With the deterioration of the current global
environment, particularly in developing countries, three common
gynecological tumors (cervical cancer, endometrial cancer and
ovarian cancer) have become major threats to women's health
(1). Therefore, it is important to
actively investigate the underlying molecular mechanisms and
potential therapeutic targets associated with such tumors.
Phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) is one
of the signaling pathways proven to serve an important role in
regulating cell proliferation, the cell cycle and apoptosis
(2). A number of studies suggest
that the PI3K/AKT signaling pathway is associated with certain
gynecological tumors (3–5), and the rational design of molecular
targets for the PI3K/AKT signaling pathway is also an important
option for the treatment of tumors. Therefore, the present study
reveals the association between the PI3K/AKT signaling pathway and
the occurrence and development of certain gynecological tumors by
summarizing relevant research and drawing conclusions.
PI3K is a member of the lipid kinases family, which
are activated by phosphorylating the 3-hydroxyl group of the
inositol ring of phosphatidylinositol (PtdIns) lipids in the plasma
membrane (6). PI3K may be divided
into three categories according to its preference for lipid
substrates and different structures (7). Among them, type I PI3K serves an
important role in tumors, and is the subtype that has been studied
most thoroughly. The type I PI3K is composed of p110. A heterodimer
consisting of a catalytic subunit of p110 and a regulatory subunit
p85, wherein there are seven regulatory subunits produced by
different genes and gene combinations: p85α, p85β, p55α, p55γ,
p50α, p101 and p87. The catalytic subunit (p110) has four subtypes,
p110α, p110β, p110γ, and p110δ (7). Typically, the catalytic subunit
(p110) binds to the regulatory subunit to stabilize the protein
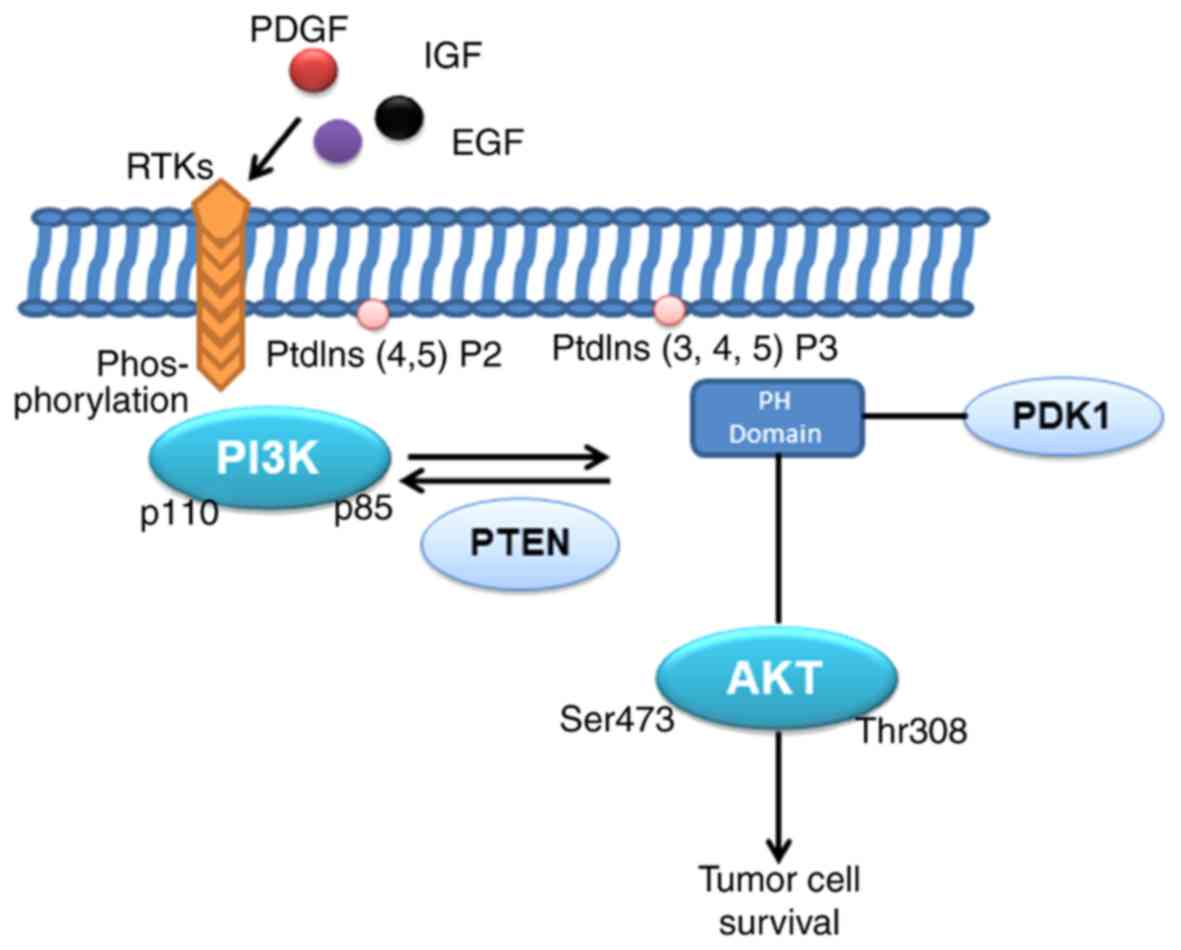
heterodimer and inhibit the activation of PI3K (8). PI3K is normally activated by
extracellular signals under physiological conditions, and there are
two principal activation pathways, involving interaction with a
factor receptor of a phosphorylated tyrosine residue to induce
heterodimer conformational changes and activation (9). Various stimuli, including growth
factors, cytokines and hormones, have been reported to activate
PI3K. In particular, epidermal growth factor (EGF),
platelet-derived growth factor and insulin-like growth factor
(10,11) bind to the corresponding
transmembrane receptor tyrosine kinase (RTK) region of the
N-terminal extracellular domain, resulting in autophosphorylation
of tyrosine residues in the cytoplasmic region of the RTK and the
linker molecule, followed by p85SH2. The interaction between the
domain and the phospho-Tyr residue on the RTK complex recruits PI3K
to RTKs, resulting in the allosteric activation of PI3K (8). In addition to RTKs, G-protein coupled
receptors are another important class of classical upstream
regulators that activate PI3K, the most common being p110β
(12). Furthermore, PI3K may be
directly or indirectly activated by the catalytic subunit (p110) in
combination with small GTPases, including Ras and Ras-related
protein Rab-5A (9,12).
