Introduction
As a white, colorless and crystalline powder, sodium
azide (NaN3) is classed as a highly toxic substance. Its
toxic effects are similar to those of cyanide, and it injures the
nervous and cardiovascular systems, eyes and skin. This toxic
element becomes active rapidly following ingestion, and its major
effects occur several hours following oral intake, depending on the
amount ingested (1,2). Exposure to NaN3 may induce
a number of symptoms within minutes, including nausea, vomiting,
headache, restlessness, dizziness, weakness, rapid breathing and
rapid heartbeat (3). High amounts
of this toxic element immediately induce convulsions, loss of
consciousness, low heart rate and blood pressure, and respiratory
failure, eventually leading to mortality (3). Among the demonstrated action
mechanisms of NaN3, the most relevant one is cytochrome
c oxidase-respiratory chain complex-inhibition (4). Previous studies have revealed that
NaN3, an inhibitor of complex IV (Cox IV), may induce
apoptosis in primary cortical neurons, which is caspase-3 dependent
and associated with the release of cytochrome c (5).
In mitochondrial biosynthesis, the promoter of
respiratory chain Cox IV may be activated by nuclear respiratory
factors (Nrf)-1/2. Concomitantly, Nrf may be adjusted by regulating
the activity of genes encoding mitochondrial transcription factor A
(Tfam) to indirectly regulate the expression levels of respiratory
chain genes. Nrf-1/2, Tfam, peroxisome proliferator-activated
receptor γ co-activator 1-α (Pgc-1α) and other co-activators
constitute the Pgc-1α signal cascade, which serves a central role
in a regulatory network governing the transcriptional control of
mitochondrial biogenesis and respiratory function. In this signal
cascade, Pgc-1α first activates Nrf-1/2 as opposed to directly
binding to the mitochondrial DNA, and Nrf-1/2 induces the
activation of Tfam in combination with the promoter of Tfam and
triggers the transcription and replication of mitochondrial DNA,
leading to increased expression levels of mitochondrial proteins
(6). Concurrently, Pgc-1α may be
activated by CaN-, Ca2+/calmodulin-dependent protein
kinase (CaMK)-, mitogen-activated protein kinase (MAPK)- and
cyclin-dependent kinase-mediated signaling pathways (7). In the present study, PC12 cells were
used to generate a dopamine neuron model, and the effects and
mechanism of NaN3 on the Pgc-1α-associated pathways in
PC12 cells were investigated, to identify whether NaN3
induced toxicity in cultured PC12 cells and the underlying
mechanisms involved in these effects.
Materials and methods
Materials
Rat pheochromocytoma PC12 cells were purchased from
the Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences (Shanghai, China). Stock solution of NaN3
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was dissolved in
the sterile saline to make a 1 M stock solution, which was
subsequently diluted to desired concentrations prior to
experimentation. A one-step TUNEL Apoptosis Assay kit (C1090),
Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit
(C1063), Enhanced adenosine 5′-triphosphate (ATP) Assay kit
(S0027), JC-1 Mitochondrial Membrane Potential Assay kit (C2006)
and Reactive Oxygen Species Assay kit (S0033) were provided by
Beyotime Institute of Biotechnology (Haimen, China).
Cell culture and viability assay
PC12 cells were maintained in Dulbecco's modified
Eagle's medium (DMEM; Hyclone; GE Healthcare Life Sciences, Logan,
Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1%
Antibiotic Antimycotic solution consisting of 10,000 U penicillin
and 10,000 U streptomycin. The cells were incubated at 37°C in
humidified atmosphere of 5% CO2 and always used at
70–80% confluence.
The viability of PC12 cells was determined using a
Cell Counting Kit (CCK-8; Dojindo Molecular Technologies, Inc.,
Shanghai, China). Cells were seeded into 96-well plates at a
density of 1×105/well. After 24 h culture, cells were
treated with NaN3 (0–80 mM) and incubated for 12, 24, 48
and 72 h to establish the cell injury model. Subsequently, 10 µl
CCK-8 solution (Dojindo Molecular Technologies, Inc.) was added to
each well and incubated for an additional 2 h under the standard
conditions (37°C and 5% CO2). The absorbance at a
wavelength of 450 nm was determined by ELx808 Absorbance Microplate
Reader (BioTek Instruments, Inc., Winooski, VT, USA). The cell
viability was calculated according to the mean optical density of 6
wells. The experiments were conducted in triplicate. The
appropriate concentrations of NaN3 for use in subsequent
experiments were determined according the results of the cell
viability assays.
Nuclear morphology of DAPI-stained
PC12 cells
PC12 cells were exposed to 0, 10, 20 and 40 mM
NaN3 for 24 h. In order to distinguish programmed cell
death from non-apoptotic cell death, nuclei were stained with 10
µg/ml DAPI (C1005; Beyotime Institute of Biotechnology). Briefly,
cells were washed twice with PBS and then fixed with 4%
paraformaldehyde at room temperature for 30 min. Subsequent to
three washes, fixed cells were stained with DAPI (1:5,000) for 5
min and then washed with PBS. Fluorescence images were acquired
with a Leica DMI fluorescence microscope (magnification, ×400).
Measurement of apoptotic rate
An Annexin V-FITC/PI Apoptosis Detection kit
(Beyotime Institute of Biotechnology) was used to determine
apoptosis of cells according to the manufacturer's instructions.
Experiments were repeated in triplicate and were performed as
follows: The apoptotic rates of control PC12 cells (0 mM
NaN3) and PC12 cells exposed to 10, 20 and 40 mM
NaN3 for 24 h was measured by flow cytometry (FC500;
Beckman Coulter, Inc., Brea, CA, USA). Statistical analyses were
conducted with SPSS statistical software v.13.0 (SPSS, Inc.,
Chicago, IL, USA).
