Introduction
Acute kidney injury (AKI), characterized by a rapid
decrease in renal excretion function, is a high-risk syndrome
(1). Its morbidity and mortality
rates are progressively increasing worldwide (2), and it is estimated that there are
~13,000,000 novel cases and ~1700,000 cases of AKI-associated
mortality annually (3). This
syndrome has a prevalence of 1–2% among hospital admissions and
occurs in 2–7% patients during a hospital stay; the mortality rate
of patients with AKI in intensive care units may be as high as
50–70% (4). Furthermore, due to
the lack of effective treatments and pharmaceutical interventions,
AKI remains a serious challenge to clinicians.
A number of factors may lead to AKI, including
infection, sepsis and the use of nephrotoxic drugs (5). In addition, AKI is considered to be a
systematic inflammatory response to ischemia-reperfusion (I/R) in
various remote organs, including the liver, myocardium, skeletal
muscle, intestine and brain (6–10).
It was demonstrated that I/R may cause tissue injury by inducing
apoptosis. In addition, remote organ I/R-associated AKI is
clinically common and intractable; the mechanisms underlying injury
induced by organ I/R are multifactorial, including oxidative
stress, the generation of free radicals, necrosis, loss of cell
polarity, dedifferentiation and the proliferation of viable cells
(11,12). Apart from these factors, autophagy
is currently an area of interest and has become the focus of an
increasing number of studies, providing a novel direction for
investigating injury induced by organ I/R.
Autophagy was initially defined as ‘self-eating’; in
actuality, it is a defense mechanism employed against environmental
stress and is critical to a variety of physiological and
pathological processes. As the entire process is strictly
regulated, it is a conserved process. Autophagy is one type of
presentation, which is closely associated with cell survival,
although it may additionally be viewed as a type of programmed cell
death. Among the various cell death pathways, autophagy is
considered an inducible and adjustable process that determines cell
survival or death (13).
Experiments have suggested that I/R-induced injury is a potent
trigger of autophagy (1,14), and it has been observed that a high
level of autophagy under high atherogenic shear stress may inhibit
endothelial cell death and inflammation, thus preventing the
development of atherosclerosis. However, whether autophagy has a
protective or invasive role in AKI remains controversial. To date,
limited previous studies have suggested that the induction of
autophagy may cause cell death during AKI; the majority of studies
have demonstrated that the activation of autophagy protects against
AKI. The appearance of this ‘dual role’ may depend on the
experimental procedures used to investigate organ I/R insults
(15).
Therefore, the present study investigated the
effects of CIR on the kidneys of rats, and examined the role of
rapamycin (an autophagy inducer) in renal injury induced by CIR
using a rat model. The aim was to elucidate the mechanism by which
AKI develops following CIR, in order to identify novel approaches
to its treatment.
Materials and methods
Animals and environmental
conditions
A total of 30 male Sprague-Dawley rats (6 weeks old)
weighing 200–250 g were purchased from the Center of Experimental
Animals of Wuhan University (Wuhan, China). All animals were
randomly assigned to polypropylene cages (n=5 per cage) and raised
in a specific pathogen-free environment with a natural light-dark
cycle (12±1 h light and 12±1 h dark). The environmental temperature
was maintained at 20–25°C and the humidity was maintained at
50–52%. All rats had free access to sterile food and water. The
experimental protocol was performed in accordance with the
principles and guidelines of the Guide for the Care and Use of
Laboratory Animals of the National Institutes of Health (Bethesda,
MD, USA). The present study was approved by the Ethics Committee of
Renmin Hospital of Wuhan University (Wuhan, China).
Experimental treatment and model
construction
All rats were randomly divided into three groups,
with 10 rats in each group: Control group (no cerebral ischemia);
model group [1.5 h of middle cerebral artery occlusion (MCAO) and
24 h of reperfusion]; and the pre-treatment group [intraperitoneal
injection of 1 mg/kg rapamycin (Aladdin Biochemical Technology,
Shanghai, China) 0.5 h prior to CIR]. Focal cerebral ischemia was
completed under anesthesia with isoflurane (0.5–3%). During
surgery, rectal temperature was maintained at 37±1°C using a
heating lamp and a heating pad. The MCAO was performed using a
4.0-monofilament nylon wire (Ethicon, Inc., Cincinnati, OH, USA).
Prior to use, the monofilament tip was rounded by heating in a
flame. The nylon filaments were held in a suitable location close
to the blood vessels for 1.5 h, and subsequently gently retracted
to allow reperfusion. In the sham group (control group), the
external carotid artery of each rat was exposed, and the incision
was sutured immediately without contact with the internal carotid
artery. Finally, all rats were sacrificed with excess carbon
dioxide, and samples of kidney tissues and blood were collected for
further investigation.
Assessment of renal function
Cardiac blood samples (6 ml for each) were acquired
from the rats in each group. The sera were collected by
centrifugation at 4°C and 2,000 × g for 15 min and subsequently
stored at −20°C prior to analysis. The serum creatinine and blood
urea nitrogen (BUN) levels were measured using a Hitachi 7170s
Automatic Biochemical Detector (Hitachi, Ltd., Tokyo,
Japan).
Histological examination
The rat kidneys were excised, fixed in 4%
paraformaldehyde for 24 h at room temperature, embedded in paraffin
and cut into 4-µm sections for histological staining. The kidney
sections were subsequently mounted on glass slides, and
hematoxylin-eosin (H&E) staining was performed for 3–5 min at
room temperature for histopathological evaluation. The observed
pathological lesions primarily included renal tubular epithelial
cell flattening, brush border falling off, cell membrane bleb
formation, peritubular/proximal tubule leukocyte infiltration,
interstitial edema, cytoplasmic vacuolization, tubular necrosis and
tubular lumen obstruction. Pathological scores of 0–5 points, based
on the estimated injury area (%), were as follows: 0, normal; 1,
injury area <10%; 2, injury area >10% but <25%; 3, injury
area >25% but <50%; 4, injury area >50% but <75%; 5,
injury area >75%. For each section, 10 areas were randomly
selected to quantitatively assess the extent of AKI using a light
microscope (magnification, ×200; Olympus Corporation, Tokyo,
Japan).