According to previous studies, among the four type I
catalytic subunits, only phosphatidylinositol-4,5-bisphosphate
3-kinase catalytic subunit alpha (PIK3CA) is frequently mutated in
human cancer (13,14). Although there are numerous
mutations in PIK3CA, only two sites have been found to cause an
increase in PI3K activity through different mechanisms (15). The ubiquitously expressed PIK3CB is
less frequently mutated (16),
likely due to the unique regulatory pattern present between the
catalytic subunit and the regulatory subunit (17). Mutations in the type I regulatory
subunit gene (PIK3R1 or PIK3R2) have been identified in cancer
cells and cause an increase in PI3K activity, and it has been
demonstrated through cell transformation experiments that PIK3CA
serves a key role in the potential carcinogenesis of PIK3R1 mutants
(18–20). Moreover, p110α serves an important
role in inducing tumor angiogenesis. Studies have demonstrated that
after knocking out the p110α gene, embryonic development is slow
and fetal vascular formation is defective in the second trimester
(21–23). In cancer cells with the wild-type
PI3K gene, which causes constitutive signal transduction by PI3K,
there is usually carcinogenic damage caused by the upstream
tyrosine kinase or Ras (14).
Phosphatase and tensin homolog (PTEN) lipid phosphatase or inositol
polyphosphate-4-phosphatase type II B deletion is another pathway
for elevated PI3K lipid products, but the inactivation of these
tumor suppressors is not mutually exclusive with mutations in the
PI3K or Ras genes (12,19). Mouse models have also demonstrated
that the loss of PTEN and the PIK3CA mutation together may lead to
the development of ovarian cancer (24). There are a number of polymerization
targets that receive signals generated by the PI3K downstream
cascade, but the most important mediator is the serine/threonine
kinase AKT (8); AKT serves a
dominant role in the signal transduction of the entire PI3K
pathway.
There are three subtypes of the serine/threonine
kinase AKT in mammals, AKT1, AKT2 and AKT3, which are key molecules
in the PI3K signal transduction pathway (25). The amino acid structure of AKT,
from the N-terminus to the C-terminus, includes a plekstrin
homology (PH) domain, the central catalytic domain and the
carboxy-terminal regulatory domain. The PH domain primarily
mediates membrane translocation following AKT activation; the
catalytic domain contains ATP binding sites. Activation of AKT is
dependent on the phosphorylation of its internal Thr308 site; the
regulatory domain at the carboxy terminus contains a large amount
of proline, containing another phosphorylated Ser473 site required
for AKT activation (26–28). In the classical PI3K/AKT activation
mode, AKT and the upstream 3-phosphoinositol-dependent protein
kinase-1 (PDK1) are recruited into the cell membrane via the
interaction of the PH domain with PtdIns (3–5) P3
(PIP3) in PI3K, and phosphorylation of AKT by activation of the
Thr308 site in the T-loop by PDK1 (29). The activated phosphorylated AKT is
transported from the cell membrane to other regions of the cell to
phosphorylate multiple downstream substrates to achieve AKT
function (30). Phosphorylation of
AKT is considered to be isoform-specific (31). Furthermore, activation and
stabilization of AKT is regulated by the phosphorylation of
multiple sites, apart from the two major sites, Thr308 and Ser473.
AKT1 has 31 potential phosphorylation sites, AKT2 has 22
phosphorylation sites, and AKT3 has 18 phosphorylation sites; with
future studies, the number of potential phosphorylation sites for
AKT may increase even further.
The PI3K/AKT signaling pathway is frequently in a
dysregulated state in tumors, and has now become an important
anticancer target (32). The
PI3K/AKT signaling pathway itself serves a major role in regulating
cell physiology and pathology, including cell proliferation,
survival and invasion (Fig. 1).
Some of the activating mutations in PI3K/AKT are also common in
human tumors, and thus may promote tumor growth (2,33).
Additionally, the replacement of E17K by a single amino acid in the
PH domain of AKT results in the recruitment of constitutive AKT to
the cell membrane (34).
Therefore, in recent years, drug targets for PI3K or AKT have been
widely developed and clinical trials have been conducted. It
follows that the PI3K/AKT signaling pathway is closely associated
with tumor development and has garnered much attention.