Measurement of mitochondrial membrane
potential (ΔΨm)
ΔΨm is a significant parameter of mitochondrial
function. It was assessed using staining with JC-1, a fluorescent
probe. Experiments were repeated in triplicate, and were performed
as follows: Control PC12 cells were treated with 0 mM
NaN3; and experimental PC12 cells were exposed to 10, 20
and 40 mM NaN3 for 24 h. Subsequently, according to the
manufacturer's protocol (C2006; Mitochondrial Membrane Potential
Assay kit; Beyotime Institute of Biotechnology, Haimen, China),
cells were incubated with the medium containing JC-1 (1X) at 37°C
for 20 min, the cells were washed three times with wash buffer and
collected with fresh medium without serum. Concomitantly, the
positive control was treated with carbonyl cyanide
3-chlorophenylhydrazone (CCCP), an inhibitor, (10 µM) at 37°C for
20 min. Then, the red/green fluorescence was determined by FCM
(FC500; Beckman Coulter, Inc.). The ratios of red fluorescence
intensity over green fluorescence intensity represented the levels
of ΔΨm.
Measurement of reactive oxygen species
(ROS) production
ROS in PC12 cells was assessed using a Reactive
Oxygen Species Assay kit (S0033; Beyotime Institute of
Biotechnology, Haimen, China). Intracellular ROS generation was
assessed by means of 2′,7′-dichlorofluorescein diacetate (DCFH-DA),
a fluorescent probe. Intracellular ROS oxidizes DCFH-DA, yielding
the fluorescent compound 2′,7′-dichlorofluorescein (DCF), and DCF
fluorescence intensity is considered to be parallel to the amount
of formed ROS, according to the instructions of the ROS assay kit.
Experiments were repeated in triplicate and performed as follows:
Positive control were treated with specific concentration of Rosup
(50 mg/ml); control PC12 cells were treated with 0 mM
NaN3; and experimental PC12 cells were exposed to 10, 20
and 40 mM NaN3 for 24 h. In addition, PC12 cells were
treated with DCFH-DA (10 mM) dissolved in serum-free DMEM (1:1,000)
for 20 min at 37°C and then washed three times with serum-free
DMEM. The positive control was treated with Rosup, to induce ROS
production. The ROS production was determined by FCM (FC500;
Beckman Coulter, Inc.). The Mean fluorescence intensities (MFI)
represented the levels of ROS.
Measurement of cellular ATP content in
PC12 cells
Experiments were repeated in triplicate and
performed as follows: Control PC12 cells were treated with 0 mM
NaN3; and experimental PC12 cells were exposed to
NaN3 at different concentrations (10, 20 and 40 mM) for
24 h. Subsequently, the cellular ATP content was determined using a
Firefly Luciferase ATP Assay kit (Beyotime Institute of
Biotechnology) according to the protocol of the manufacturer.
Western blot analysis
PC12 cells were exposed to 0, 10, 20 and 40 mM
NaN3 for 24 h, lysed in radioimmunoprecipitation assay
lysis buffer (Beyotime Institute of Biotechnology) containing a
protease inhibitor and centrifuged at 13,362 × g for 10 min at 4°C
in order to collect the supernatants. Subsequently, the protein
concentrations were determined using BCA kit (Pierce; Thermo Fisher
Scientific, Inc.). Equal amounts of proteins (90 µg) were subjected
to 10 and 12% SDS-PAGE, and then transferred onto polyvinylidene
fluoride membranes (0.45 µm) using a Semidry Electro-transfer Unit
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Following blocking
with 5% bovine serum albumin in TBS containing 0.1% Tween-20 (TBST)
for 2 h at room temperature, the membranes were incubated with
primary antibodies against Pgc-1α (Abcam, Cambridge, MA, USA;
1:500), Nrf-2 (Abcam; 1:500), Cox IV (Abcam; 1:1,000), Tfam (Abcam;
1:1,000), procaspase-3 (Abcam; 1:500), Nrf-1 (Cell Signaling
Technology, Inc., Danvers, MA, USA, 1:500), pan-calcineurin A (CaN;
Cell Signaling Technology Inc.; 1:1,000), phosphorylated (p)-CaMKII
(Cell Signaling Technology; 1:1,000), p-p38 MAPK (Cell Signaling
Technology, Inc.; 1:1,000), p-extracellular signal-regulated kinase
(Erk)1/2 (Cell Signaling Technology, Inc.; 1:1,000), B-cell
lymphoma-2 (Bcl-2)-associated X protein (Bax; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA; 1:200), Bcl-2 (Santa Cruz
Biotechnology, Inc.; 1:200) and cytochrome c (Santa Cruz
Biotechnology, Inc.; 1:200) at 4°C overnight. The membranes were
then washed with TBST and incubated with horse-radish peroxidase
(HRP)-conjugated secondary antibodies (rabbit; cat. no. A0208;
1:1,000; or mouse; cat. no. A0216; 1;1,000; both Beyotime Institute
of Biotechnology) for 1 h at room temperature. Immunoreactive bands
were visualized by the enhanced chemiluminescence system (Clinx
Science Instruments Co., Ltd., Shanghai, China) and quantitatively
analyzed with Image J version l.32 J (National Institutes of
Health, Bethesda, MD USA), and β-actin was selected as the loading
control.
Statistical analysis
All statistical analyses were conducted with SPSS
statistical software v.13.0 (SPSS, Inc., Chicago, IL, USA). Data
are expressed as the mean ± standard deviation standard error of
the mean. The statistical significance of differences between
groups was determined by one-way analysis of variance followed by
Bonferroni post-hoc tests. P<0.05 was considered to indicate a
statistically significant difference. Each experiment was repeated
least three times.