Immunohistochemistry
The paraffin-embedded sections were placed in an
oven at 65°C for 2 h, dewaxed in xylene and rehydrated. The
sections were placed in EDTA buffer for antigen retrieval.
Following washing with PBS, the sections were placed in 3% hydrogen
peroxide solution and incubated at room temperature for 10 min, and
subsequently blocked with 5% bovine serum albumin (BSA;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 20 min at room
temperature following PBS-washing and drying. The BSA solution was
removed, and 50 µl diluted primary antibodies against TNF-α (cat.
no. ab6671) and IL-1β (cat. no. ab9722; both 1:1,00; Abcam,
Cambridge, UK) were added and incubated overnight at 4°C.
Subsequently, 50–100 µl biotin-conjugated SP9000 goat-anti-rabbit
immunoglobulin G (IgG) secondary antibody (cat. no. TA130016;
1:1,000; OriGene Technologies, Inc., Beijing, China) was added to
each section and incubated at 37°C for 50 min. Following washing
with PBS, 50–100 µl DAB solution was added to each section for 5–10
min at room temperature for the observation of the color under a
microscope. Subsequently, the sections were rinsed with distilled
water, re-dyed with hematoxylin for 2 min at room temperature,
differentiated with 1% hydrochloric acid alcohol, and the nucleus
was stained blue with ammonia for 10 min at room temperature. The
sections were subsequently placed through a graded ethanol series
for 10 min each time, dehydrated and dried, dewaxed with xylene and
sealed with neutral gum. Finally, the sections were viewed under a
light microscope (magnification, ×200; Olympus Corporation).
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) assay
The TUNEL assay was performed according to the
manufacturer's protocol (Roche Diagnostics, Indianapolis, IN, USA).
The renal tissue sections were placed in proteinase K solution (20
µg/ml), hydrolyzed for 15 min at room temperature to remove the
tissue protein, and subsequently placed into 10 mM sodium citrate
buffer (pH 6.0) for 10 min for antigen retrieval. The sections were
incubated with 3% hydrogen peroxide for 30 min to block endogenous
peroxidase activity. Following washing with PBS, the sections were
incubated with 1.5% normal goat serum (Beijing Solarbio Science
& Technology, Ltd., Beijing, China) for 30 min at room
temperature, followed by incubation with TUNEL reaction mixture
overnight at 4°C. Following washing with PBS, the sections were
incubated with 0.05% DAB for 5 min at room temperature. Finally,
the sections were mounted with neutral balsam, and the results were
examined under a light microscope (magnification, ×200; Olympus
Corporation). The cells with positive TUNEL staining (brown
staining in the nucleus) were counted in 10 different fields per
section. The results are expressed as the mean number of
TUNEL-positive cells in each group.
Western blot analysis
The renal tissue proteins were extracted from the
samples of each group. The tissues were homogenized in a lysis
buffer provided by Shanghai Biyuntian Bio-Technology Co., Ltd.
(Shanghai, China) with a polytron homogenizer (IKA GmbH,
Königswinter, Germany) on ice. The lysates were subsequently
collected, and the concentrations of protein were detected with a
bicinchoninic acid protein assay. Equal quantities of total protein
(40 µg) were loaded into each well, resolved via 15% SDS-PAGE,
fractionated by electrophoresis and transferred onto polyvinylidene
difluoride membranes. The membranes were blocked with 5% non-fat
milk dissolved in Tris-buffered saline with Tween 20 for 1 h at
room temperature. The following primary antibodies were incubated
with the membranes overnight at 4°C: Anti-microtubule-associated
protein 1 light chain 3β (LC3B; cat. no. ab192890), anti-Beclin-1
(cat. no. ab207612), anti-p62 (cat. no. ab155686), anti-caspase-9
(cat. no. ab2013), anti-caspase-3 (cat. no. ab184787), anti-cleaved
caspase-3 (cat. no. ab184787), anti-B-cell lymphoma (Bcl)-2 (cat.
no. ab196495; all 1:2,000; Abcam), anti-autophagy-related 13
(Atg13; cat. no. 13273), anti-mammalian target of rapamycin complex
1 (mTORC1; cat. no. 2587), anti-unc-51 like autophagy activating
kinase 1 (ULK1; cat. no. 8054) and anti-phosphorylated (p-)ULK1
(cat. no. 14202; all 1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA). Subsequently, a goat-anti-rabbit
fluorescently-labeled secondary antibody (cat. no. C51007;
1:15,000; LI-COR Biosciences, Lincoln, NE, USA) conjugated to
horseradish peroxidase was used for 1 h at room temperature to
identify the primary antibodies. The protein bands were detected
with a two-color infrared imaging system (Odyssey; LI-COR
Biosciences, Lincoln, NE, USA). The relative band intensity was
quantified using Quantity One 4.6.2 software (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). GAPDH was used as an internal
reference.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA was isolated from the kidney tissues using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), following the manufacturer's protocol. RT (37°C
for 15 min and 85°C for 5 sec) was performed using a PrimeScript RT
Reagent kit (Takara Bio, Inc., Otsu, Japan). Amplification was
performed with a 7500 Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The reaction mixture (total 20 µl)
contained 2 µl cDNA, 10 µM primers and 10 µl 2X SYBR Premix Ex Taq
II (Takara Bio, Inc.). The primer sequences were designed using the
Primer Express 2.0 software package (Applied Biosystems; Thermo
Fisher Scientific, Inc.), and were as follows: TNF-α forward,
5′-CTTCTCATTCCTGCTCGTGG-3′ and reverse, 5′-CGGGCTTGTCACTCGAGTTT-3′;
IL-1β forward, 5′-GGCAGTGTCACTCATTGTGG-3′ and reverse,
5′-CTAGCAGGTCGTCATCATCCC-3′; GAPDH forward,
5′-CGCTAACATCAAATGGGGTG-3′ and reverse,
5′-TTGCTGACAATCTTGAGGGAG-3′.
The thermocycling conditions were as follows: 95°C
for 30 sec, 40 cycles of denaturation at 95°C for 5 sec and
extension at 60°C for 40 sec. All samples were run in triplicate,
and the melting curves of all products were analyzed. Quantitative
measurements were determined using the 2−ΔΔCq method
(16). GAPDH was used as the
internal control.