Specifically, >80% of endometrial cancer cases
have at least one somatic alteration that affects signaling
pathways, and the PI3K/AKT signaling pathway is one of the most
frequently altered biochemical pathways in endometrial cancer
(35). Statistics indicate that
~90% of young endometrial cancer patients have high progesterone
receptor expression and exhibit resistance to progesterone therapy
(36), and it has also been
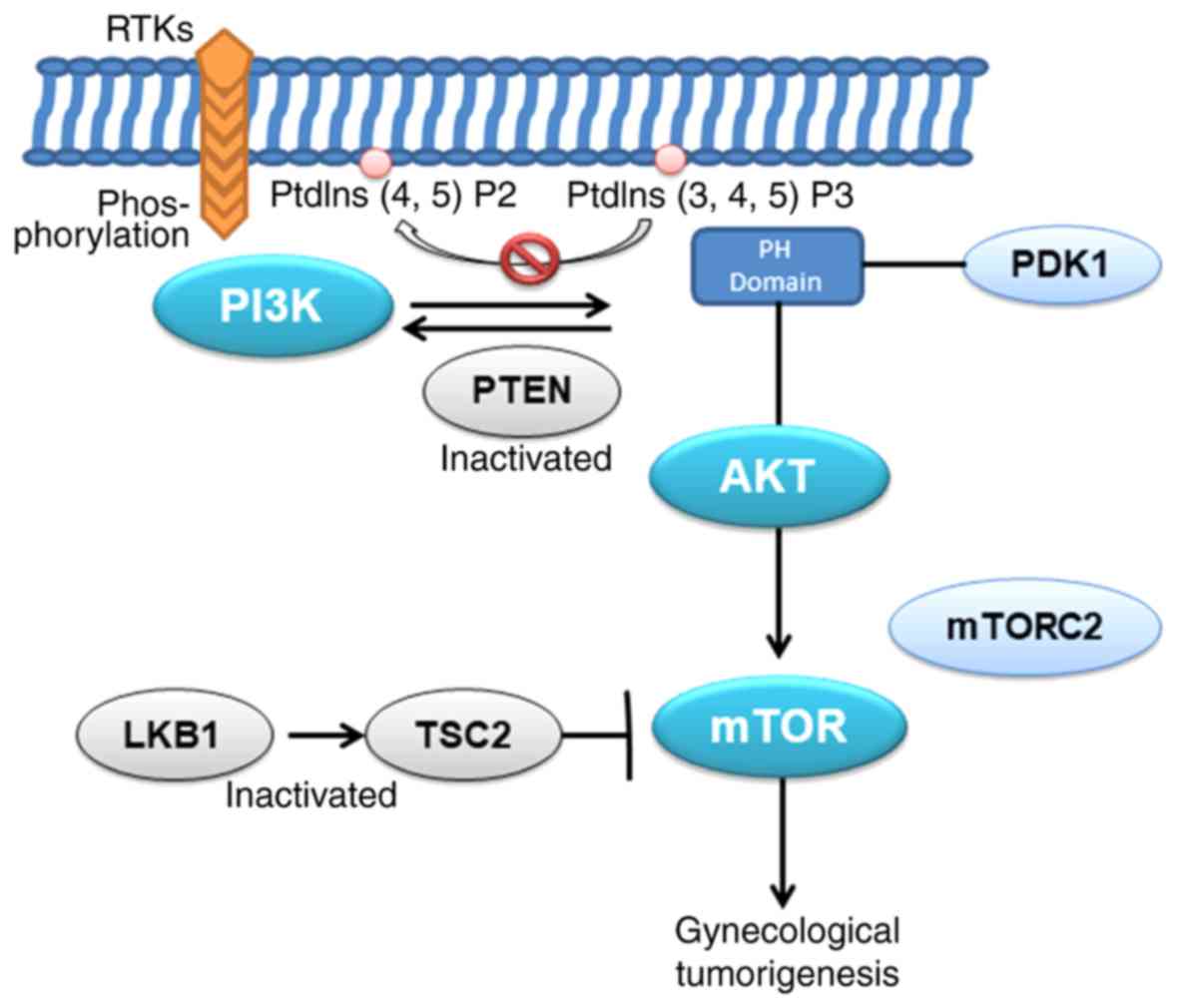
demonstrated that high activation of the PI3K/AKT/mechanistic
target of rapamycin kinase (mTOR) signaling pathway is essential
for the progression of endometrial cancer (37). When inhibiting the phosphorylation
of mTOR, it has a marked inhibitory effect on the proliferation of
endometrial cancer (38). In
addition, studies have suggested that the dysregulation of mTOR in
primary endometrial cancer may also be associated with the loss of
TSC complex subunit 2 (TSC2) and serine/threonine kinase 11 (LKB1)
expression (39). Other studies
also demonstrated that activation of mTOR complex 2 (mTORC2) and
the phosphorylation of AKT are also upregulated in endometrial
cancer, suggesting that the rapamycin-insensitive mTORC2 pathway
may be involved in the development of endometrial cancer (40). These conclusions suggest that the
PI3K/AKT signaling pathway serves a key role in endometrial
cancer.
According to the molecular spectrum of endometrial
cancer reported by The Cancer Genome Atlas in 2013 (41), four major molecular spectra were
analyzed and identified in the genome, transcriptome and proteome
based on array and sequencing technology: DNA polymerase-ε (POLE;
super mutation), microsatellite instability (hypermutation), low
copy number (endometrioid) and high copy number (serous). In
general, the mutation rates of PI3KCA, PIK3R1, AKT1 and PTEN in
endometrial cancer in all cases collected were 59.7, 33, 3.2 and
66%, respectively, and the PIK3R1, AKT1 and PTEN mutation rates in
the POLE group were the highest, reaching 94, 71 and 65%,
respectively.
An important negative regulator in the PI3K pathway
is PTEN, which is located on chromosome 10q23 and encodes a protein
with tyrosine kinase functions (42). PTEN has lipid and protein
phosphatase activity, which leads to cell cycle arrest at the G2/S
checkpoint and inhibits PI3 phosphorylation by dephosphorylation of
PIP3 to phosphatidylinositol 4,5-bisphosphate, leading to a
decrease in intracellular PtdIns levels and affecting downstream
AKT signaling pathways (43);
simultaneously, the PTEN-encoded protein phosphatase inhibits cell
proliferation and migration, and thus the loss of PTEN activity may
result in abnormal cell growth and apoptosis escape. Inactivation
of PTEN may be due to gene mutations, promoter methylation or
protein degradation, leading to loss of expression or a mild loss
of heterozygosity (44–48). However, according to statistics,
20% of cases of endometrial hyperplasia, 55% of precancerous
lesions, and 35–80% of endometrial cancer cases have PTEN mutations
(49). The molecular mechanisms
involved in PTEN mutations may further suggest that the PI3K/AKT
signaling pathway is involved in the early events of endometrial
cancer and promotes the conversion of precancerous lesions into
tumors.
The presence of AKT hyperphosphorylation in cervical
cancer specimens suggests a constitutive activation of the PI3K/AKT
pathway in cervical cancer (50).
The existence of an association between HPV and cervical cancer has
long been under consensus, and mTOR inhibitors blocked the
phosphorylation of eukaryotic translation initiation factor
4E-binding protein 1 and markedly reduced the expression level of
human papillomavirus E7 protein in an in vitro model,
leading to the aggregation of cells in the G1 phase and the
induction of apoptosis (51).