Results
NaN3 inhibits the growth of
PC12 cells
The effect of NaN3 exposure on the
proliferation of PC12 cells was assessed by CCK-8 assay. Cells were
challenged with different concentrations (0–80 mM) of
NaN3 for 12–72 h. Table
I and Fig. 1A-D indicate that
the cell viability was decreased by NaN3 in a
concentration-dependent manner. Almost 100% of the cells died
following exposure to 80 mM NaN3 for 72 h, indicating
that NaN3 markedly induced cell death, and the
cytotoxicity of NaN3 was detected in a dose- and
time-dependent manner. Exposure to NaN3 at a
concentration of 20 mM for 24 h caused marked cell death in the
PC12 cells (50%). Therefore, the cells cultured for 24 h were used
for subsequent experiments.
 | Figure 1.NaN3 suppresses cell
viability in cultured PC12 cells. Following exposure to 0, 5, 10,
20, 40 and 80 mM NaN3 for 24 h, the survival rate was
decreased to 100, 39.33±3.99, 34.86±7.98, 24.89±3.33, 16.89±0.73
and 7.35±2.55%, respectively. The cytotoxicity in PC12 cells was
detected at (A) 12, (B) 24, (C) 48 and (D) 72 h by CCK-8 assay.
*P<0.05 and **P<0.01 vs. the control group. NaN3,
sodium azide. |
 | Table I.NaN3 suppresses the growth
of PC12 cells (n=6). |
Table I.
NaN3 suppresses the growth
of PC12 cells (n=6).
|
| Time interval, h (%
viability) |
|---|
|
|
|
|---|
| NaN3
concentration (mmol/l) | 12 | 24 | 48 | 72 |
|---|
| 0 | 100 | 100 | 100 | 100 |
| 5 | 98.21±7.84 | 39.33±3.99 | 33.69±3.34 | 27.29±12.99 |
| 10 | 95.94±1.47 | 34.86±7.98 | 26.06±3.22 | 19.7±9.91 |
| 20 | 90.57±2.87 | 24.89±3.33 | 12.99±2.83 | 7.52±4.65 |
| 40 | 87.21±0.46 | 16.89±0.73 | 4.40±1.36 | 2.19±1.49 |
| 80 | 74.78±2.97 | 7.35±2.55 | 1.37±0.73 | 0.65±1.44 |
Cell morphology
In DAPI staining, the morphological changes of PC12
cells were observed by fluorescence microscopy following exposure
to different concentrations of NaN3. The fragmentation
of cell nuclei exposed to NaN3 for 24 h was observed,
and cell nuclei shrinkage and chromatin condensation were increased
slightly with the concentration of NaN3 in PC12 cells as
compared to the control. In these cells, apoptotic cells were
smaller and brighter compared with normal cells. Chromatin
condensation and nuclear fragmentation were also observed, whereas
blue nuclei of viable cells were identified in the control group.
In addition, the number of apoptotic cells and the intensity of
green fluorescence were increased in cells exposed to
NaN3 (Fig. 2).
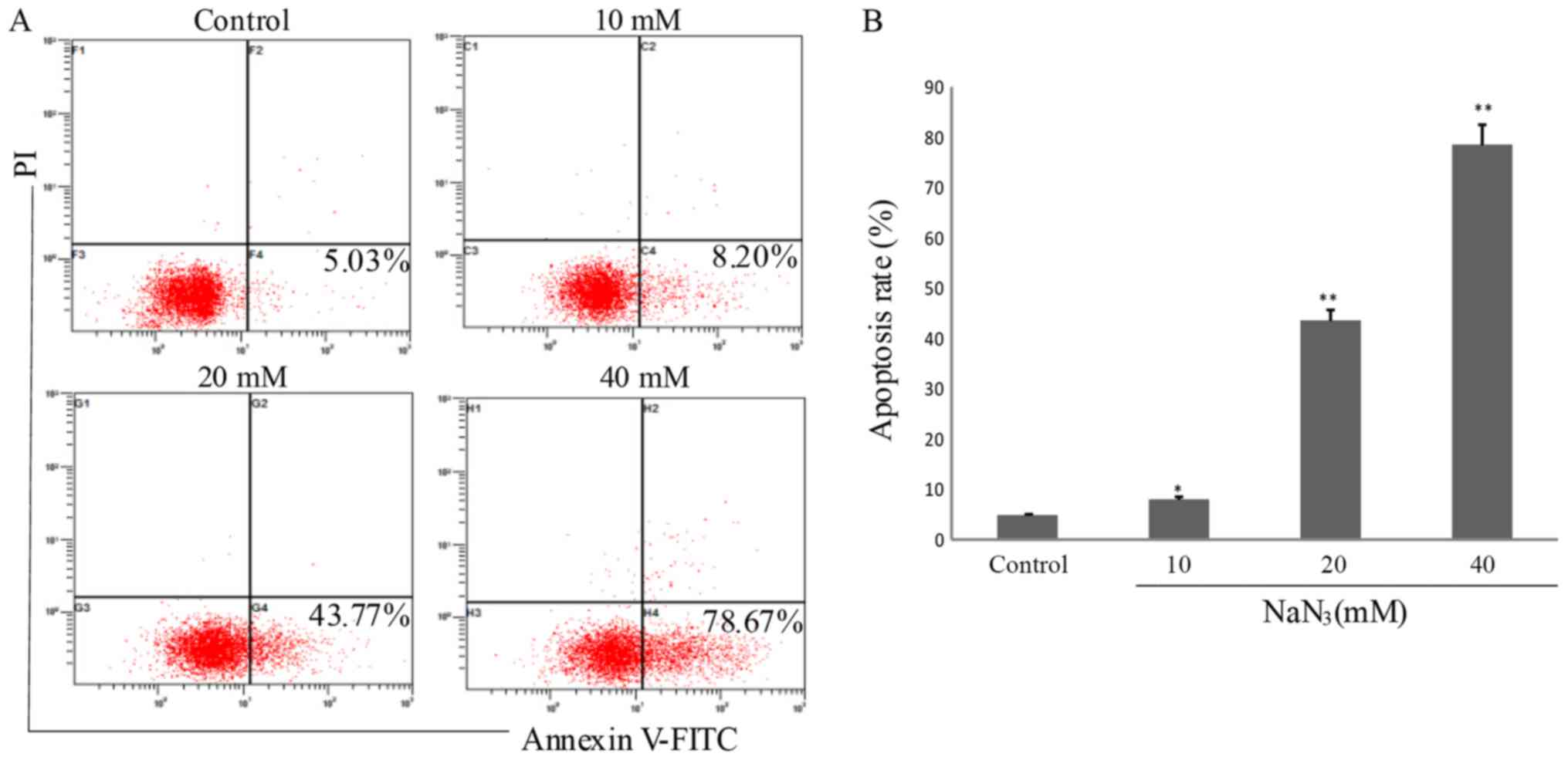
Apoptotic rate determination by