Immunofluorescence
The paraffin-embedded sections were fixed in 100%
acetone for 20 min at room temperature. Following washing with PBS
and antigen retrieval, the sections were placed in a 3%
H2O2-methanol solution and incubated at room
temperature for 10 min. The sections were subsequently blocked with
5% BSA (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 20 min
at room temperature following PBS-washing and drying. The renal
tissue sections were incubated with a monoclonal primary antibody
against LC3B (cat. no. ab48394; 1:1,00; Abcam) at 4°C overnight
following PBS washing. The sections were subsequently incubated
with a fluorescein isothiocyanate-labeled goat-anti-rabbit IgG
(cat. no. ab6717; 1:2,000; Abcam) at 37°C for 1 h in the dark.
Finally, the sections were sealed with glycerol and observed using
a fluorescence microscope (magnification, ×200; Olympus
Corporation). The quantification was analyzed using the Image
Pro-Plus 6.0 system (Media Cybernetics, Inc., Rockville, MD,
USA).
Statistical analysis
All experiments were repeated three times
independently. Data are presented as the mean ± standard deviation.
GraphPad Prism v5.0 (GraphPad Software, Inc., La Jolla, CA USA) was
used to analyze the results using one-way analysis of variance and
Student's t-test. Multiple comparisons between the groups were
performed using Tukey's method as the post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
CIR causes AKI in rats
To assess renal alterations following CIR in the
present study, histopathological alterations were assessed in the
kidneys of the rats, and indices reflective of renal function were
measured, including serum creatinine and BUN, following 90 min of
MCAO and 24 h of reperfusion. The results demonstrated that the
levels of serum creatinine and BUN were significantly increased
following CIR (P<0.05; Table I;
Fig. 1A and B). In addition,
compared with the control group, falling off the brush border of
renal tubular epithelial cells, renal tubular dilation, tube type
in lumen, renal tubular necrosis and increased inflammatory cell
infiltration around the tubules were observed in the CIR group, as
observed in the H&E-stained sections (Fig. 1C). The histological score was
additionally significantly increased in the CIR group compared with
the control group (P<0.05; Fig.
1D).
 | Table I.Serum creatinine and BUN levels of
rats from each group. |
Table I.
Serum creatinine and BUN levels of
rats from each group.
| Parameter | CON | CIR | RAPA |
|---|
| Scr, µmol/l | 51.80±9.88 |
70.69±11.94a |
58.07±10.08b |
| BUN, mmol/l | 2.85±0.56 |
4.03±0.78a |
3.09±0.61b |
Rapamycin ameliorates CIR-induced
renal dysfunction in rats
To determine the role of rapamycin in CIR-induced
AKI in rats, rapamycin was administered via intraperitoneal
injection to the rats prior to CIR. It was identified that,
compared with the CIR group, the levels of serum creatinine and BUN
in the rapamycin pre-treatment group were significantly reduced
(P<0.05; Fig. 1A and B). It was
additionally demonstrated that the kidney histopathological
alterations were markedly improved in the rapamycin pre-treatment
group compared with the CIR group (Fig. 1C); the histological score was
additionally significantly decreased (P<0.05; Fig. 1D).
Rapamycin alleviates CIR-induced renal
inflammation in rats
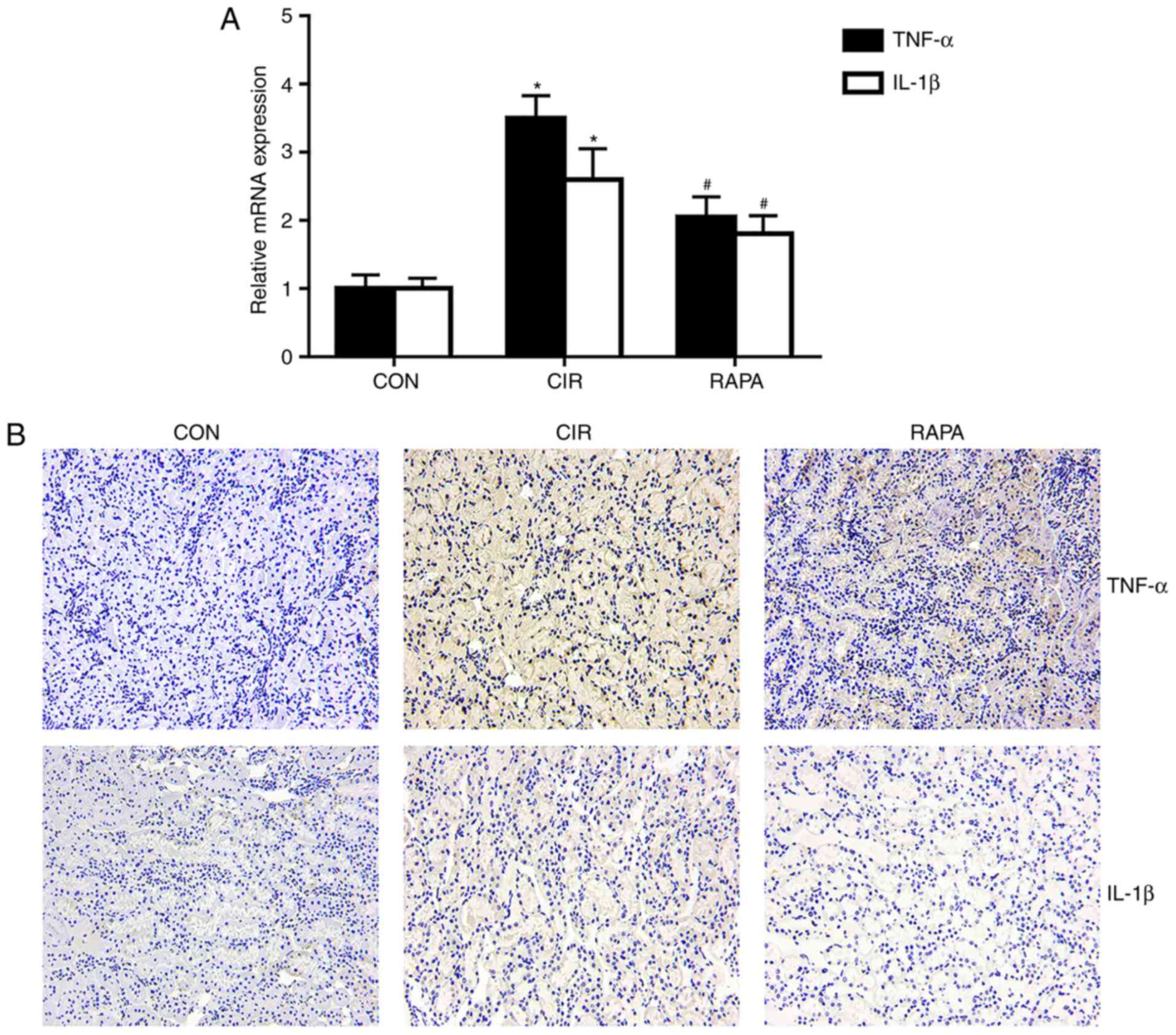
Renal injury is closely associated with the
expression of certain inflammatory mediators. In order to determine
the effects of CIR on renal inflammation in rats and assess whether
rapamycin may regulate this inflammation, the present study
examined how the expression of TNF-α and IL-1β altered with
rapamycin pre-treatment. The results of the RT-qPCR analysis
(P<0.05; Fig. 2A) and
immunohistochemistry (Fig. 2B)
demonstrated that the expression levels of TNF-α and IL-1β in the
kidney tissues were significantly increased in the CIR group
compared with the control group, whereas rapamycin pre-treatment
significantly reversed these effects (P<0.05; Fig. 2).
