medical key discipline construction (grant no.
2017-51-07), Zhejiang provincial foundation of natural science
(grant no. LQ16H310004), Public-service technology research plan of
Zhejiang province (grant nos. 2015C33269 and 2016C33210),
High-level talents coming back from abroad innovation and
entrepreneurship program in Hangzhou (grant no. 2051), Zhejiang
provincial program for the cultivation of high-level innovative
health talents (grant no. 2010-190-4), Scientific and technological
developing scheme of Hangzhou city (grant nos. 20150733Q14 and
20140633B03).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
Y-LL, XH, Q-YL, FW, BZ, KD and B-QT performed the
experiments. N-ML and CZ conceived and designed the study. CZ wrote
the manuscript. All authors read and approved the manuscript.
Ethics approval and consent to
participate
All animal manipulations were performed in
accordance with the National institutes of health guide for the
care and use of laboratory animals, and were approved by the
Zhejiang University City College animal care and use committee.
This article does not contain any studies with human participants
performed by any of the authors.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Davies J, Patel M, Gridelli C, de Marinis
F, Waterkamp D and McCusker ME: Real-world treatment patterns for
patients receiving second-line and third-line treatment for
advanced non-small cell lung cancer: A systematic review of
recently published studies. PLoS One. 12:e01756792017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen Z, Fillmore CM, Hammerman PS, Kim CF
and Wong KK: Non-small-cell lung cancers: A heterogeneous set of
diseases. Nat Rev Cancer. 14:535–546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang T, Qiao M, Zhou F, Ren S, Su C and
Zhou C: Effect of combined therapy inhibiting EGFR and VEGFR
pathways in non-small-cell lung cancer on progression-free and
overall survival. Clin Lung Cancer. 18:421–431.e3. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takeda M and Nakagawa K: Toxicity profile
of epidermal growth factor receptor tyrosine kinase inhibitors in
patients with epidermal growth factor receptor gene
mutation-positive lung cancer. Mol Clin Oncol. 6:3–6. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Han W and Du Y: Recent development of the
second and third generation irreversible epidermal growth factor
receptor inhibitors. Chem Biodivers. 14:2017. View Article : Google Scholar :
|
|
6
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu CQ, da Cunha Santos G, Ding K,
Sakurada A, Cutz JC, Liu N, Zhang T, Marrano P, Whitehead M, Squire
JA, et al: Role of KRAS and EGFR as biomarkers of response to
erlotinib in national cancer institute of canada clinical trials
group study BR.21. J Clin Oncol. 26:4268–4275. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jeung YJ, Kim HG, Ahn J, Lee HJ, Lee SB,
Won M, Jung CR, Im JY, Kim BK, Park SK, et al: Shikonin induces
apoptosis of lung cancer cells via activation of FOXO3a/EGR1/SIRT1
signaling antagonized by p300. Biochim Biophys Acta.
1863:2584–2593. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou Z, Lu B, Wang C, Wang Z, Luo T, Piao
M, Meng F, Chi G, Luo Y and Ge P: RIP1 and RIP3 contribute to
shikonin-induced DNA double-strand breaks in glioma cells via
increase of intracellular reactive oxygen species. Cancer Lett.
390:77–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Komi Y, Suzuki Y, Shimamura M, Kajimoto S,
Nakajo S, Masuda M, Shibuya M, Itabe H, Shimokado K, Oettgen P, et
al: Mechanism of inhibition of tumor angiogenesis by
beta-hydroxyisovalerylshikonin. Cancer Sci. 100:269–277. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liang W, Cai A, Chen G, Xi H, Wu X, Cui J,
Zhang K, Zhao X, Yu J, Wei B and Chen L: Shikonin induces
mitochondria-mediated apoptosis and enhances chemotherapeutic
sensitivity of gastric cancer through reactive oxygen species. Sci
Rep. 6:382672016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gara RK, Srivastava VK, Duggal S, Bagga
JK, Bhatt M, Sanyal S and Mishra DP: Shikonin selectively induces
apoptosis in human prostate cancer cells through the endoplasmic
reticulum stress and mitochondrial apoptotic pathway. J Biomed Sci.
22:262015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li W, Liu J, Jackson K, Shi R and Zhao Y:
Sensitizing the therapeutic efficacy of taxol with shikonin in
human breast cancer cells. PLoS One. 9:e940792014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song J, Zhao Z, Fan X, Chen M, Cheng X,
Zhang D, Wu F, Ying X and Ji J: Shikonin potentiates the effect of
arsenic trioxide against human hepatocellular carcinoma in vitro
and in vivo. Oncotarget. 7:70504–70515. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao Q, Kretschmer N, Bauer R and Efferth
T: Shikonin and its derivatives inhibit the epidermal growth factor
receptor signaling and synergistically kill glioblastoma cells in
combination with erlotinib. Int J Cancer. 137:1446–1456. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li X, Fan XX, Jiang ZB, Loo WT, Yao XJ,
Leung EL, Chow LW and Liu L: Shikonin inhibits gefitinib-resistant
non-small cell lung cancer by inhibiting TrxR and activating the
EGFR proteasomal degradation pathway. Pharmacol Res. 115:45–55.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li YL, Sun J, Hu X, Pan YN, Yan W, Li QY,
Wang F, Lin NM and Zhang C: Epothilone B induces apoptosis and
enhances apoptotic effects of ABT-737 on human cancer cells via
PI3K/AKT/mTOR pathway. J Cancer Res Clin Oncol. 142:2281–2289.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang C, Shi J, Mao SY, Xu YS, Zhang D,
Feng LY, Zhang B, Yan YY, Wang SC, Pan JP, et al: Role of p38 MAPK
in enhanced human cancer cells killing by the combination of
aspirin and ABT-737. J Cell Mol Med. 19:408–417. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Niu FY and Wu YL: Novel agents and
strategies for overcoming EGFR TKIs resistance. Exp Hematol Oncol.