Annexin V-FITC staining
Dead cells or late apoptotic cells that have lost
cell membrane integrity may be stained by propidium iodide. Due to
the loss of this cell membrane integrity, Annexin V-FITC may enter
into the cytoplasm and combine with the phosphatidylserine inside
of the cell membrane, exhibiting green fluorescence in dead cells.
Fig. 3 demonstrates that the
numbers of apoptotic cells were 5.03, 8.02, 43.77 and 78.67%
(P<0.05) in the PC12 cells following exposure to NaN3
at different concentrations (0, 10, 20 and 40 mM) for 24 h,
respectively. These results additionally confirmed that
NaN3 induced cell apoptosis in a dose-dependent
manner.
Changes in mitochondrial membrane
potential (ΔΨm)
Mitochondria are generally considered key regulatory
organelles involved in cell viability. Therefore, ΔΨm was used as
the indicator of mitochondrial function. PC12 cells were stained
with JC-1, a cationic dye that exhibits potential-dependent
accumulation in mitochondria. Red fluorescence (J-aggregates),
representing active mitochondria with stable membrane potential,
was observed in a small number of cells exposed to increasing
concentrations of NaN3. By contrast, green fluorescence
(J-monomer), representing apoptotic mitochondria with damaged ΔΨm,
was observed in a number of cells. The ratio of red and green
fluorescence gradually decreased in the control group (Fig. 4).
NaN3-induced accumulation
of mitochondrial ROS
It is well known that mitochondria are the primary
source of cellular ROS, and ROS serve an important role in
activation of apoptotic signaling. Therefore, the role of ROS in
NaN3-induced PC12 cell death was additionally
investigated. Fig. 5 indicates
that the fluorescence intensities in control cells and cells
exposed to NaN3 at various concentrations (0, 10, 20 and
40 mM) for 24 h were 3.65, 6.99, 18.63 and 39.35, respectively,
indicating that NaN3 exposure significantly increased
the production of mitochondrial ROS compared with the control group
(P<0.05). These results suggested that NaN3-induced
apoptosis improved the production of intracellular ROS.
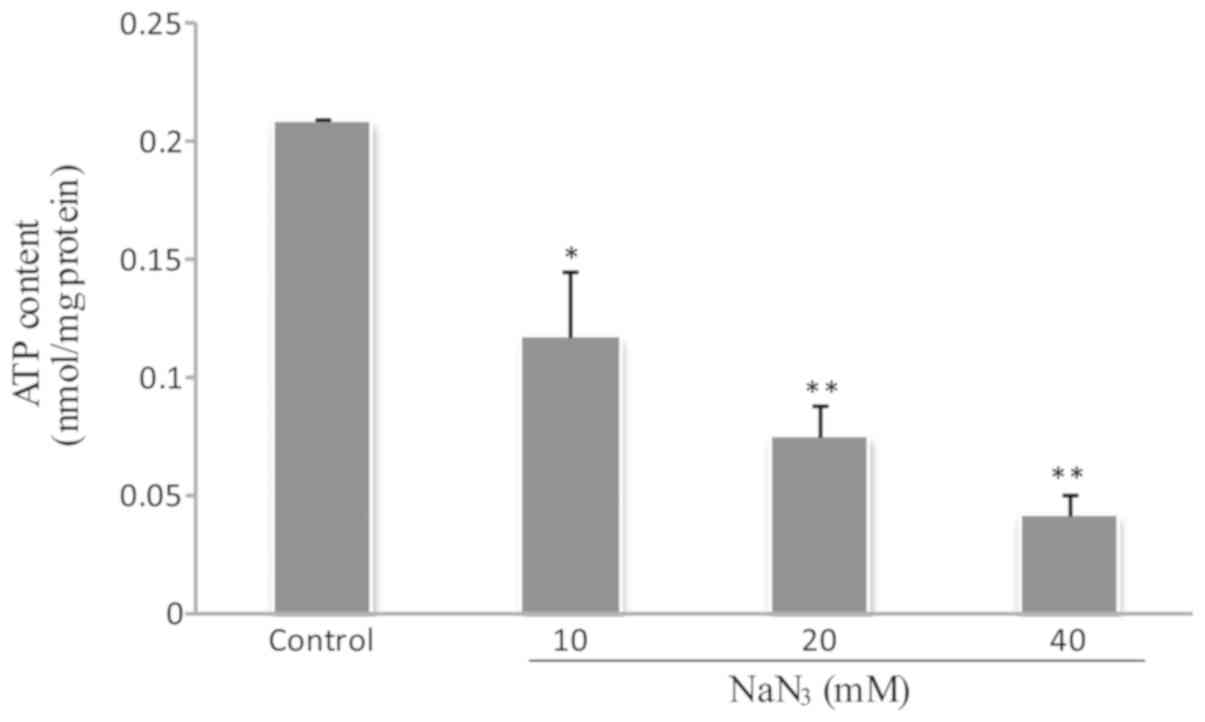
NaN3 downregulates the
mitochondrial energy production of cellular ATP
To evaluate the ATP content in PC12 cells exposed to
NaN3, the relative luminescence unit (RLU) was
quantitatively determined by a luminometer. Fig. 6 indicates that NaN3
inhibited mitochondrial ATP production and gradually decreased the
cellular ATP level (P<0.05).