Rapamycin suppresses CIR-induced renal
apoptosis in rats
Apoptosis is a pathological process involved in the
development of AKI. To examine the effects of CIR on renal
apoptosis in rats and the role of rapamycin in AKI following CIR,
the present study assessed the protein expression of Bcl-2 and
cleaved caspase-3, which are commonly used markers of apoptosis,
and the expression of caspase-9 and caspase-3. The western blot
analysis demonstrated that, compared with the control group, the
expression of cleaved caspase-3 was significantly increased and the
expression of Bcl-2 was significantly decreased in the CIR group
(P<0.05; Fig. 3A-E). However, a
significant increase in the expression of Bcl-2 and significant
decrease in the expression of cleaved caspase-3 was observed in the
rapamycin pre-treatment group, compared with the CIR group
(P<0.05; Fig. 3A-E). In
addition, the TUNEL staining results demonstrated that the number
of TUNEL-positive cells in the CIR group was higher compared than
that in the control group (Fig.
3F), which was corroborated by quantitative analysis
(P<0.05; Fig. 3G). However,
rapamycin pre-treatment significantly decreased the number of
TUNEL-positive cells, compared with the number in the CIR group
(P<0.05; Fig. 3F and G).
Rapamycin enhances CIR-induced renal
autophagy in rats
To elucidate the effects of CIR on renal autophagy
in rats, and further assess the role of rapamycin in CIR-induced
AKI, rapamycin was administered via intraperitoneal injection to
the rats prior to CIR and the expression of numerous critical
autophagy markers, including LC3, Beclin-1 and p62, were measured.
As presented in Fig. 4A-D, the
expression levels of LC3B, a more common form of LC3, and Beclin-1
were significantly increased and the expression of p62 was
significantly decreased in the CIR group, compared with the control
group, as determined by western blot analysis (P<0.05). A
similar trend was observed for the expression of LC3B in the kidney
tissues of rats via fluorescence microscopy and quantitative
analysis (P<0.05; Fig. 4E and
F). Additionally, the expression levels of LC3B and Beclin-1
were increased further and the expression of p62 was decreased
further in the rapamycin pre-treatment group compared with the CIR
group, as determined by western blot analysis (P<0.05; Fig. 4A-D). Similarly, compared with the
CIR group, fluorescence microscopy and quantitative analysis
demonstrated that LC3B levels were further significantly increased
in the rapamycin pre-treatment group (P<0.05; Fig. 4E and F).
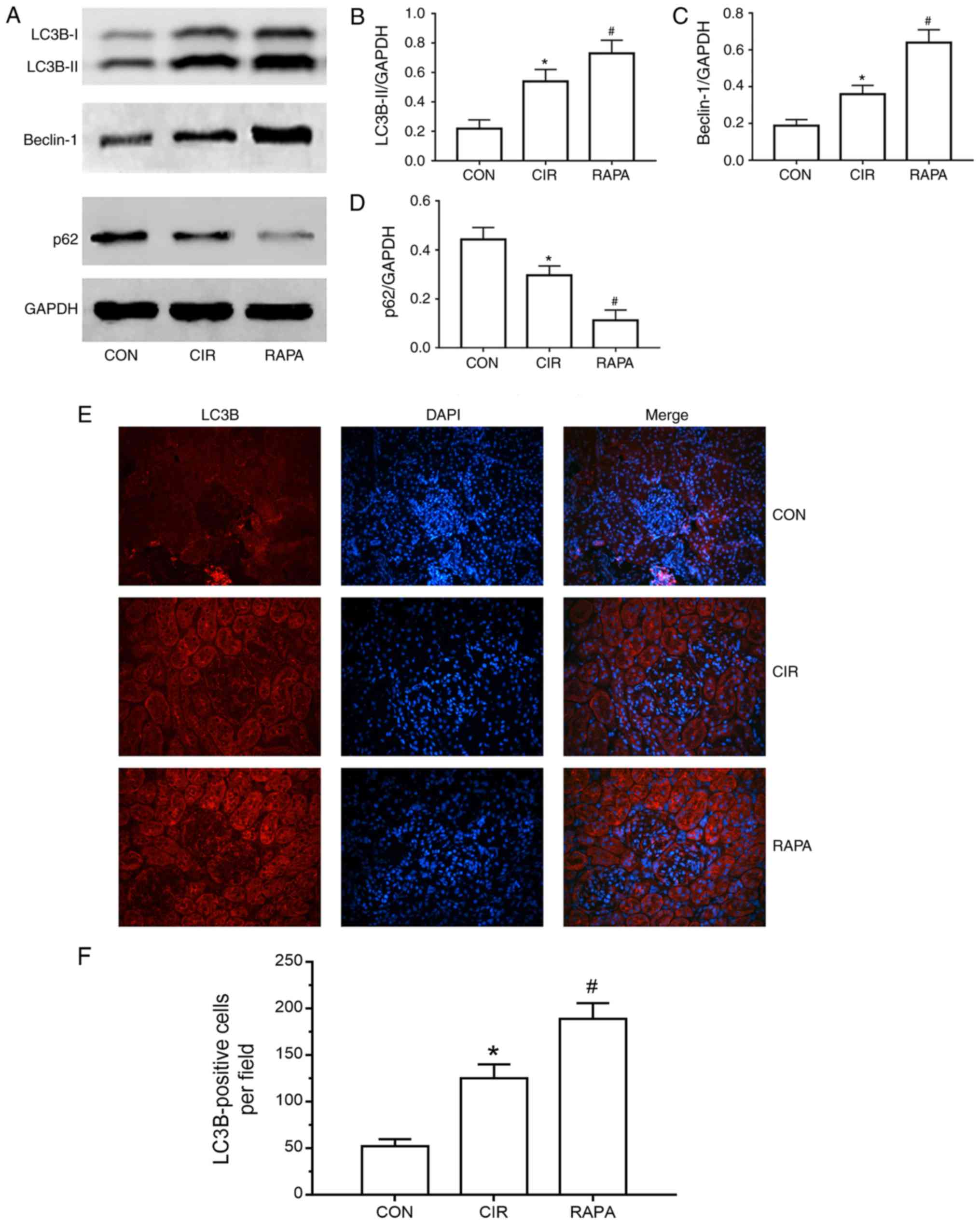 | Figure 4.Renal cell autophagy in rats from
each group. (A) Representative images of the protein expression of
LC3B, Beclin-1 and p62, as determined by western blot analysis.