Moreover, studies have reported that changes in genes related to
the PI3K/AKT pathway are associated with an incomplete metabolic
response following chemoradiation in cervical cancer, and that
PIK3CA-activating mutations are associated with long-term survival
post-radiotherapy (52,53). The most common PIK3CA mutation in
cervical cancer is in E545K, a tumor-associated mutation site in
the helical domain of the p110α catalytic subunit of PI3K, which
may lead to constitutive PI3K activation and enhance tumorigenicity
(54–56). C420R in PIK3CA may also induce
oncogenic conversion by promoting the membrane binding of p110α
(57). Activation of the PI3K/AKT
pathway is ubiquitous in various cancer types, and the carcinogenic
effects of PIK3CA mutations have been widely accepted as evidence
for preclinical diagnosis; however, the PIK3CA mutation is not
effective as a biomarker in obese patients with cervical cancer,
which may be due to obesity-associated factors affecting the
transduction of relevant molecules in the PI3K signaling pathway
(58,59).
Ovarian cancer is a type of malignancy that poses a
serious threat to female health worldwide, according to recent
statistics (65,66). In general, the PI3K/AKT signaling
pathway is dysregulated in ovarian cancer, and ~12% of ovarian
cancer cases present with mutations in PIK3CA (67,68).
High-grade serous ovarian cancer is the most important subtype of
ovarian cancer, and ~50% of patients have activations inPIK3CA. In
addition, mutations in PI3K also appear to be associated with the
histology of ovarian cancer. In cases analyzed previously, 20% had
amplifications in PIK3CA, and 5% of the three AKT subtypes
presented with PTEN deletion (67,69).
Studies in a mouse model have demonstrated that ovarian cancer is
triggered by the activation of PIK3CA mutations and a PTEN
deletion, and that inhibition of PI3K/mTOR may prolong tumor growth
and survival. The expression levels of p-AKT and PIK3CA are
associated with ovarian cancer survival, and the activation status
of the PI3K/AKT pathway is considered to be an independent
prognostic marker in ovarian cancer (as measured by AKT and mTOR
phosphorylation levels) (70,71);
PIK3CA mutations predict the response to PI3K and mTOR inhibitors
(72). A study also reported that
alterations in the gene copy number of the catalytic subunits p110α
and p110β of PI3K are associated with poor prognosis in ovarian
cancer (73).
Polycyclic aromatic hydrocarbons, which are
environmental toxins, are known to be reproductive toxins, which
cause primary follicle atresia and premature ovarian failure. It
has been reported that treatment with the PI3K inhibitor LY294002
may prevent follicular atresia (74). In the face of toxic follicle
destruction, follicles attempt to induce cell survival by
upregulating the PI3K/AKT/mTOR pathway, which in turn leads to
increased follicle proliferation, consuming reserves of primitive
ovarian follicles (75).
Therefore, the PI3K/AKT/mTOR pathway in ovarian cancer may be used
as a predictor of damage to primordial follicles during normal
oocyte maturation (75). The
function of the PI3K/AKT pathway in ovarian cancer is very complex,
with two major alterations: Various alterations in the PI3K/AKT
pathway itself, and different effects on the PI3K/AKT pathway.
Through these modifications, PI3K/AKT has been demonstrated to
serve a key role in ovarian cancer development, progression and
chemoresistance. This complexity, which begins with PI3K/AKT
dysregulation, may be due to overactivation and mutations in the
catalytic domain and regulatory domain mutations, or the
modification of downstream targets of PI3K (76). In the development and progression
of ovarian cancer, it is clear that the PI3K/AKT pathway serves an
important role in the complex phenotype of ovarian cancer in unique
ways, many of which may be highly invasive. In serous carcinoma or
low-grade endometrial ovarian cancer, mutations in the pathway may
contribute to cell proliferation, invasion and migration by
modifying cell cycle inhibitors and matrix metalloproteinases
(77).
To date, various PI3K-associated inhibitors with
isomeric properties have been tested in various tumors, although a
number of these drugs have not yet been used in clinical practice,
including GDC0941, XL147, BKM120, NVP-BYL719 (p110α specificity),
SAR260301 (P110β specificity) and TGR-1202 (p110β specificity)
(12,78). PI3K and mTOR are structurally
similar, and there are currently many studies in progress on dual
inhibitors of PI3K and mTOR, such as NVP-BEZ235, GDC-0980 and
XL765, which may be able to overcome the limitations of single
kinase inhibition via the feedback loop (12,78–80).
In addition, different AKT subtypes mediate different
pathophysiological changes in tumors, including AKT1, which
primarily mediates tumorigenesis and early development, whereas
AKT2 appears to primarily promote tumor metastasis (81,82).
Thus the investigation of specific inhibitors of AKT isoforms is
also a promising strategy for the treatment of tumors. Up to now,
the standard drugs targeting the PI3K/AKT pathway, widely used in
clinical practice, have been downstream mTOR blockers, including
temsirolimus and everolimus. Comprehensive clinical studies have
demonstrated that the overall efficiency of mTOR blockers varies
from 4–24% (83). In clinical
trials of ovarian cancer, it was reported in five cases that
sirolimus may be more effective in the treatment of clear cell
ovarian cancer (84). The second
generation of mTOR blockers is also under development. It is
reported that second-generation mTOR blockers contain two protein
complexes, TORC1 and TORC2, which simultaneously inhibit mTORC1 and
mTORC2, thereby reducing the influence of negative feedback AKT
phosphorylation; with this, the therapeutic effect is likely to be
better, and less resistance will occur (85). With ongoing research, the prospect
of drug targets for the PI3K/AKT pathway is becoming a focus for
tumor treatment.
