Introduction
Abnormalities involving sex chromosomes account for
approximately 0.5% of live births. Individuals with mosaic
structural aberrations of the X and Y chromosomes exhibit
complicated and variable phenotypes. The phenotypes of sex
chromosome mosaicism vary from females with Turner syndrome to
males with infertility, and include individuals with ambiguous
genitalia (1).
Cytogenetically visible Y chromosome anomalies
involve deletions, translocations, rings and isochromosomes among
others. The Y chromosome comprises highly repetitive sequences,
including direct repeats, inverted repeats and palindromes
(2). The highly repetitive
structures of the Y chromosome are mainly located in the
azoospermia factor (AZF) regions, where rearrangements occur in
high frequency to form delY, idicY and so forth. These
rearrangements lead not only to loss, but also gain of specific
genes (3,4). The locus for AZF in Yq11.2 is
subdivided into the AZFa, AZFb and AZFc regions, which serve an
important role in spermatogenesis and fertility (5).
Notably, the majority of Y chromosome anomalies have
been reported in a mosaic form, usually in association with a 45,X
cell line (6,7). In the process of gonadal development,
the percentage of 45 cells has an important role in sex
determination. A dicentric Y chromosome is a common abnormal
structural rearrangement between sister chromatids and is unstable
during cell division; therefore, it is highly likely to generate
various cell lines, including 45,X and delY cell lines. Previous
studies have mostly described individuals with ambiguous genitalia
and mixed gonadal dysgenesis who were diagnosed with sex chromosome
mosaicism postnatally (1,8–16).
However, there are few studies reporting prenatally diagnosed
fetuses, particularly with three different cell lines.
The present study reports the case of a fetus with a
45,X/46,X,del(Y)(q11.222)/46,X,idic(Y)(q11.222) karyotype. The
fetus carried a derivative Y chromosome with the deletion of AZFb
and AZFc regions, and a dicentric Y chromosome with a break point
located in the Yq11.222. The results were confirmed through
cytogenetic, single nucleotide polymorphism (SNP) microarray and
fluorescence in situ hybridization (FISH) detections. The
comprehensive use of multiple technologies was beneficial for
accurately diagnosing the karyotype, identifying the origin of the
marker chromosome and preparing effective genetic counseling.
Materials and methods
Case report
A 24-year-old pregnant female (gravida 1 para 0) was
enrolled into the present study. The patient experienced regular
menstruation (4 days per 28 days), was in good health, had no
abnormal family history, and had not been exposed to teratogenic
agents prior to or during the pregnancy. She refused serum
screening and underwent noninvasive prenatal testing (NIPT) at 16
weeks and 4 days of gestation. The NIPT results suggested
suspicious abnormalities of the fetal sex chromosomes.
Subsequently, amniocentesis was performed at 21 weeks and 4 days of
gestation, and 20-ml amniotic fluid was sampled under ultrasound
guidance for routine amniotic fluid cell culture and karyotype
analysis. The karyotype analysis results indicated that the fetus
exhibited sex chromosome mosaicism with two marker chromosomes.
Therefore, further assays were recommended. Amniotic fluid (20 ml)
was sampled again at 23 weeks and 4 days of gestation for analysis
by SNP microarray and FISH assay. The molecular results confirmed
the existence of two Y chromosomes, deletions in Yq and mosaicism.
The patient terminated the pregnancy at 27 weeks of gestation.
Karyotype analysis of peripheral blood samples obtained from the
father and mother of the fetus was identified as 46,XY and 46,XX,
respectively. This study was approved by the Medical Ethics
Committee of the Maternity and Child Health Care Hospital
(Yancheng, China), and written informed consent was obtained from
the two participants.
Cytogenetics
Amniotic fluid cell culture was performed according
to standard techniques (17).
Routine G-bands were obtained using trypsin and Giemsa staining
(GTG analysis) at 400 band resolution, and this analysis was used
to prepare the amniotic cell chromosome specimens. The peripheral
blood lymphocytes obtained from the parents were cultured, followed
by karyotyping analysis. GTG-banding was also performed according
to the standard protocol.
FISH analysis
In order to ascertain the origins of the marker
chromosomes and mosaic proportions, FISH analysis was performed on
the amniotic specimens using the DXZ1/DYZ3/D18Z1 probes located at
the centromeric region of chromosomes X, Y and 18 (Vysis; Abbott
Laboratories, Abbott Park, IL, USA), according to the
manufacturer's protocol. Human chromosomes were stained by
4′,6-diamidino-2-phenylindole (Vysis; Abbott Laboratories) in the
dark for 10–15 min at room temperature and exhibited bright
fluorescence at the desired locations for detection.