Relative protein expression of (B) LC3B-II, (C) Beclin-1 and (D)
p62, as determined through quantitative analysis. (E)
Representative fluorescence microscopy images of LC3B
(magnification, ×200). (F) Number of LC3B-positive cells in each
group (quantitative analysis). The results are presented as the
mean ± standard deviation. n=10/group. *P<0.05 CIR, vs. CON;
#P<0.05 RAPA, vs. CIR. CON, control group; CIR,
cerebral ischemia-reperfusion; RAPA, rapamycin pre-treatment prior
to CIR; LC3B, microtubule-associated protein 1 light chain 3β. |
Rapamycin regulates renal autophagy in
rats via the mTORC1/ULK1/Atg13 signaling pathway
To further examine the potential mechanism involved
in rapamycin relieving AKI following CIR via the activation of
autophagy, the levels of a number of crucial autophagy-associated
proteins, including mTORC1, Atg13, ULK1 and p-ULK1, were assessed.
The results of the western blot and quantitative analyses suggested
that the expression levels of Atg13 and ULK1 were significantly
increased, and the expression levels of mTORC1 and p-ULK1 were
significantly decreased in the rapamycin pre-treatment group,
compared with expression levels in the CIR group (P<0.05;
Fig. 5).
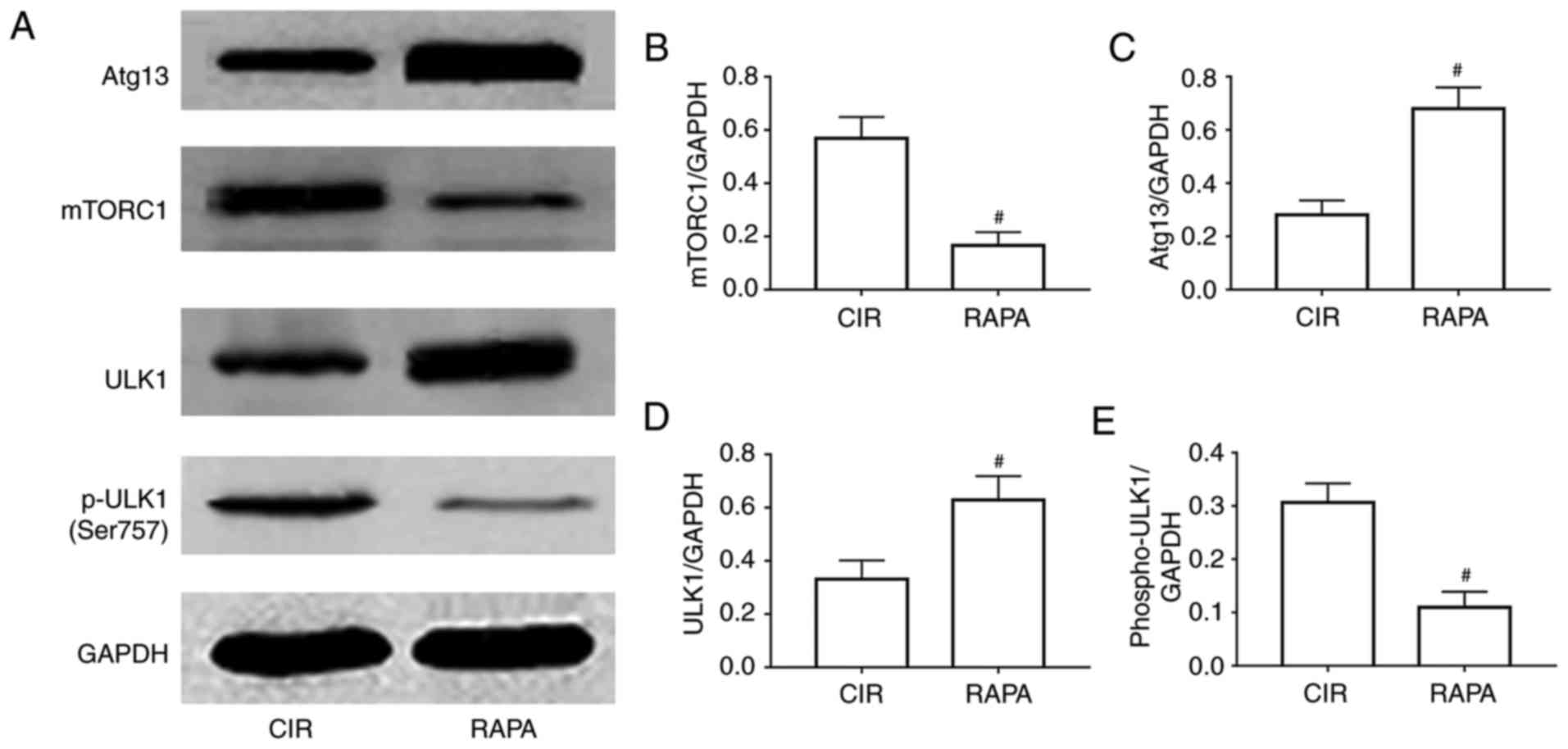 | Figure 5.Involvement of a relevant signaling
pathway in rapamycin relieving CIR-induced acute kidney injury
through the activation of autophagy. (A) Representative images of
the protein expression of mTORC1, Atg13, ULK1 and p-ULK1, as
determined by western blot analysis. Relative expression of (B)
mTORC1, (C) Atg13, (D) ULK1 and (E) p-ULK1, as determined via
quantitative analysis. The results are presented as the mean ±
standard deviation. n=10/group. #P<0.05 RAPA vs. CIR.