With the development of new knowledge, in addition
to the aforementioned more classical mTOR inhibitors, the targeted
treatment of PI3K/AKT has been gradually enriched. Previous studies
have identified multiple genetic alterations in gynecological
tumors, including PTEN loss, ARID1A mutations, ERBB2
overexpression, and the mutation of PI3K/AKT (86). Excessive activation of PI3K/AKT may
produce resistance to tumors, including endometrial cancer,
targeting RTK inhibitors of EGFR and VEGF, thus drug sensitivity
increases markedly following targeted reduction of this resistance
(87–89). Targeted inhibitors of PI3K, BKM120
and MK-2206, have demonstrated antitumor activity, while MK-2260
exhibits limited single-agent activity in wild-type and mutant
PIK3CA, and this positive role is undoubtedly encouraging (90). In addition, comprehensive genomic
analysis of metastatic cervical cancer has produced important
findings for the identification of novel therapeutic targets and
the targeted treatment of cervical cancer; studies have
demonstrated that the use of FGFR tyrosine kinase inhibitors in
patients with FGFR3-TACC3 fusion expression has achieved good
results and it has been suggested that FGFR3-TACC3 gene fusion
activation is associated with the HPV-induced
carcinogenesis-related FGFR pathway, and these molecular
alterations involve the PI3K/AKT/mTOR and RAF proto-oncogene/MEK
pathways (91,92). Since 2012, CRISPR has been an
active part of cancer research as a powerful gene editing
technique, used for cutting target DNA. Studies have demonstrated
that in PTEN wild-type endometrial cancer cells, the use of
CRISPR/Cas9 to cut PTEN, as a negative regulator of PI3K/AKT, may
enhance the inhibition of cells by a combination of PARP/PI3K
(93); this reveals that
CRISPR/Cas9 may be a useful technology in cancer therapy.
PI3K/AKT is one of most important signal
transduction pathways, which is involved in cell proliferation,
cell cycle regulation, apoptosis and other relevant
pathophysiological processes, and serves a key role in the
occurrence and development of tumors (Fig. 2). Therefore, it is feasible that
relevant drug molecules may be used to block or inhibit the
PI3K/AKT signaling pathway to facilitate the identification of
anti-tumor targets. The specific function of each molecular site
and domain of the pathway in different tumors remains obscure and
requires investigation in future research; however, targeted drugs
for each subunit and site have gained further development and
verification. With the deepening elucidation of the underlying
mechanism by numerous studies, it is considered that drug targets
for the PI3K/AKT signaling pathway will become effective clinical
therapeutic approaches in cancer prevention and treatment.
The authors would like to thank Dr Ping He
(Department of Clinical Laboratory, Guizhou Medical University
Affiliated Hospital, Guizhou, China) for assistance in correcting
the manuscript.
Not applicable.
Data sharing is not applicable to this article, as
no data sets were generated or analyzed during the current
study.
XZ and DT designed the theme of the review. JW, YL
and CC retrieved the relevant literature. XS wrote and reviewed the
article.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Zeng X, Zhang Y, Yang L, Xu H, Zhang T, An
R and Zhu K: Association between RAD51 135 G/C polymorphism and
risk of 3 common gynecological cancers: A meta-analysis. Medicine
(Baltimore). 97:e112512018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rodon J, Dienstmann R, Serra V and
Tabernero J: Development of PI3K inhibitors: Lessons learned from
early clinical trials. Nat Rev Clin Oncol. 10:143–153. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Barra F, Evangelisti G, Ferro Desideri L,
Di Domenico S, Ferraioli D, Vellone VG, De Cian F and Ferrero S:
Investigational PI3K/AKT/mTOR inhibitors in development for
endometrial cancer. Expert Opin Investig Drugs. 28:131–142. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu R, Yang Z, Xu G and Yu S: miR-338
modulates proliferation and autophagy by PI3K/AKT/mTOR signaling
pathway in cervical cancer. Biomed Pharmacother. 105:633–644. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang X, Zhou Y and Gu YE: Tanshinone IIA
induces apoptosis of ovarian cancer cells in vitro and in vivo
through attenuation of PI3K/AKT/JNK signaling pathways. Oncol Lett.
17:1896–1902. 2019.PubMed/NCBI
|
|
6
|
Fruman DA, Meyers RE and Cantley LC:
Phosphoinositide kinases. Annu Rev Biochem. 67:481–507. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vanhaesebroeck B, Guillermet-Guibert J,
Graupera M and Bilanges B: The emerging mechanisms of
isoform-specific PI3K signalling. Nat Rev Mol Cell Biol.
11:329–341. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo H, German P, Bai S, Barnes S, Guo W,
Qi X, Lou H, Liang J, Jonasch E, Mills GB and Ding Z: The PI3K/AKT
pathway and renal cell carcinoma. J Genet Genomicsbao. 42:343–353.
2015. View Article : Google Scholar
|
|
9
|
Osaki M, Oshimura M and Ito H: PI3K-Akt
pathway: Its functions and alterations in human cancer. Apoptosis.
9:667–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Auger KR, Serunian LA, Soltoff SP, Libby P
and Cantley LC: PDGF-dependent tyrosine phosphorylation stimulates
production of novel polyphosphoinositides in intact cells. Cell.
57:167–175. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ruderman NB, Kapeller R, White MF and
Cantley LC: Activation of phosphatidylinositol 3-kinase by insulin.
Proc Natl Acad Sci USA. 87:1411–1415. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fruman DA and Rommel C: PI3K and cancer:
Lessons, challenges and opportunities. Nat Rev Drug Discov.
13:140–156. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Samuels Y and Ericson K: Oncogenic PI3K
and its role in cancer. Curr Opin Oncol. 18:77–82. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vadas O, Burke JE, Zhang X, Berndt A and
Williams RL: Structural basis for activation and inhibition of
class I phosphoinositide 3-kinases. Sci Signal. 4:re22011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dbouk HA, Khalil BD, Wu H, Shymanets A,
Nurnberg B and Backer JM: Characterization of a tumor-associated
activating mutation of the p110β PI 3-kinase. PLoS One.
8:e638332013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Vadas O, Perisic O, Anderson KE,
Clark J, Hawkins PT, Stephens LR and Williams RL: Structure of
lipid kinase p110β/p85β elucidates an unusual SH2-domain-mediated
inhibitory mechanism. Mol Cell. 41:567–578. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jaiswal BS, Janakiraman V, Kljavin NM,
Chaudhuri S, Stern HM, Wang W, Kan Z, Dbouk HA, Peters BA, Waring
P, et al: Somatic mutations in p85alpha promote tumorigenesis
through class IA PI3K activation. Cancer Cell. 16:463–474. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheung LW, Hennessy BT, Li J, Yu S, Myers
AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, et al:
High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer
elucidates a novel mechanism for regulation of PTEN protein
stability. Cancer Discov. 1:170–185. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun M, Hillmann P, Hofmann BT, Hart JR and
Vogt PK: Cancer-derived mutations in the regulatory subunit
p85alpha of phosphoinositide 3-kinase function through the
catalytic subunit p110alpha. Proc Natl Acad Sci USA.