SNP microarray
Human cyto12 SNP-array (Illumina, Inc., San Diego,
CA, USA) comprising around 300,000 SNP probes was applied to
perform a whole genome scan on the amniotic cell DNA of the fetus.
SNP-array tests were performed according to the manufacturer's
protocol (Illumina, Inc.), while molecular karyotype analysis was
conducted using KaryoStudio version 1.4.3.0 (Illumina, Inc.).
Databases including DECIPHER (http://decipher.sanger.ac.uk/), DGV (http://projects.tcag.ca/variation), UCSC
(http://genome.ucsc.edu/) and OMIM (http://www.ncbi.nlm.nih.gov/omim), were used as
references to evaluate the array data and analyze
genotype-phenotype correlations.
Results
Karyotype analysis
The routine G-band staining analysis indicated that
the fetus had a mosaic karyotype with two marker chromosomes in
three cell lines. The abnormal karyotype was
45,X[13]/46,X,+mar1[6]/46,X,+mar2[9], and the mosaic proportion was
46, 21 and 33% (13/28, 6/28 and 9/28 cells), respectively (Fig. 1A-C). GTG analysis was also
performed for the parents, who were both revealed to have a normal
karyotype.
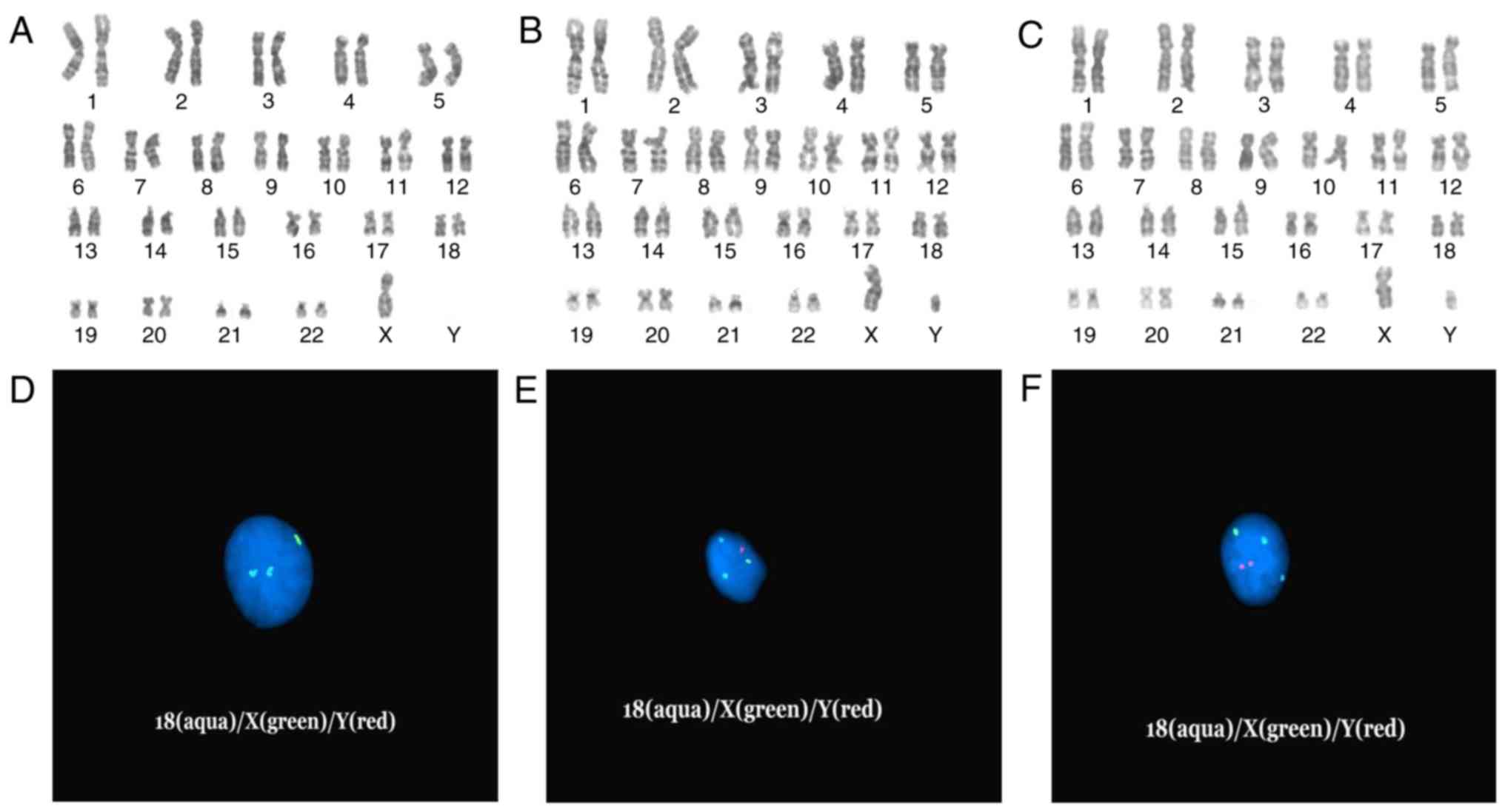 | Figure 1.(A-C) Giemsa banding karyotype
analysis of the fetus: (A) 45,X (B) 46,X,del(Y)(q11.222) (C)
46,X,idic(Y)(q11.222) and (D-F) D18Z1(aqua)/DXZ1(green)/DYZ3(red)
centromeric probe detection in interphase cell nuclei of the fetus:
(D) Two aqua fluorescent signals indicate two chr18, one green
signal indicates one chrX. (E) Two aqua signals indicate two chr18,
one green signal indicates one chrX, one red signal indicates one