Expression levels of Pgc-1α and
apoptosis-associated proteins in PC12 cells
The Pgc-1α expression at the protein level was
significantly decreased following NaN3 exposure compared
with the control (P<0.05). In addition, Nrf-1, Tfam, p-Erk1/2,
Nrf-2 and Cox IV are well-known downstream targets of Pgc-1α
dynamics in various cell types (8). The present study identified that
NaN3 exposure significantly inhibited Pgc-1α dynamics in
a dose-dependent manner (P<0.01; Fig. 7A).
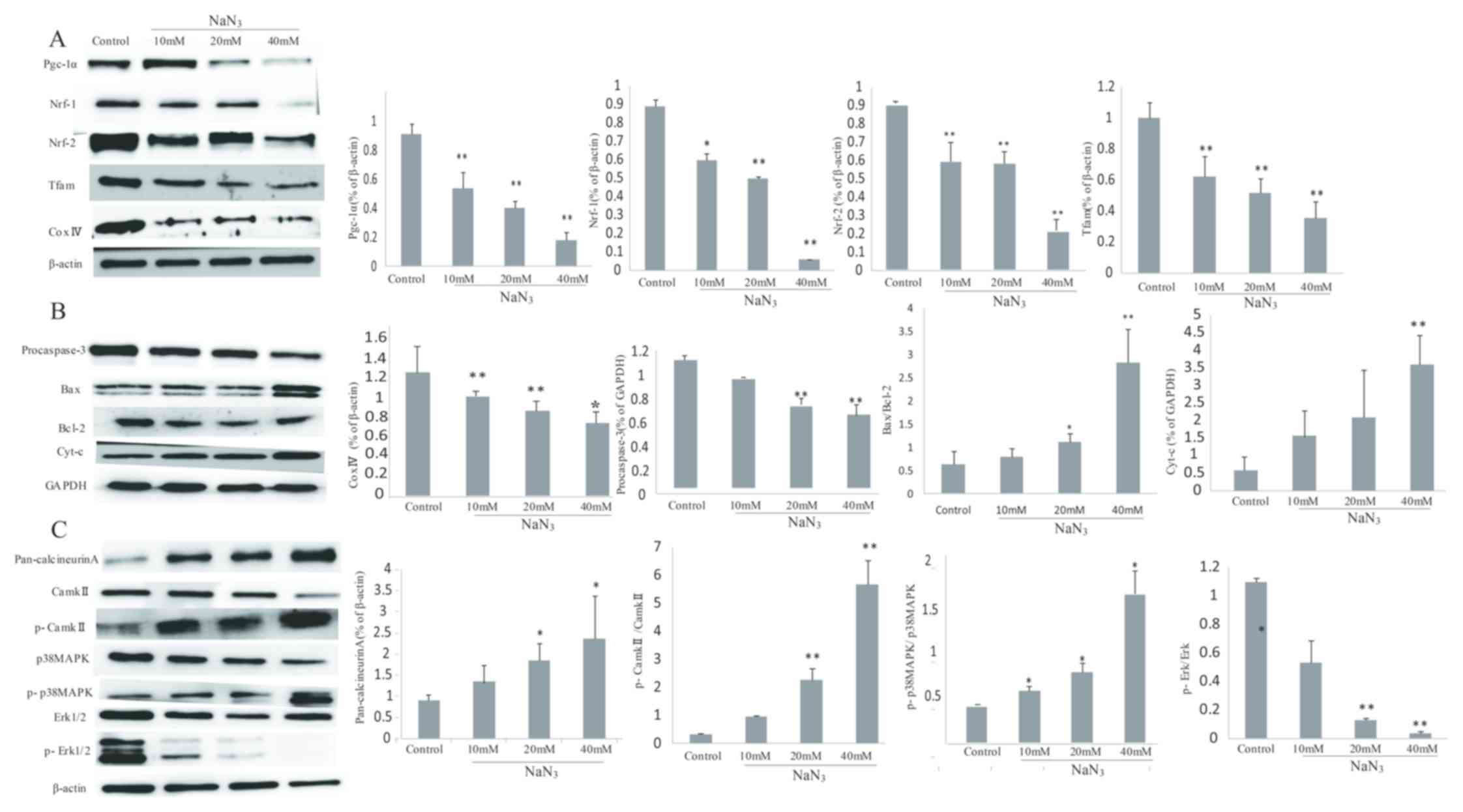 | Figure 7.NaN3 induces
mitochondria-mediated apoptosis through the expression of
Pgc-1α-associated proteins in PC12 cells. (A) Expression levels of
Pgc-1α, Nrf-1, Nrf-2, Tfam and Cox IV detected by western blot
analysis. (B) Expression levels of procaspase-3, Bax, Bcl-2 and
cyt-c detected by western blot analysis. (C) Expression levels of
pan-calcineurin A, CaMKII, p-CaMKII, p38 MAPK, p-p38 MAPK, Erk1/2
and p-Erk1/2 detected by western blot analysis. β-actin and GAPDH
were used as the internal control. Band intensity ratios for each
group are presented as mean ± standard deviation (n=3). *P<0.05
and **P<0.01 vs. the control group. Pgc-1α, peroxisome
proliferator-activated receptor γ co-activator 1-α; Nrf-1/2,
nuclear respiratory factor-1/2; Tfam. Mitochondrial transcription
factor A; Cox IV, complex IV; Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein; cyt-c, cytochrome c; CaMKII,
Ca2+/calmodulin-dependent protein kinase II; p,
phosphorylated; p38 MAPK, p38 mitogen-activated protein kinase;
Erk1/2, extracellular signal-regulated kinase 1/2. |
In addition, NaN3 exposure upregulated
the expression levels of Bax and cytochrome c, while it
downregulated the expression levels of Bcl-2 and procaspase-3
(P<0.05). The ratio of Bax/Bcl-2 was also significantly
increased compared with the control group (P<0.05; Fig. 7B).