CIR, cerebral ischemia-reperfusion; RAPA, rapamycin pre-treatment
prior to cerebral ischemia-reperfusion; mTORC1, mammalian target of
rapamycin complex; Atg-13, autophagy-related 13; ULK1, unc-51 like
autophagy activating kinase 1; p-ULK1, phosphorylated-ULK1. |
Discussion
It is well known that the injury of one organ may
cause alterations in another distal organ. The interaction between
the liver and kidney is known as hepato-renal syndrome, and that
between the heart and kidney is known as cardio-renal syndrome.
Pulmonary-renal, intestinal-kidney and oculo-cerebro-renal
syndromes have additionally been described (17–21).
Therefore, as a systematic response, CIR is considered not only to
cause brain tissue damage; however, additionally to induce damage
to distant organs, including the kidney. Tsagalis et al
(22) identified that AKI was a
common complication following acute stroke, including ischemic
stroke, and demonstrated that AKI was an independent predictor of
early and long-term mortality following acute stroke. Khatri et
al (23) observed that renal
dysfunction was induced by acute ischemic stroke, and that it was
associated with a longer hospital stay and increased mortality
rate. In the present study, numerous histological alterations were
identified in rat kidneys following CIR, including widespread renal
tubular necrosis, inflammatory cell infiltration and tubular
dilatation, among others. Furthermore, rats in the CIR group
exhibited renal dysfunction, which was reflected in the significant
elevation of serum creatinine and BUN levels, compared with the
control group.
AKI is a persistent clinical problem associated with
high mortality rates and healthcare costs. The incidence of AKI has
been increasing, and is likely to increase even further in the
future due to the aging population and the emergence of
comorbidities (22). AKI may cause
an inflammatory response and apoptosis within the kidney (24). Inflammation is a primary factor
involved in the progression of AKI; the acute inflammatory response
is characterized by the activation of inflammatory cells and the
excessive secretion of pro-inflammatory cytokines, including TNF-α
and IL-1β (25). Nongnuch et
al (26) demonstrated that
acute cerebral injury may cause AKI and trigger an inflammatory
cascade in the kidney. In the present study, compared with the
control group, increased inflammatory cell infiltration was
identified in the kidney sections from the CIR group, as determined
by H&E staining, and increased secretion of TNF-α and IL-1β was
observed in the CIR group, as demonstrated by immunohistochemistry.
Apoptosis is another central mechanism in AKI; it is an organized
process regulating the development and homeostasis of multiple
organisms, and is a type of autonomic and programmed cell death
pathway regulated by genes (27).
Apoptosis is critical in various physiological processes and
pathological conditions, and involves the expression of
apoptosis-associated genes, including Bcl-2 and caspase-3 (28). These proteins either promote or
inhibit apoptosis, and the imbalance between pro- and
anti-apoptotic genes may be a decisive factor. Bcl-2 family
proteins are potent regulators of apoptosis; it is increasingly
believed that Bcl-2 may inhibit cell death from a wide variety of
pathogenic stimuli. It may additionally inhibit mitochondrial
membrane potential and decrease caspase-3 activation, in addition
to inhibiting apoptosis via its binding to pro-apoptotic proteins
(28). Bcl-2 is a substrate of
caspase-3, and may thus be hydrolyzed by caspase-3. Regarding the
activation of proteases, a proteolytic cascade of effector caspases
is directly responsible for the execution phase of apoptosis
(28). The ‘executioner’ caspase-3
is activated by the ‘initiator’ caspase-9, resulting in cell death;
therefore, caspase-3 may promote apoptosis (29,30).
In the present study, it was identified that CIR increased the
protein expression of cleaved caspase-3 (an activated form of
caspase-3) and inhibited the protein expression of Bcl-2 in rat
kidney tissues, compared with the control group, as determined by
western blot analysis. Furthermore, the numbers of TUNEL-positive
cells were significantly increased in the CIR group.
The basic pathogenesis of AKI is multifactorial,
including ischemia, hypoxia, nutrient and growth factor
deprivation, energy depletion, oxidant injury, endoplasmic
reticulum stress and other factors; these stimuli may drive
autophagy (31). Among those that
are activated as part of the renal stress response to organ I/R,
autophagy has become the focus of numerous investigations (31). Autophagy is an evolutionarily
conserved multistep process that involves the degradation of
intracellular organelles, proteins and other macromolecules by
lysosomal hydrolytic enzymes (32). The degraded cellular contents are
utilized for the synthesis of novel macromolecules and organelles.
Under normal physiological conditions, a basal level of autophagy
maintains cellular homeostasis (32). Under pathological conditions,
external stressors contribute to the induction of autophagy
(33). In cell and animal models
of I/R-induced injury, it has been demonstrated that autophagy is
activated (34). As no available
effective therapies for AKI are available, one of the increasingly
recognized and potential therapeutic targets is cellular autophagy.