107:15547–15552. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Graupera M, Guillermet-Guibert J, Foukas
LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J,
Cutillas PR, et al: Angiogenesis selectively requires the p110alpha
isoform of PI3K to control endothelial cell migration. Nature.
453:662–666. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Graupera M and Potente M: Regulation of
angiogenesis by PI3K signaling networks. Exp Cell Res.
319:1348–1355. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hirsch E, Ciraolo E, Franco I, Ghigo A and
Martini M: PI3K in cancer-stroma interactions: Bad in seed and ugly
in soil. Oncogene. 33:3083–3090. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kinross KM, Montgomery KG, Kleinschmidt M,
Waring P, Ivetac I, Tikoo A, Saad M, Hare L, Roh V, Mantamadiotis
T, et al: An activating Pik3ca mutation coupled with Pten loss is
sufficient to initiate ovarian tumorigenesis in mice. J Clin
Invest. 122:553–557. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Toulany M, Maier J, Iida M, Rebholz S,
Holler M, Grottke A, Jüker M, Wheeler DL, Rothbauer U and Rodemann
HP: Akt1 and Akt3 but not Akt2 through interaction with DNA-PKcs
stimulate proliferation and post-irradiation cell survival of
K-RAS-mutated cancer cells. Cell Death Discov. 3:170722017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carnero A, Blanco-Aparicio C, Renner O,
Link W and Leal JF: The PTEN/PI3K/AKT signalling pathway in cancer,
therapeutic implications. Curr Cancer Drug Targets. 8:187–198.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tokunaga E, Oki E, Egashira A, Sadanaga N,
Morita M, Kakeji Y and Maehara Y: Deregulation of the Akt pathway
in human cancer. Curr Cancer Drug Targets. 8:27–36. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Manning BD and Toker A: AKT/PKB signaling:
Navigating the network. Cell. 169:381–405. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alessi DR, James SR, Downes CP, Holmes AB,
Gaffney PR, Reese CB and Cohen P: Characterization of a
3-phosphoinositide-dependent protein kinase which phosphorylates
and activates protein kinase Balpha. Curr Biol. 7:261–269. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Andjelković M, Alessi DR, Meier R,
Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM and
Hemmings BA: Role of translocation in the activation and function
of protein kinase B. J Biol Chem. 272:31515–31524. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Girardi C, James P, Zanin S, Pinna LA and
Ruzzene M: Differential phosphorylation of Akt1 and Akt2 by protein
kinase CK2 may account for isoform specific functions. Biochim
Biophys Acta. 1843:1865–1874. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:7–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wong KK, Engelman JA and Cantley LC:
Targeting the PI3K signaling pathway in cancer. Curr Opin Genet
Dev. 20:87–90. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Carpten JD, Faber AL, Horn C, Donoho GP,
Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage
S, et al: A transforming mutation in the pleckstrin homology domain
of AKT1 in cancer. Nature. 448:439–444. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mazloumi Gavgani F, Smith Arnesen V,
Jacobsen RG, Krakstad C, Hoivik EA and Lewis AE: Class I
phosphoinositide 3-Kinase PIK3CA/p110α and PIK3CB/p110β isoforms in
endometrial cancer. Int J Mol Sci. 19(pii): E39312018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Prat J, Gallardo A, Cuatrecasas M and
Catasús L: Endometrial carcinoma: Pathology and genetics.
Pathology. 39:72–87. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee TY, Martinez-Outschoorn UE, Schilder
RJ, Kim CH, Richard SD, Rosenblum NG and Johnson JM: Metformin as a
therapeutic target in endometrial cancers. Front Oncol. 8:3412018.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hua L, Zhang L, Zhang X and Cui Z:
PI3K/AKT/mTOR pathway promotes progestin resistance in endometrial
cancer cells by inhibition of autophagy. Onco Targets Ther.
10:2865–2871. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lu KH, Wu W, Dave B, Slomovitz BM, Burke
TW, Munsell MF, Broaddus RR and Walker CL: Loss of tuberous
sclerosis complex-2 function and activation of mammalian target of
rapamycin signaling in endometrial carcinoma. Clin Cancer Res.
14:2543–2550. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shen Q, Stanton ML, Feng W, Rodriguez ME,
Ramondetta L, Chen L, Brown RE and Duan X: Morphoproteomic analysis
reveals an overexpressed and constitutively activated phospholipase
D1-mTORC2 pathway in endometrial carcinoma. Int J Clin Exp Pathol.
4:13–21. 2010.PubMed/NCBI
|
|
41
|
Cancer Genome Atlas Research Network, ;
Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H,
Robertson AG, Pashtan I, Shen R, et al: Integrated genomic
characterization of endometrial carcinoma. Nature. 497:67–73. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shi B, Wang Y, Zhao R, Long X, Deng W and
Wang Z: Bone marrow mesenchymal stem cell-derived exosomal miR-21
protects C-kit+ cardiac stem cells from oxidative injury through
the PTEN/PI3K/Akt axis. PLoS One. 13:e01916162018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pavlidou A and Vlahos NF: Molecular
alterations of PI3K/Akt/mTOR pathway: A therapeutic target in
endometrial cancer. ScientificWorldJournal. 2014:7097362014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sun H, Enomoto T, Fujita M, Wada H,
Yoshino K, Ozaki K, Nakamura T and Murata Y: Mutational analysis of
the PTEN gene in endometrial carcinoma and hyperplasia. Am J Clin
Pathol. 115:32–38. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kanamori Y, Kigawa J, Itamochi H, Shimada
M, Takahashi M, Kamazawa S, Sato S, Akeshima R and Terakawa N:
Correlation between loss of PTEN expression and Akt phosphorylation
in endometrial carcinoma. Clin Cancer Res. 7:892–895.