The protein expression levels of other important
members, including CaN, CaMKII, p-CaMKII, p38 MAPK and p-p38 MAPK,
were also assessed. The results indicated that NaN3
increased the expression of CaN and the phosphorylation of CaMKII
and p38 MAPK compared with the control group, while the expression
levels of total CaMKII and p38 MAPK were not changed. In addition,
the ratios of p-CaMKII/CaMKII and p-p38/p38 MAPK in the
NaN3 group were significantly increased compared with
those in the control group (P<0.01; Fig. 7C).
Discussion
To determine whether NaN3 inhibited the
proliferation of PC12 cells, the number of treated cells in the
logarithmic phase was compared with the number of non-treated
control cells. Cell growth was inhibited by ~75% after 24 h of
exposure to 20 mM NaN3. Therefore, this concentration
was used for subsequent experiments. Apoptosis involves changes in
cellular morphology, including membrane blebbing, cell shrinkage,
chromatin condensation, nuclear fragmentation and DNA fragmentation
(9). To additionally analyze
nuclear morphology of apoptosis and apoptotic rate, cells were
challenged with 0, 10, 20 and 40 mM NaN3 for 24 h.
Following treatment, cells were then stained with DAPI and Annexin
V-FITC/PI, and the distribution of the nuclei was analyzed. The
results confirmed that NaN3 exposure induced apoptosis
in PC12 cells in a concentration-dependent manner.
Mitochondria are involved in numerous metabolic
functions, including maintenance of intracellular pH and production
of ROS, which promote and regulate cell apoptosis (10). The hallmarks of cell apoptosis are
preceded by mitochondrial alterations, including a loss of ΔΨm, a
decrease in energy production (ATP) and an increase in permeability
of the mitochondrial membrane. Previous studies have suggested that
ROS trigger damage to the mitochondrial respiratory chain and
induce the loss of ΔΨm, which are the factors that mediate or
amplify the neuronal dysfunction during the course of the
neurodegeneration, consequently leading to the development of
neurodegenerative diseases (11,12).
NaN3 is known to drive degeneration and excitotoxicity
by increasing the permeability potential of the mitochondrial
membrane through lipid peroxidation (13–15),
and NaN3 exerts its primary toxic action by inhibiting
the function of cytochrome oxidase in the mitochondrial electron
transport chain and preventing the ATP production (4). In the present study, FCM was used to
identify ΔΨm and ROS production in PC12 cells. In addition, the ATP
synthesis in mitochondria was examined following exposure to
various concentrations of NaN3. The disruption of the
plasma membrane, the increase of mitochondrial ROS production and
the decrease in cellular ATP content observed suggested that
NaN3 exposure induced the apoptosis in PC12 cells.
Mitochondrial cell death may be activated by
multiple stimuli, including the developmental program, DNA damage,
endoplasmic reticulum stress, growth factor and nutrient
deprivation, viral infection and oxidative stress (16). As a member of the ever-growing
family of nuclear co-regulators, Pgc-1α may activate a large set of
genes and regulate the expression levels of genes involved in
energy metabolism in response to signaling pathways that mediate
thermogenesis, gluconeogenesis, muscle fiber type switching and
mitochondrial biogenesis (17).
These co-regulators exist and function in large multi-protein
complexes, in which rather than binding to DNA, they regulate
Nrf-1/2 and Tfam and modulate their transcriptional potency by
promoting the subsequent biochemical interactions required for
induction or repression of gene transcription (8). In addition, Nrf-1/2 also indirectly
controls the expression of mitochondrial DNA-encoded genes by
potently inducing the nuclear-encoded Tfam A, B1 and B2 (Tfam,
Tfb1m and Tfb2m, respectively), which are the regulators of the
transcription and replication of the mitochondrial genome (18). Previous studies have demonstrated
that NaN3 is an inhibitor of the mitochondrial
respiratory chain complex IV, which is frequently affected in
primary mitochondrial disorders (19,20).
In the present study, the expression of Pgc-1α signal cascade,
including Pgc-1α family proteins (Pgc-1α, Nrf-1, Nrf-2 and Tfam)
and Cox IV, was first examined in PC12 cells to verify whether the
signaling events were involved in NaN3-induced
apoptosis. The results suggested that the NaN3-induced
apoptosis was associated with the expression levels of Pgc-1α
family proteins and Cox IV in mitochondria-mediated signaling
pathway. When the concentrations of NaN3 were increased,
the expression levels of these proteins were significantly
decreased, indicating that they may be involved in the activation
and development of apoptosis.