3:22014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Engelman JA and Jänne PA: Mechanisms of
acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in non-small cell lung cancer. Clin Cancer Res.
14:2895–2899. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li YL, Pan YN, Wu WJ, Mao SY, Sun J, Zhao
YM, Dong JY, Zhang DY, Pan JP, Zhang C and Lin NM: Evodiamine
induces apoptosis and enhances apoptotic effects of erlotinib in
wild-type EGFR NSCLC cells via S6K1-mediated Mcl-1 inhibition. Med
Oncol. 33:162016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oxnard GR, Thress KS, Alden RS, Lawrance
R, Paweletz CP, Cantarini M, Yang JC, Barrett JC and Jänne PA:
Association between plasma genotyping and outcomes of treatment
with osimertinib (AZD9291) in advanced non-small-cell lung cancer.
J Clin Oncol. 34:3375–3382. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Suda K, Rivard CJ, Mitsudomi T and Hirsch
FR: Overcoming resistance to EGFR tyrosine kinase inhibitors in
lung cancer, focusing on non-T790M mechanisms. Expert Rev
Anticancer Ther. 17:779–786. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hsieh YS, Liao CH, Chen WS, Pai JT and
Weng MS: Shikonin inhibited migration and invasion of human lung
cancer cells via suppression of c-Met-mediated
epithelial-to-mesenchymal transition. J Cell Biochem.
118:4639–4651. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gwon SY, Ahn JY, Jung CH, Moon BK and Ha
TY: Shikonin suppresses ERK 1/2 phosphorylation during the early
stages of adipocyte differentiation in 3T3-L1 cells. BMC Complement
Altern Med. 13:2072013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Regales L, Gong Y, Shen R, de Stanchina E,
Vivanco I, Goel A, Koutcher JA, Spassova M, Ouerfelli O,
Mellinghoff IK, et al: Dual targeting of EGFR can overcome a major
drug resistance mutation in mouse models of EGFR mutant lung
cancer. J Clin Invest. 119:3000–3010. 2009.PubMed/NCBI
|
|
28
|
Jean-Philippe J, Paz S and Caputi M: hnRNP
A1: The Swiss army knife of gene expression. Int J Mol Sci.
14:18999–19024. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yin SY, Efferth T, Jian FY, Chen YH, Liu
CI, Wang AH, Chen YR, Hsiao PW and Yang NS: Immunogenicity of
mammary tumor cells can be induced by shikonin via direct
binding-interference with hnRNPA1. Oncotarget. 7:43629–43653.
2016.PubMed/NCBI
|
|
30
|
Cammas A, Lacroix-Triki M, Pierredon S, Le
Bras M, Iacovoni JS, Teulade-Fichou MP, Favre G, Roché H, Filleron
T, Millevoi S and Vagner S: hnRNP A1-mediated translational
regulation of the G quadruplex-containing RON receptor tyrosine
kinase mRNA linked to tumor progression. Oncotarget. 7:16793–16805.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu DW, Chen TC, Huang HS and Lee H:
TC-N19, a novel dual inhibitor of EGFR and cMET, efficiently
overcomes EGFR-TKI resistance in non-small-cell lung cancer cells.
Cell Death Dis. 7:e22902016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sano R and Reed JC: ER stress-induced cell
death mechanisms. Biochim Biophys Acta. 1833:3460–3470. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Corazzari M, Gagliardi M, Fimia GM and
Piacentini M: Endoplasmic reticulum stress, unfolded protein
response, and cancer cell fate. Front Oncol. 7:782017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Terai H, Kitajima S, Potter DS, Matsui Y,
Quiceno LG, Chen T, Kim TJ, Rusan M, Thai TC, Piccioni F, et al: ER
stress signaling promotes the survival of cancer ‘persister cells’
tolerant to EGFR tyrosine kinase inhibitors. Cancer Res.
78:1044–1057. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: A vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rutkowski DT and Kaufman RJ: That which
does not kill me makes me stronger: Adapting to chronic ER stress.
Trends Biochem Sci. 32:469–476. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Verfaillie T, Rubio N, Garg AD, Bultynck
G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A
and Agostinis P: PERK is required at the ER-mitochondrial contact
sites to convey apoptosis after ROS-based ER stress. Cell Death
Differ. 19:1880–1891. 2012. View Article : Google Scholar : PubMed/NCBI
|



