chrY. (F) Two aqua signals indicate two chr18, one green signal
indicates one chrX, two red signals indicate idicY (combined with
the karyotype C). Chr, chromosome. |
FISH analysis
The DXZ1/DYZ3/D18Z1 probes were successfully
hybridized to interphase cell nuclei. There was one signal and two
signals in the centromeric regions of chromosomes X and 18,
respectively (Fig. 1D-F). As
displayed in Fig. 1D, there was no
Y centromeric signal. One red signal revealed one Y centromere of
mar1 (Fig. 1E), and two signals
revealed two Y centromeres of mar2 (Fig. 1F). In addition, FISH analysis on
uncultured amniocytes revealed the karyotype
45,X,ish(DXZ1+,DYZ3-,D18Z1++)[5]/46,X,+mar1,ish
(DXZ1+,DYZ3+,D18Z1++)[11]/46,X,+mar2,ish(DXZ1+,DYZ3++,D18Z1++)[14].
The mosaic proportion of this karyotype sequence was 17, 37 and 46%
(5/30, 11/30 and 14/30 cells), respectively.
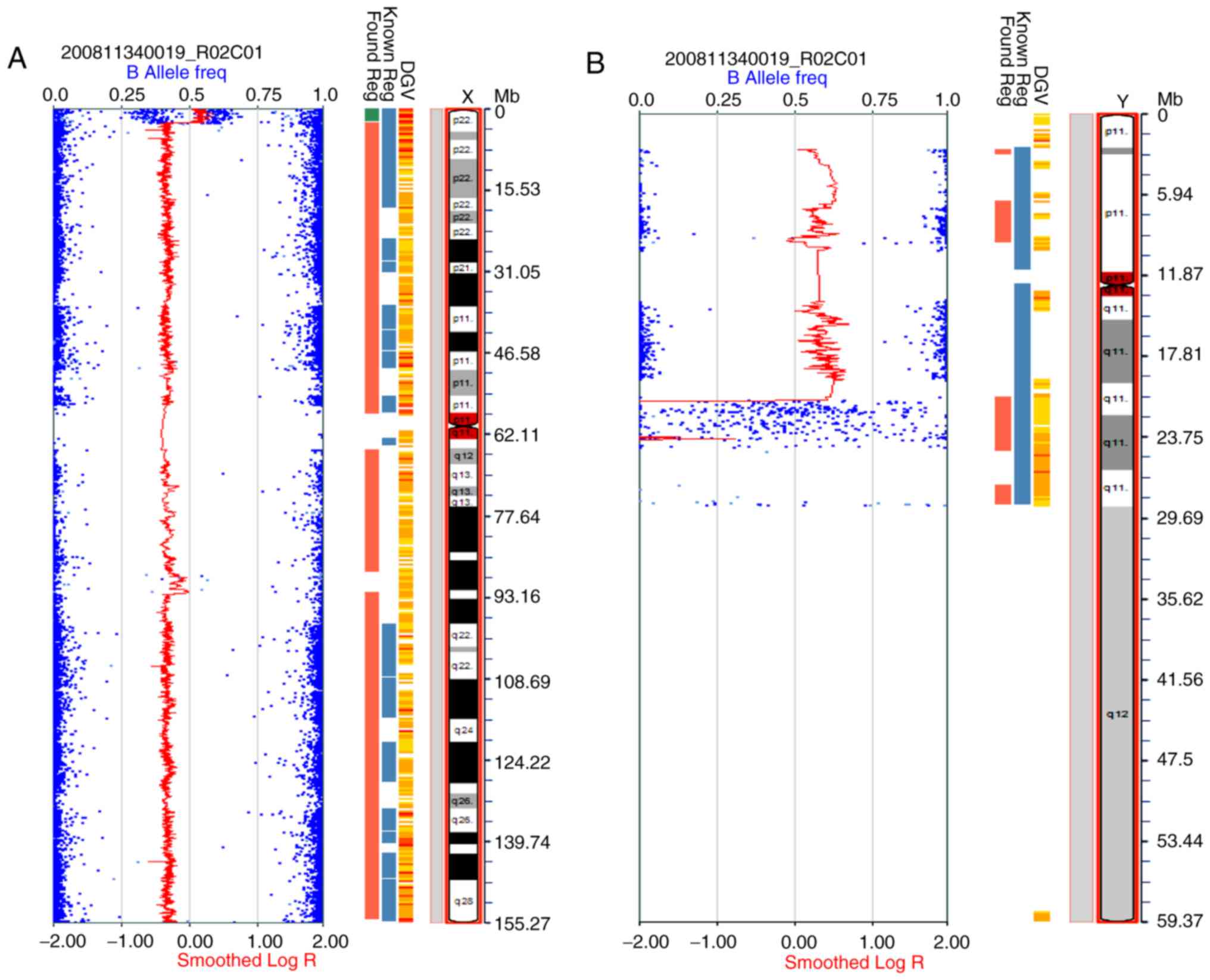
SNP microarray
The SNP microarray analysis performed on the DNA
extracted from amniotic fluid revealed two Y chromosomes and 7.8-Mb
deletions involving the region in Yq11.222q12. The molecular
karyotype was arr[hg19] (X)x1, (Y)x2,
Yq11.222q12(21,032,051–28,786,812)x0 (Fig. 2A and B). The deletion parts were
located at AZFb and AZFc regions in the long arm of the Y
chromosome, including a number of OMIM genes, such as the CDY,
HSFY, RBMY, PRY, BPY2 and DAZ gene families (Fig. 3).
Prenatal diagnosis
Combining the results of chromosome karyotype
analysis, FISH and SNP-array analysis, it was possible to identify