Conversely, complex IV may trigger apoptosis in
primary cortical neurons, which is caspase3-dependent and
associated with the release of cytochrome c (5). It is well known that mitochondria are
key regulators of cell apoptosis. The Bcl-2 family proteins are the
important initiators of the mitochondrial apoptotic pathway. This
family includes the pro-apoptotic proteins Bax, Bcl-2 homologous
antagonist/killer and Bcl-2-associated agonist of cell death and
anti-apoptotic proteins Bcl-2 and Bcl-extra large (Bcl-xL). In
healthy cells, Bax is an inactive cytosolic protein, but it is
translocated into the mitochondria during apoptosis. It exerts its
pro-apoptotic effects by forming a pore in the mitochondrial outer
membrane, through which cytochrome c is released into the
cytoplasm, leading to the activation of caspase 3 (21). The anti-apoptotic proteins Bcl-2
and Bcl-xL suppress the function of Bax by maintaining integrity of
mitochondrial membrane, which prevents the release of cytochrome c
and the activation of caspase 3 (22). In the present study,
NaN3 increased the levels of pro-apoptotic Bax and
cytochrome c and decreased levels of anti-apoptotic Bcl-2 and
procaspase-3, indicating that NaN3 initiated
mitochondrial apoptosis signaling in PC12 cells.
In addition, the expression levels of Pgc-1α
co-activators are highly inducible at the transcriptional level via
a variety of upstream signaling pathways. For example, the
expression of Pgc-1α is induced by exercise and cold exposure under
the control of stress signaling via cellular Ca2+ and
cyclic adenosine 5′-monophosphate (cAMP) signaling (23). The transcription of Pgc-1α may be
affected by CaMK, calcineurin, β-adrenergic receptor/cAMP and p38
MAPK (24–26). In addition, p38 MAPK belonging to
the MAPK family serves a pivotal function in cell proliferation,
differentiation, transformation and apoptosis, since the activation
of apoptosis may induce Pgc-1α via direct phosphorylation (27). In the present study, the expression
levels of proteins in Ca2+ signaling pathways
(pan-calcineurin A and p-CaMKII/CaMKII) and p38 MAPK pathway
(p-p38MAPK/p38 MAPK and p-Erk1/2) were examined to investigate the
effects of NaN3 on intracellular Ca2+
homeostasis and p38 MAPK. The data indicated that the protein
levels of pan-calcineurin A, and the p-CaMKII/CaMKII and p-p38
MAPK/p38 MAPK ratios were increased and p-Erk1/2 level was
decreased in the NaN3-treated group, suggesting that
NaN3 triggered the apoptosis of PC12 cells in a
dose-dependent manner, and such an activation was associated with
the Ca2+ and p38 MAPK pathways. Taken together, these
experimental results confirmed that NaN3 may induce the
apoptosis of PC12 cells by activating Ca2+ and p38 MAPK
pathways. To the best of our knowledge, the present study revealed
for the first time that NaN3 induced
mitochondria-mediated apoptosis in PC12 cells through
Pgc-1α-associated signaling pathways, including
Ca2+/p-CaMKII and p38 MAPK.
In summary, the present study demonstrated that
NaN3 may induce the apoptosis of PC12 cells. In order to
elucidate the underlying toxic mechanism of NaN3
exposure, the expression levels of a series of pro-apoptotic
proteins (Bax and cytochrome c) and anti-apoptotic proteins (Bcl-2,
procaspase-3, p-p38 MAPK, p-CaMKII and Pgc-1α) were examined. The
results confirmed that pro-apoptotic proteins exerted pro-apoptotic
effects on PC12 cells via activation and phosphorylation of CaMKII
and p38 MAPK, which stimulated the activation of Pgc-1α and
procaspase-3 in PC12 cells. The data provide a basis for subsequent
studies investigating NaN3 mechanisms of action at the
molecular level. Future studies may include the addition of
protective agents, for example mitochondrial division inhibitor 1,
a derivative of quinazolinone that is a newly-identified
mitochondrial division inhibitor, prior to NaN3
treatment, in order to investigate the neuroprotective effects of
the protective agent in attenuating NaN3-induced
apoptosis in PC12 cells, and to additionally elucidate the
underlying mechanism.
Acknowledgements
Not applicable.
Funding
The present study was financially supported by the
grants from the National Natural Science Foundation of China (grant
no. 81571848) and the Priority Academic Program Development of
Jiangsu Higher Education Institutions.