A number of previous studies demonstrated the role of autophagy in
I/R-induced AKI. Chien et al (35) identified that autophagy may
ameliorate AKI caused by I/R. Hsiao et al (36) suggested that autophagy is
beneficial in AKI due to sepsis by cecal ligation and puncture, and
that the decline of autophagy contributed to proximal tubular
dysfunction in late-stage sepsis. Kimura et al (37) used proximal tubule-specific
Atg5-knockout mice to demonstrate that autophagy was
reno-protective following I/R. Additionally, Sun et al
(38) observed that octreotide may
reduce AKI following hepatic I/R in a rat model via the induction
of autophagy. Zhang et al (39) demonstrated that niclosamide may
attenuate inflammation by inducing autophagy in a rat model of
renal I/R.
As the consequences of autophagy in CIR-induced AKI
have not been investigated, to the best of our knowledge, and based
on the aforementioned studies, it was hypothesized that autophagy
may additionally be important in CIR-induced AKI. However, whether
it provides protection or aggravates AKI following CIR remains to
be elucidated. A frequently used autophagy inducer is rapamycin, a
macrolide antibiotic originally used for antifungal therapy.
However, it may additionally be used to regulate autophagy and
maintain cell metabolism. Zhang et al (14) demonstrated that rapamycin may be a
promising therapy for I/R and AKI. Cui et al (40) suggested that rapamycin may
ameliorate gentamicin-induced AKI by enhancing autophagy in
miniature pig models. Luo et al (41) identified that rapamycin inhibited
vascular smooth muscle cell senescence via inducing autophagy. In
the present study, it was identified that rapamycin pre-treatment
prior to CIR attenuated renal pathological alterations and improved
renal function, and the number of inflammatory cells around the
renal tubules was significantly reduced in the rapamycin
pre-treatment group compared with the CIR group. Furthermore,
rapamycin pre-treatment suppressed the expression of TNF-α and
IL-1β in rat kidney tissues. Compared with the CIR group, rapamycin
pre-treatment increased the protein expression of Bcl-2 and
decreased the protein expression of cleaved caspase-3;
additionally, the rapamycin pre-treatment group exhibited fewer
TUNEL-positive cells compared with the CIR group. It was
additionally observed that the protein expression levels of LC3B
and Beclin-1 were significantly increased, whereas the protein
expression of p62 was significantly inhibited in the CIR group,
compared with the control group; this effect of autophagy was
considered to be a limited self-protection response to stress.
Rapamycin pre-treatment followed by CIR resulted in the increased
induction of LC3B and Beclin-1 proteins and inhibition of p62
protein, mediated via the mTORC1/ATG13/ULK1 signaling pathway.
Fluorescence microscopy analysis additionally demonstrated that the
expression of LC3B in rat kidney tissues was induced by CIR and
further enhanced by rapamycin. Therefore, the results demonstrated
that CIR may cause AKI, and that rapamycin pre-treatment may
improve renal function, reduce renal inflammation and apoptosis,
and further activate CIR-induced autophagy in the kidneys of rats
via the mTORC1/ATG13/ULK1 signaling pathway.
In conclusion, rapamycin may relieve CIR-induced AKI
by activating autophagy through the mTORC1/ATG13/ULK1 signaling
pathway. These results are likely to assist in further elucidating
the pathogenesis of AKI following CIR and may provide a promising
treatment approach for this condition. However, the specific
underlying mechanisms require further investigation.
Acknowledgements
The authors would like to thank Professor Zhihua
Wang of the Central Laboratory of Renmin Hospital of Wuhan
University for providing relevant experimental facilities and
technical support.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81470923, 81770078
and 81770688).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YS, XC, CL and JZ designed the study. YS, JL and PG
performed the experiments. JL and XC gathered the experimental
data. XC and YS analyzed the experimental data. YS drafted the
manuscript. XC, CL and JZ revised the paper for intellectual
content. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental protocol was performed in
accordance with the principles and guidelines of the Guide for the
Care and Use of Laboratory Animals of the National Institutes of
Health. The present study was approved by the Ethics Committee of
Renmin Hospital of Wuhan University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Melk A, Baisantry A and Schmitt R: The yin
and yang of autophagy in acute kidney injury. Autophagy.
12:596–597. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ronco C and Chawla LS: Acute kidney
injury: Kidney attack must be prevented. Nat Rev Nephrol.
9:198–199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mehta RL, Cerdá J, Burdmann EA, Tonelli M,
Garcia-Garcia G, Jha V, Susantitaphong P, Rocco M, Vanholder R,
Sever MS, et al: International Society of Nephrology's 0by25
initiative for acute kidney injury (zero preventable deaths by
2025): A human rights case for nephrology. Lancet. 385:2616–2643.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bellomo R, Kellum JA and Ronco C: Acute
kidney injury. Lancet. 380:756–766. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dirkes S: Sepsis and inflammation: Impact
on acute kidney injury. Nephrol Nurs J. 40:125–132. 2013.PubMed/NCBI
|
|
6
|
Lee HT, Park SW, Kim M and D'Agati VD:
Acute kidney injury after hepatic ischemia and reperfusion injury
in mice. Lab Invest. 89:196–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aydin SI, Seiden HS, Blaufox AD, Parnell
VA, Choudhury T, Punnoose A and Schneider J: Acute kidney injury
after surgery for congenital heart disease. Ann Thorac Surg.
94:1589–1595. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garbaisz D, Turoczi Z, Aranyi P, Fulop A,
Rosero O, Hermesz E, Ferencz A, Lotz G, Harsanyi L and Szijarto A:
Attenuation of skeletal muscle and renal injury to the lower limb
following ischemia-reperfusion using mPTP inhibitor NIM-811. PLoS
One. 9:e1010672014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun Q, Meng QT, Jiang Y and Xia ZY:
Ginsenoside Rb1 attenuates intestinal ischemia reperfusion induced
renal injury by activating Nrf2/ARE pathway. Molecules.
17:7195–7205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nadkarni GN, Patel AA, Konstantinidis I,
Mahajan A, Agarwal SK, Kamat S, Annapureddy N, Benjo A and Thakar
CV: Dialysis requiring acute kidney injury in acute cerebrovascular
accident hospitalizations. Stroke. 46:3226–3231. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sheridan AM and Bonventre JV: Cell biology
and molecular mechanisms of injury in ischemic acute renal failure.