2001.PubMed/NCBI
|
|
46
|
Kong D, Suzuki A, Zou TT, Sakurada A, Kemp
LW, Wakatsuki S, Yokoyama T, Yamakawa H, Furukawa T, Sato M, et al:
PTEN1 is frequently mutated in primary endometrial carcinomas. Nat
Genet. 17:143–144. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Quddus MR, Ologun BA, Sung CJ, Steinhoff
MM and Lawrence WD: Utility of PTEN expression of endometrial
‘surface epithelial changes’ and underlying atypical endometrial
hyperplasia. Int J Gynecol Pathol. 28:471–476. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mutter GL: Pten, a protean tumor
suppressor. Am J Pathol. 158:1895–1898. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bansal N, Yendluri V and Wenham RM: The
molecular biology of endometrial cancers and the implications for
pathogenesis, classification, and targeted therapies. Cancer
Control. 16:8–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bertelsen BI, Steine SJ, Sandvei R, Molven
A and Laerum OD: Molecular analysis of the PI3K-AKT pathway in
uterine cervical neoplasia: Frequent PIK3CA amplification and AKT
phosphorylation. Int J Cancer. 118:1877–1883. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Oh KJ, Kalinina A, Park NH and Bagchi S:
Deregulation of eIF4E: 4E-BP1 in differentiated human
papillomavirus-containing cells leads to high levels of expression
of the E7 oncoprotein. J Virol. 80:7079–7088. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schwarz JK, Payton JE, Rashmi R, Xiang T,
Jia Y, Huettner P, Rogers BE, Yang Q, Watson M, Rader JS and
Grigsby PW: Pathway-specific analysis of gene expression data
identifies the PI3K/Akt pathway as a novel therapeutic target in
cervical cancer. Clin Cancer Res. 18:1464–1471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Rashmi R, DeSelm C, Helms C, Bowcock A,
Rogers BE, Rader JL, Grigsby PW and Schwarz JK: AKT inhibitors
promote cell death in cervical cancer through disruption of mTOR
signaling and glucose uptake. PLoS One. 9:e929482014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Isakoff SJ, Engelman JA, Irie HY, Luo J,
Brachmann SM, Pearline RV, Cantley LC and Brugge JS: Breast
cancer-associated PIK3CA mutations are oncogenic in mammary
epithelial cells. Cancer Res. 65:10992–11000. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bader AG, Kang S and Vogt PK:
Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc
Natl Acad Sci USA. 103:1475–1479. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhao JJ, Liu Z, Wang L, Shin E, Loda MF
and Roberts TM: The oncogenic properties of mutant p110alpha and
p110beta phosphatidylinositol 3-kinases in human mammary epithelial
cells. Proc Natl Acad Sci USA. 102:18443–18448. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gymnopoulos M, Elsliger MA and Vogt PK:
Rare cancer-specific mutations in PIK3CA show gain of function.
Proc Natl Acad Sci USA. 104:5569–5574. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ocana A, Vera-Badillo F, Al-Mubarak M,
Templeton AJ, Corrales-Sanchez V, Diez-Gonzalez L, Cuenca-Lopez MD,
Seruga B, Pandiella A and Amir E: Activation of the PI3K/mTOR/AKT
pathway and survival in solid tumors: Systematic review and
meta-analysis. PLoS One. 9:e952192014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Grigsby P, Elhammali A, Ruiz F, Markovina
S, McLellan MD, Miller CA, Chundury A, Ta NL, Rashmi R, Pfeifer JD,
et al: Clinical outcomes and differential effects of PI3K pathway
mutation in obese versus non-obese patients with cervical cancer.
Oncotarget. 9:4061–4073. 2017.PubMed/NCBI
|
|
60
|
Bosch FX, Lorincz A, Muñoz N, Meijer CJ
and Shah KV: The causal relation between human papillomavirus and
cervical cancer. J Clin Pathol. 55:244–265. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Libby G, Donnelly LA, Donnan PT, Alessi
DR, Morris AD and Evans JM: New users of metformin are at low risk
of incident cancer: A cohort study among people with type 2
diabetes. Diabetes Care. 32:1620–1625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Eskander RN and Tewari KS: Exploiting the
therapeutic potential of the PI3K-AKT-mTOR pathway in enriched
populations of gynecologic malignancies. Expert Rev Clin Pharmacol.
7:847–858. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ko J, Lee YH, Hwang SY, Lee YS, Shin SM,
Hwang JH, Kim J, Kim YW, Jang SW, Ryoo ZY, et al: Identification
and differential expression of novel human cervical cancer oncogene
HCCR-2 in human cancers and its involvement in p53 stabilization.
Oncogene. 22:4679–4689. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cho GW, Shin SM, Namkoong H, Kim HK, Ha
SA, Hur SY, Kim TE, Chai YG and Kim JW: The phosphatidylinositol
3-kinase/Akt pathway regulates the HCCR-1 oncogene expression.
Gene. 384:18–26. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gui T and Shen K: The epidermal growth
factor receptor as a therapeutic target in epithelial ovarian
cancer. Cancer Epidemiol. 36:490–496. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Mabuchi S, Kuroda H, Takahashi R and
Sasano T: The PI3K/AKT/mTOR pathway as a therapeutic target in
ovarian cancer. Gynecol Oncol. 137:173–179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Levine DA, Bogomolniy F, Yee CJ, Lash A,
Barakat RR, Borgen PI and Boyd J: Frequent mutation of the PIK3CA
gene in ovarian and breast cancers. Clin Cancer Res. 11:2875–2878.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Andorfer P, Heuwieser A, Heinzel A, Lukas
A, Mayer B and Perco P: Vascular endothelial growth factor A as
predictive marker for mTOR inhibition in relapsing high-grade
serous ovarian cancer. BMC Syst Biol. 10:332016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Cheaib B, Auguste A and Leary A: The
PI3K/Akt/mTOR pathway in ovarian cancer: Therapeutic opportunities
and challenges. Chin J Cancer. 34:4–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mabuchi S, Kawase C, Altomare DA,
Morishige K, Sawada K, Hayashi M, Tsujimoto M, Yamoto M,
Klein-Szanto AJ, Schilder RJ, et al: mTOR is a promising
therapeutic target both in cisplatin-sensitive and
cisplatin-resistant clear cell carcinoma of the ovary. Clin Cancer
Res. 15:5404–5413. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Di Nicolantonio F, Arena S, Tabernero J,
Grosso S, Molinari F, Macarulla T, Russo M, Cancelliere C, Zecchin
D, Mazzucchelli L, et al: Deregulation of the PI3K and KRAS
signaling pathways in human cancer cells determines their response
to everolimus. J Clin Invest. 120:2858–2866. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Altomare DA, Hui QW, Skele KL, De Rienzo
A, Klein-Szanto AJ, Godwin AK and Testa JR: AKT and mTOR
phosphorylation is frequently detected in ovarian cancer and can be
targeted to disrupt ovarian tumor cell growth. Oncogene.