the precise breakage of the mar1 and mar2 on Yq11.222. According to
these results, the molecular karyotype of the fetus was identified
as 45,X,ish(DXZ1+,DYZ3-,D18Z1++)[5]/46,X,
del(Y)(q11.222),ish(DXZ1+,DYZ3+,D18Z1++)[11]/46,X,idic(Y)(q11.222),ish(DXZ1+,DYZ3++,D18Z1++)[14].
SNP-array analysis revealed two Y chromosomes and 7.8-Mb deletions
in Yq11.222q12 located at AZFb and AZFc regions. The deletion
regions included DAZ, RBMY and PRY genes, which could cause
spermatogenesis obstacle and sterility. Although the proportions of
45,X cell lines were discrepant between the karyotype and FISH
analysis results, it could serve a role in sex determination and
gonad development. Following genetic consultation, the parents
decided to terminate the pregnancy, and labor was induced at 27
weeks of gestation.
Discussion
In the present study, a fetus was found to have a
mosaic karyotype of three cell lines
45,X[13]/46,X,+mar1[6]/46,X,+mar2[9] using the GTG method, while
FISH and SNP-array analyses were then conducted to identify the
mosaic percentage and origins of the two marker chromosomes. The
mosaic percentage differences between the karyotype and FISH
analysis results may be caused by the amniotic cell culture
selective growth and cell counting (18,19).
Although SNP-array analysis had a high level of resolution, low
mosaicism could not be detected with this analysis. In addition, it
was ascertained that mar1 was del(Y)(q11.222) with a 7.8-Mb
deletion from Yq11.222 to the long-arm end and mar2 was
idic(Y)(q11.222) with two mar1s connected in Yq11.222 with two Y
centromeres.