Curr Opin Nephrol Hypertens. 9:427–434. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nicoud IB, Knox CD, Jones CM, Anderson CD,
Pierce JM, Belous AE, Earl TM and Chari RS: 2-APB protects against
liver ischemia-reperfusion injury by reducing cellular and
mitochondrial calcium uptake. Am J Physiol Gastrointest Liver
Physiol. 293:G623–G630. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lenoir O, Tharaux PL and Huber TB:
Autophagy in kidney disease and aging: Lessons from rodent models.
Kidney Int. 90:950–964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang YL, Zhang J, Cui LY and Yang S:
Autophagy activation attenuates renal ischemia-reperfusion injury
in rats. Exp Biol Med (Maywood). 240:1590–1598. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Decuypere JP, Ceulemans LJ, Agostinis P,
Monbaliu D, Naesens M, Pirenne J and Jochmans I: Autophagy and the
Kidney: Implications for Ischemia-Reperfusion Injury and Therapy.
Am J Kidney Dis. 66:699–709. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Davenport A: AKI in a patient with
cirrhosis and ascites. Clin J Am Soc Nephrol. 7:2041–2048. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bagshaw SM, Cruz DN, Aspromonte N,
Daliento L, Ronco F, Sheinfeld G, Anker SD, Anand I, Bellomo R,
Berl T, et al: Epidemiology of cardio-renal syndromes: Workgroup
statements from the 7th ADQI Consensus Conference. Nephrol Dial
Transplant. 25:1406–1416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
West SC, Arulkumaran N, Ind PW and Pusey
CD: Pulmonary-renal syndrome: A life threatening but treatable
condition. Postgrad Med J. 89:274–283. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ritz E: Intestinal-renal syndrome: Mirage
or reality? Blood Purif. 31:70–76. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lowe M: Structure and function of the Lowe
syndrome protein OCRL1. Traffic. 6:711–719. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsagalis G, Akrivos T, Alevizaki M, Manios
E, Theodorakis M, Laggouranis A and Vemmos KN: Long-term prognosis
of acute kidney injury after first acute stroke. Clin J Am Soc
Nephrol. 4:616–622. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Khatri M, Himmelfarb J, Adams D, Becker K,
Longstreth WT and Tirschwell DL: Acute kidney injury is associated
with increased hospital mortality after stroke. J Stroke
Cerebrovasc Dis. 23:25–30. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ranganathan P, Jayakumar C, Mohamed R,
Weintraub NL and Ramesh G: Semaphorin 3A inactivation suppresses
ischemia-reperfusion-induced inflammation and acute kidney injury.
Am J Physiol Renal Physiol. 307:F183–F194. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rabb H, Griffin MD, McKay DB, Swaminathan
S, Pickkers P, Rosner MH, Kellum JA and Ronco C: Acute Dialysis
Quality Initiative Consensus XIII Work Group: Inflammation in AKI:
Current understanding, key questions and knowledge gaps. J Am Soc
Nephrol. 27:371–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nongnuch A, Panorchan K and Davenport A:
Brain-kidney crosstalk. Crit Care. 18:2252014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin X, Zhang Y, Li X, Zhang J and Xu D:
C-type natriuretic peptide ameliorates ischemia/reperfusion-induced
acute kidney injury by inhibiting apoptosis and oxidative stress in
rats. Life Sci. 117:40–45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Havasi A and Borkan SC: Apoptosis and
acute kidney injury. Kidney Int. 80:29–40. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ma P, Zhang S, Su X, Qiu G and Wu Z:
Protective effects of icariin on cisplatin-induced acute renal
injury in mice. Am J Transl Res. 7:2105–2114. 2015.PubMed/NCBI
|
|
31
|
He L, Livingston MJ and Dong Z: Autophagy
in acute kidney injury and repair. Nephron Clin Pract. 127:56–60.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Parzych KR and Klionsky DJ: An overview of
autophagy: morphology, mechanism and regulation. Antioxid Redox
Signal. 20:460–473. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kaushal GP and Shah SV: Autophagy in acute
kidney injury. Kidney Int. 89:779–791. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiang M, Liu K, Luo J and Dong Z:
Autophagy is a renoprotective mechanism during in vitro hypoxia and
in vivo ischemia-reperfusion injury. Am J Pathol. 176:1181–1192.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chien CT, Shyue SK and Lai MK: Bcl-xL
augmentation potentially reduces ischemia/reperfusion induced
proximal and distal tubular apoptosis and autophagy.
Transplantation. 84:1183–1190. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hsiao HW, Tsai KL, Wang LF, Chen YH,
Chiang PC, Chuang SM and Hsu C: The decline of autophagy
contributes to proximal tubular dysfunction during sepsis. Shock.
37:289–296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kimura T, Takabatake Y, Takahashi A,
Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T,
Soga T, et al: Autophagy protects the proximal tubule from
degeneration and acute ischemic injury. J Am Soc Nephrol.
22:902–913. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun H, Zou S, Candiotti KA, Peng Y, Zhang
Q, Xiao W, Wen Y, Wu J and Yang J: Octreotide attenuates acute
kidney injury after hepatic ischemia and reperfusion by enhancing
autophagy. Sci Rep. 7:427012017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang LX, Zhao HJ, Sun DL, Gao SL, Zhang
HM and Ding XG: Niclosamide attenuates inflammatory cytokines via
the autophagy pathway leading to improved outcomes in renal
ischemia/reperfusion injury. Mol Med Rep. 16:1810–1816. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cui J, Bai XY, Sun X, Cai G, Hong Q, Ding
R and Chen X: Rapamycin protects against gentamicin-induced acute
kidney injury via autophagy in mini-pig models. Sci Rep.
5:112562015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Luo Z, Xu W, Ma S, Qiao H, Gao L, Zhang R,
Yang B, Qiu Y, Chen J, Zhang M, et al: Moderate autophagy inhibits
vascular smooth muscle cell senescence to stabilize progressed
atherosclerotic plaque via the mTORC1/ULK1/ATG13 signal pathway.
Oxid Med Cell Longev. 2017:30181902017. View Article : Google Scholar : PubMed/NCBI
|