Availability of data and materials
The datasets used or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ and SZ conceived and designed the study. YZ, JH,
HX, TG and YW acquired the data. YZ, JH and HX analyzed and
interpreted the data, and drafted the manuscript. All authors
critically revised the manuscript, and read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NaN3
|
sodium azide
|
|
Cox IV
|
complex IV
|
|
Tfam
|
mitochondrial transcription factor
A
|
|
Nrf-1/2
|
nuclear respiratory factor-1/2
|
|
CaN
|
pan-calcineurin A
|
|
Pgc-1α
|
peroxisome proliferator-activated
receptor γ co-activator 1-α
|
|
PC12
|
rat pheochromocytoma
|
|
ΔΨm
|
mitochondrial membrane potential
|
|
CCCP
|
carbonyl cyanide
3-chlorophenylhydrazone
|
|
ROS
|
reactive oxygen species
|
|
FCM
|
flow cytometry
|
|
MAPK
|
mitogen-activated protein kinase
|
References
|
1
|
Herbold M, Schmitt G, Aderjan R and Pedal
I: Fatal sodium azide poisoning in a hospital: A preventable
accident. Arch Kriminol. 196:143–148. 1995.(In German). PubMed/NCBI
|
|
2
|
Marquet P, Clément S, Lotfi H, Dreyfuss
MF, Debord J, Dumont D and Lachâtre G: Analytical findings in a
suicide involving sodium azide. J Anal Toxicol. 20:134–138. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang S and Lamm SH: Human health effects
of sodium azide exposure: A literature review and analysis. Int J
Toxicol. 22:175–186. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leary SC, Hills BC, Lyons CN, Carison CG,
Michaud D, Kraft CS, Ko K, Glerum DM and Moyes CD: Chronic
treatment with azide in situ leads to an irreversible loss of
cytochrome c oxidase activity via holoenzyme dissociation. J Biol
Chem. 277:11321–11328. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grammatopoulos TN, Morris K, Bachar C,
Moore S, Andres R and Weyhenmeyer JA: AngiotensinII attenuates
chemical hypoxia-induced caspase-3 activation in primary cortical
neuronal cultures. Brain Res Bull. 62:297–303. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scarpulla RC: Metabolic control of
mitochondrial biogenesis through the PGC-1 family regulatory
network. Biochim Biophys Acta 1813. 1269–1278. 2011.
|
|
7
|
Qian G, Guo JB and Li L: The role of
Pgc-1α and mitochondrial regulation in cardiovascular disease.
Chinese Pharmacol Bull. 29:1–5. 2013.
|
|
8
|
Jones AW, Yao Z, Vicencio JM,
Karkucinska-Wieckowska A and Szabadkai G: PGC-1 family coactivators
and cell fate: Roles in cancer, neurodegeneration, cardiovascular
disease and retrograde mitochondria-nucleus signaling.
Mitochondrion. 12:86–99. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kerr JFR, Winterford CM and Harmon BV:
Apoptosis: Its significance in cancer and cancer therapy. Cancer.
73:2013–2026. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chan DC: Mitochondria: Dynamic organelles
in disease, ageing and development. Cell. 125:1241–1252. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Islam MT: Oxidative stress and
mitochondrial dysfunctionlinked neurodegenerative disorders. Neurol
Res. 39:73–82. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chaturvedi RK and Flint BM: Mitochondrial
diseases of the brain. Free Radic Biol Med. 63:1–29. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Van Laar VS, Roy N, Liu A, Raiprohat S,
Arnold B, Dukes AA, Holbein CD and Berman SB: Glutamate
excitotoxicity in neurons triggers mitochondrial and endoplasmic
reticulum accumulation of Parkin and in the presence of N-acetyl
cysteine, mitophagy. Neurobiol Dis. 74:180–193. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu L, Peritore C, Ginsberg J, Shinh J,
Arun S and Donmez G: Protective role of SIRT5 against motor deficit
and dopaminergic degeneration in MPTP-induced mice model of
Parkinson's disease. Behav Brain Res. 281:215–221. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Palomo GM and Manfredi G: Exploring new
pathways of neurodegeneration in ALS: The role of mitochondria
quality control. Brain Res 1607. 36–46. 2015. View Article : Google Scholar
|
|
16
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vercauteren K, Pasko RA, Gleyzer N, Marino
VM and Scarpulla RC: PGC-1-related coactivator: Immediate early
expression and characterization of a CREB/NRF-1 binding domain
associated with cytochrome c promoter occupancy and respiratory
growth. Mol Cell Biol. 26:7409–7419. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Scarpulla RC: Transcriptional paradigms in
mammalian mitochondrial biogenesis and function. Physiol Rev.
88:611–638. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Betts J, Lightowlers RN and Turnbull DM:
Neuropathological aspects of mitochondrial DNA disease. Neurochem
Res. 29:505–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tanji K, Kunimatsu T, Vu TH and Bonilla E:
Neuropathological features of mitochondrial disorders. Semin Cell
Dev Biol. 12:429–439. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aluvila S, Mandal T, Hustedt E, Fajer P,
Choe JY and Oh KJ: Organization of the mitochondrial apoptotic BAK
pore: Oligomerization of the BAK homodimers. J Biol Chem.
289:2537–2551. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang E, Zha J, Jockel J, Boise LH,
Thompson CB and Korsmeyer SJ: Bad, a heterodimeric partner for
Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell.
80:285–291. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hock MB and Kralli A: Transcriptional
control of mitochondrial biogenesis and function. Annu Rev Physiol.
71:177–203. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Handschin C, Rhee J, Lin J, Tarr PT and
Spieglman BM: An autoregulatory loop controls peroxisome
proliferator-activated receptor gamma coactivator 1alpha expression
in muscle. Proc Natl Acad Sci USA. 100:7111–7116. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schaeffer PJ, Wende AR, Magee CJ, Neilson
JR, Leone TC, Chen F and Kelly DP: Calcineurin and
calcium/calmodulin-dependent protein kinase activate distinct
metabolic gene regulatory programs in cardiac muscle. J Biol Chem.
279:593–603. 2004. View Article : Google Scholar
|
|
26
|
Rohas LM, St-Pierre J, Uldry M, Jäger S,
Handschin S and Spiegelman BM: A fundamental system of cellular
energy homeostasis regulated by PGC-1 alpha. Proc Natl Acad Sci
USA. 104:7933–7938. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Puigserver P, Rhee J, Lin J, Wu Z, Yoon
JC, Zhang CY, Krauss S, Mootha VK, Lowell BB and Spiegelman BM:
Cytokine stimulation of energy expenditure through p38 MAP kinase
activation of PPARgamma coactivator-1. Mol Cell. 8:971–982. 2001.
View Article : Google Scholar : PubMed/NCBI
|