23:5853–5857. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Borman SM, Christian PJ, Sipes IG and
Hoyer PB: Ovotoxicity in female Fischer rats and B6 mice induced by
low-dose exposure to three polycyclic aromatic hydrocarbons:
Comparison through calculation of an ovotoxic index. Toxicol Appl
Pharmacol. 167:191–198. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Sobinoff AP, Nixon B, Roman SD and
McLaughlin EA: Staying alive: PI3K pathway promotes primordial
follicle activation and survival in response to 3MC-induced
ovotoxicity. Toxicol Sci. 128:258–271. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Cancer Genome Atlas Research Network, .
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Dobbin ZC and Landen CN: The importance of
the PI3K/AKT/MTOR pathway in the progression of ovarian cancer. Int
J Mol Sci. 14:8213–8227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Cho D: Novel targeting of
phosphatidylinositol 3-kinase and mammalian target of rapamycin in
renal cell carcinoma. Cancer J. 19:311–315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
O'Reilly KE, Rojo F, She QB, Solit D,
Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al:
mTOR inhibition induces upstream receptor tyrosine kinase signaling
and activates Akt. Cancer Res. 66:1500–1508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Figlin RA, Kaufmann I and Brechbiel J:
Targeting PI3K and mTORC2 in metastatic renal cell carcinoma: New
strategies for overcoming resistance to VEGFR and mTORC1
inhibitors. Int J Cancer. 133:788–796. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Dillon RL, Marcotte R, Hennessy BT,
Woodgett JR, Mills GB and Muller WJ: Akt1 and akt2 play distinct
roles in the initiation and metastatic phases of mammary tumor
progression. Cancer Res. 69:5057–5064. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Endersby R, Zhu X, Hay N, Ellison DW and
Baker SJ: Nonredundant functions for Akt isoforms in astrocyte
growth and gliomagenesis in an orthotopic transplantation model.
Cancer Res. 71:4106–4116. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Behbakht K, Sill MW, Darcy KM, Rubin SC,
Mannel RS, Waggoner S, Schilder RJ, Cai KQ, Godwin AK and Alpaugh
RK: Phase II trial of the mTOR inhibitor, temsirolimus and
evaluation of circulating tumor cells and tumor biomarkers in
persistent and recurrent epithelial ovarian and primary peritoneal
malignancies: A gynecologic oncology group study. Gynecol Oncol.
123:19–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Takano M, Kikuchi Y, Kudoh K, Goto T,
Furuya K, Kikuchi R, Kita T, Fujiwara K, Shiozawa T and Aoki D:
Weekly administration of temsirolimus for heavily pretreated
patients with clear cell carcinoma of the ovary: A report of six
cases. Int J Clin Oncol. 16:605–609. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Pétignylechartier C, Duboc C, Jebahi A,
Louis MH, Abeilard E, Denoyelle C, Gauduchon P, Poulain L and
Villedieu M: The mTORC1/2 Inhibitor AZD8055 strengthens the
efficiency of the mek inhibitor trametinib to reduce the Mcl-1/[Bim
and Puma] ratio and to sensitize ovarian carcinoma cells to
ABT-737. Mol Cancer Ther. 16:102–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Itamochi H, Oishi T, Oumi N, Takeuchi S,
Yoshihara K, Mikami M, Yaegashi N, Terao Y, Takehara K, Ushijima K,
et al: Whole-genome sequencing revealed novel prognostic biomarkers
and promising targets for therapy of ovarian clear cell carcinoma.
Br J Cancer. 117:717–724. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Slomovitz BM and Coleman RL: The
PI3K/AKT/mTOR pathway as a therapeutic target in endometrial
cancer. Clin Cancer Res. 18:5856–5864. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Sahoo SS, Lombard JM, Ius Y, O'Sullivan R,
Wood LG, Nahar P, Jaaback K and Tanwar PS: Adipose-Derived
VEGF-mTOR signaling promotes endometrial hyperplasia and cancer:
Implications for obese women. Mol Cancer Res. 16:309–321. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Philip CA, Laskov I, Beauchamp MC, Marques
M, Amin O, Bitharas J, Kessous R, Kogan L, Baloch T, Gotlieb WH and
Yasmeen A: Inhibition of PI3K-AKT-mTOR pathway sensitizes
endometrial cancer cell lines to PARP inhibitors. BMC Cancer.
17:6382017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Han C, Altwerger G, Menderes G, Haines K,
Feinberg J, Lopez S, Manzano A, Varughese J and Santin AD: Novel
targeted therapies in ovarian and uterine carcinosarcomas. Discov
Med. 25:309–319. 2018.PubMed/NCBI
|
|
91
|
Tamura R, Yoshihara K, Saito T, Ishimura
R, Martínez- Ledesma JE, Xin H, Ishiguro T, Mori Y, Yamawaki K,
Suda K, et al: Novel therapeutic strategy for cervical cancer
harboring FGFR3-TACC3 fusions. Oncogenesis. 7:42018. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Carneiro BA, Elvin JA, Kamath SD, Ali SM,
Paintal AS, Restrepo A, Berry E, Giles FJ and Johnson ML:
FGFR3-TACC3: A novel gene fusion in cervical cancer. Gynecol Oncol
Rep. 13:53–56. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Bian X, Gao J, Luo F, Rui C, Zheng T, Wang
D, Wang Y, Roberts TM, Liu P, Zhao JJ and Cheng H: PTEN deficiency
sensitizes endometrioid endometrial cancer to compound PARP-PI3K
inhibition but not PARP inhibition as monotherapy. Oncogene.
37:341–351. 2018. View Article : Google Scholar : PubMed/NCBI
|