The abnormal Y chromosomes of the fetus were a
result of rearrangements at the meiosis phase between sister
chromatids or intrachromosome during spermatogenesis in the father.
Another cause probably occurred during the mitosis stages (20). Structural aberrations of the Y
chromosome result in predisposition to subsequent chromosome
instability and loss of the abnormal Y chromosome, leading to
mosaic 45,X. A fetus with the presence of a 45,X cell line has a
risk of being a phenotypic female with Turner syndrome
manifestations or having ambiguous external genitalia, whether the
other cell line is Yp, Yq, Yp plus Yq, or even a free Y chromosome
(6,20).
Although to the best of our knowledge there are no
identical case reports in the literature, there are a number of
cases with 45,X/46,X,idic(Y) and 45,X/46,X,del(Y)(q11.2).
Phenotypes associated with the two sex chromosomal mosaicism vary
from females with Turner syndrome to males with infertility, and
include individuals with ambiguous genitalia (1). Their phenotypic spectrums are very
broad and variable, and are attributed to variable locations of the
breakpoints and to the proportion of 45,X cells distributed over
the different tissues (9,21,22).
As in the majority of previous studies with idicY
and delY (1,5,7,8,12–16),
the breakpoint in the fetus is in the long arm of chromosome Y,
which results in the duplication of the entire short arm and
centromere, and a deletion of the distal Yq. A review by Hsu
(6) reported 74 cases with
mos45,X/46,X,idic(Y)(q11). Among them, 20 cases (27%) involved
phenotypic males with abnormal testes and azoospermia, 17 cases
(23%) had ambiguous external genitalia with mixed gonads and short
height, and 37 cases (50%) were phenotypic females with streak
gonads and Ullrich-Turner syndrome changes. The aforementioned
review also reported 38 postnatal cases of mos45,X/46,X,del(Y)(q11)
manifesting as phenotypic males with hypospadias and azoospermia in
13 cases (34.2%), intersex individuals with ambiguous external
genitalia in 18 cases (47.4%) and phenotypic females with streak
gonads and short stature in 7 cases (18.4%) (6).
To date, few prenatal cases of the karyotype
45,X/46,X,idic(Y)(q11) and 45,X/46,X,del(Y)(q11.2) have been
reported. Telvi et al (23)
reported substantial differences between prenatally and postnatally
diagnosed cases of 45,X/46,XY mosaicism. A normal male phenotype
was detected in 90% of prenatally diagnosed cases, whereas the
postnatally diagnosed cases exhibited a wide spectrum of
phenotypes. This 10% risk of an abnormal outcome in prenatally
diagnosed cases requires further attention (23). In the present study, the prenatal
ultrasound of the fetus was unremarkable, because the symptoms of
sex chromosome mosaicism would be clinically evident following
birth or puberty.
The Y chromosome is the shortest chromosome of the
human genome, but accumulates male-related genes, including the
sex-determining region of Y-chromosome (SRY) and several
spermatogenesis-associated genes. The long arm of chromosome Y is
enriched with palindromes that have recently been demonstrated to
mediate rearrangements between the arms of sister chromatids
(24). The rearrangements lead to
not only loss, but also gain of specific genes. Loss of
Y-chromosome sequences has been detected in men with azoospermia or
severe oligospermia, leading to the definition of the azoospermia
factor region. The region has been mapped to Yq11.22-23 and
consists of three sub-regions termed AZFa, AZFb and AZFc (3–5).
Gain of Y-chromosome sequences may generate the idicYp chromosomes
and other attachments.
In the current study, the idicY chromosome of the
fetus had two SRY genes. The deletion parts of the fetus were
located at AZFb and AZFc regions in the long arm of Y chromosome,
including the OMIM genes of the CDY, HSFY, RBMY1, PRY, BPY2 and DAZ
gene families. A study by Lahn and Page (25) identified the CDY, PRY and BPY2
testes-specific gene families. Furthermore, Skaletsky et al
(26) determined that the HSFY
gene is exclusively expressed in the testes, and there are two
palindromic copies of the HSFY gene within palindrome 4 of
chromosome Yq. A literature review on the RBMY1 gene family
reported by Delbridge et al (27) indicated that the RBMY1 gene family
may have an additional role in germ cell development. The DAZ gene
encodes an RNA-binding protein with a role in spermatogenesis
(28). Taken together, the
deletion regions can cause spermatogenesis obstacle and
sterility.
In conclusion, multiple studies have observed that
patients with 45,X/46,X,idic(Y)(q11) or 45,X/46,X,del(Y)(q11.2)
exhibit ambiguous external genitalia, azoospermia or Turner
syndrome (8–16). Therefore, it is likely that the
fetus in the present study would suffer similar syndromes upon
maturation. The comprehensive use of cytogenetic, SNP-array and
FISH detections in the current study provided adequate genetic
counseling to the patient and her family, and the family decided to
terminate the pregnancy as the fetus would have been born with
birth defects. Thus, the present study may provide guidance for
future pregnancy and birth decisions.
Acknowledgements
Not applicable.
Funding
No funding was received for this study.
Availability of data and materials
All data generated or analyzed during this study are
included in the article.
Authors' contributions
JZ drafted the paper and interpreted the SNP-array
and FISH data. XY drafted part of the discussion and interpreted
the cytogenetic and molecular data. YG, FY and MX were responsible
for the conventional cytogenetic analysis. ML and YZ collected the
data and provided clinical consultation. XJ, YW and PH were
involved in the SNP-array and FISH analysis. JZ and HL designed the
study and gave the final approval of the manuscript. All of the
authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Medical Ethics
Committee of Maternity and Child Health Care Hospital (Yancheng,
China), and written informed consent was obtained from the two
participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Al-Achkar W, Wafa A, Liehr T, Klein E and
Moassass F: Detailed analysis of an idic(Y)(q11.21) in a mosaic
karyotype. Mol Med Rep. 6:293–296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li Z, Haines CJ and Han Y:
‘Micro-deletions’ of the human Y chromosome and their relationship
with male infertility. J Genet Genomics. 35:193–199. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuroda-Kawaguchi T, Skaletsky H, Brown LG,
Minx PJ, Cordum HS, Waterston RH, Wilson RK, Silber S, Oates R,
Rozen S and Page DC: The AZFc region of the Y chromosome features
massive palindromes and uniform recurrent deletions in infertile
men. Nat Genet. 29:279–286. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Repping S, Skaletsky H, Lange J, Silber S,
Van Der Veen F, Oates RD, Page DC and Rozen S: Recombination
between palindromes P5 and P1 on the human Y chromosome causes
massive deletions and spermatogenicfailure. Am J Hum Genet.
71:906–922. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vogt PH, Edelmann A, Kirsch S, Henegariu
O, Hirschmann P, Kiesewetter F, Köhn FM, Schill WB, Farah S, Ramos
C, et al: Human Y chromosome azoospermia factors(AZF) mapped to
different subregions in Yq11. Hum Mol Genet. 5:933–943. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hsu LY: Phenotype/karyotype correlations
of Y chromosome aneuploidy with emphasis on structural aberrations
in postnatally diagnosed cases. Am J Med Genet. 53:108–140. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jaruzelska J, Korcz A, Wojda A,
Jedrzejczak P, Bierla J, Surmacz T, Pawelczyk L, Page DC and
Kotecki M: Mosaicism for 45,X cell line may accentuate the severity
of spermatogenic defects in men with AZFc deletion. J Med Genet.
38:798–802. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reshmi SC, Miller JL, Deplewski D, Close
C, Henderson LJ, Littlejohn E, Schwartz S and Waggoner DJ: Evidence
of a mechanism for isodicentric chromosome Y formation in a
45,X/46,X,idic(Y)(p11.31)/46,X,del(Y)(p11.31) mosaic karyotype. Eur
J Med Genet. 54:161–164. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hernando C, Carrera M, Ribas I, Parear N,
Baraibar R, Egocue J and Fuster C: Prenatal and postnatal
characterization of Y chromosome structural anomalies by molecular
cytogenetic analysis. Prenat Diagn. 22:802–805. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jakubowski L, Jeziorowska A, Constantinou
M and Kałuzewski B: Molecular analysis of Y chromosome long arm
structural instability in patients with gonadal dysfunction. Clin
Genet. 57:291–295. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
DesGroseilliers M, Beaulieu Bergeron M,
Brochu P, Lemyre E and Lemieux N: Phenotypic variability in
isodicentric Y patients: Study of nine cases. Clin Genet.
70:145–150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee J, Park JK, Kim DS, Lee HS, Choi SI
and Cho YG: Detailed analysis of isodicentric Y in a case with
azoospermia and 45,x/46,x,idic(Y) mosaicism. Ann Clin Lab Sci.
45:206–208. 2015.PubMed/NCBI
|
|
13
|
Becker RE and Akhavan A: Prophylactic
bilateral gonadectomy for ovotesticular disorder of sex development
in a patient with mosaic 45,X/46,X,idic(Y)q11.222 karyotype. Urol
Case Rep. 5:13–16. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shinawi M, Cain MP, Vanderbrink BA,
Grignon DJ, Mensing D, Cooper ML, Bader P and Cheung SW: Mixed
gonadal dysgenesis in a child with isodicentric Y chromosome: Does
the relative proportion of the 45,X line really matter? Am J Med
Genet A. 152A:1832–1837. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang Y, Wang R, Li L, Xue L, Deng S and
Liu R: Molecular cytogenetic study of de novo mosaic karyotype
45,X/46,X,i(Yq)/46,X,idic(Yq) in an azoospermic male: Case report
and literature review. Mol Med Rep. 16:3433–3438. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Si YM, Dong Y, Wang W, Qi KY and Wang X:
Hypospadias in a male infant with an unusual mosaic 45,X/46,X,psu
idic(Y)(p11.32)/46,XY and haploinsufficiency of SHOX: A case
report. Mol Med Rep. 16:201–207. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang L, Ren M, Song G, Zhang Y, Liu XX,
Zhang X and Wang J: Prenatal diagnosis of sex chromosomal
inversion, translocation and deletion. Mol Med Rep. 17:2811–2816.
2018.PubMed/NCBI
|
|
18
|
Ghionzoli M, Repele A, Sartiani L,
Costanzi G, Parenti A, Spinelli V, David AL, Garriboli M, Totonelli
G, Tian J, et al: Human amniotic fluid stem cell differentiation
along smooth muscle lineage. FASEB J. 27:4853–4865. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fishman MC and Schaffner AE: Carotid body
cell culture and selective growth of glomus cells. Am J Physiol.
246:C106–C113. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Patsalis PC, Skordis N, Sismani C,
Kousoulidou L, Koumbaris G, Eftychi C, Stavrides G, Ioulianos A,
Kitsiou-Tzeli S, Galla-Voumvouraki A, et al: Identification of high
frequency of Y chromosome deletions in patients with sex chromosome
mosaicism and correlation with the clinical phenotype and
Y-chromosome instability. Am J Med Genet A. 135:145–149. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kotzot D, Dufke A, Tzschach A,
Baeckert-Sifeddine IT, Geppert M, Holland H, Florus JM and Froster
UG: Molecular breakpoint analysis and relevance of variable
mosaicism in a woman with short stature, primary amenorrhea,
unilateral gonadoblastoma, and a 46,X,del(Y)(q11)/45,X karyotype.
Am J Med Genet. 112:51–55. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
DesGroseilliers M, Fortin F, Lafrenière
AM, Brochu P, Lemyre E and Lemieux N: Dynamic increase of a 45,X
cell line in a patient with multicentric ring Y chromosomes.
Cytogenet Genome Res. 115:90–93. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Telvi L, Lebbar A, Del Pino O, Barbet JP
and Chaussain JL: 45,X/46,XY mosaicism: Report of 27 cases.
Pediatrics. 104:304–308. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lange J, Skaletsky H, van Daalen SK, Embry
SL, Korver CM, Brown LG, Oates RD, Silber S, Repping S and Page DC:
Isodicentric Y chromosomes and sex disorders as byproducts of
homologous recombination that maintains palindromes. Cell.
138:855–869. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lahn BT and Page DC: Functional coherence
of the human Y chromosome. Science. 278:675–680. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Skaletsky H, Kuroda-Kawaguchi T, Minx PJ,
Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J,
Bieri T, et al: The male-specific region of the human Y chromosome
is a mosaic of discrete sequence classes. Nature. 423:825–837.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Delbridge ML, Lingenfelter PA, Disteche CM
and Graves JA: The candidate spermatogenesis gene RBMY has a
homologue on the human X chromosome. Nat Genet. 22:223–224. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsui S, Dai T, Roettger S, Schempp W,
Salido EC and Yen PH: Identification of two novel proteins that
interact with germ-cell-specific RNA-binding proteins DAZ and
DAZL1. Genomics. 65:266–273. 2000. View Article : Google Scholar : PubMed/NCBI
|