Introduction
Prostate cancer is the most common malignant tumor
in men, with a mortality rate second to lung cancer (1). Nearly 1.3 million new prostate cancer
cases and 359,000 associated deaths were reported worldwide in 2018
(2). Although surgery or
radiotherapy can effectively improve the prognosis and prolong the
survival of prostate cancer patients, the recurrence and metastasis
rates remain high. Therefore, identification of novel potential
anticancer targets for the development of therapeutic treatments is
critical.
p53 is a classic tumor suppressor. Overexpression of
p53 not only inhibits the growth of prostate cancer cells but also
enhances the chemosensitivity of prostate cancer cells (3,4).
Several studies have demonstrated that peroxisome
proliferator-activated receptor γ coactivator-1α (PGC-1α) is
expressed at high levels in p53-mutant or p53-deleted tumor cells
and have revealed that PGC-1α plays a role in cancer progression.
For example, Shiota et al (5) reported that PGC-1α was highly
expressed in prostate cancer PC3 and DU145 cells with p53 deletion
or mutation and promoted tumor cell growth. Ogasawara et al
(6) revealed that PGC-1α was highly
expressed in p53-deficient chronic lymphocytic leukemia cells and
maintained normal mitochondrial function. The R72 variant of mutant
p53 promoted tumor metabolism and metastasis by enhancing the
function of PGC-1α (7). These
findings indicate that the deletion or mutation of p53 may
contribute to the enhancement of PGC-1α expression and function in
tumor cells. However, the effect of wild-type p53 on PGC-1α in
tumor cells is unclear.
PGC-1α is a member of the peroxisome
proliferator-activated receptor γ coactivator 1 family that
coordinates the activity of transcription factors to modulate
energy metabolism and other cellular processes (8). As a primary regulator of mitochondrial
biogenesis, PGC-1α functions in the activation of the nuclear
respiratory factor 1 (NRF1) transcription factor, leading to the
transcription of both nuclear-encoded mitochondrial genes (such as
SDHA) and the transcription factor A, mitochondrial
(TFAM) (9,10). PGC-1α is also involved in the
regulation of mitochondrial fission/fusion; PGC-1α promotes
mitochondrial fusion through transcriptional activation of
mitofusin (MFN) 1 and 2 and affects mitochondrial fission
via direct regulation of dynamin-related protein 1 (DRP1)
expression by binding to its promoter (11–14).
Peng et al (15) revealed
that rotenone reduces TFAM, MFN2 and DRP1 expression by inhibiting
PGC-1α in pheochromocytoma PC12 cells, resulting in decreased
mitochondrial copy number, imbalance of fission/fusion and cell
death. Thus, PGC-1α-mediated regulation of mitochondrial biogenesis
and fission/fusion not only affects mitochondrial function but also
determines the survival and death of tumor cells. These findings
indicate that PGC-1α may be a potential target for cancer
therapy.
The present study examined the effect of p53 on
PGC-1α and revealed that p53 decreased the expression of
mitochondrial biogenesis and fission/fusion-associated genes by
inhibiting PGC-1α, leading to mitochondrial dysfunction and
ultimately apoptosis. These findings revealed that PGC-1α is a
crucial target of p53-induced apoptosis in PC3 prostate cancer
cells and indicated that targeting PGC-1α may provide a new
therapeutic strategy for prostate cancer.
Materials and methods
Cell culture and reagents
Human prostate cancer cell lines PC3 and DU145 were
purchased from the American Type Culture Collection and cultured in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.) containing
10% fetal bovine serum (Invitrogen; Thermo Fisher Scientific,
Inc.). ZLN005, a transcriptional activator of PGC-1α, was purchased
from MedChemExpress LLC. Hoechst 33342 and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were purchased from Sigma-Aldrich (Merck KGaA). Anti-β-actin (cat.
no. sc-47778), anti-p53 (cat. no. sc-6243), anti-Mfn1 (cat. no.
sc-166644), anti-Mfn2 (cat. no. sc-100560), anti-DRP1 (cat. no.
sc-271583), anti-Bak (cat. no. sc-832) and anti-Bcl-2 (cat. no.
sc-509) antibodies were purchased from Santa Cruz Biotechnology,
Inc. Anti-NRF1 (cat. no. A5547) and anti-TFAM (cat. no. A1926)
antibodies were purchased from ABclonal Biotech Co., Ltd.
Anti-PGC-1α (cat. no. 66369-Ig) and anti-SDHA (cat. no. 14865-1-AP)
antibodies were purchased from ProteinTech Group, Inc.
Anti-cleaved-caspase-3 (product no. 9664) was purchased from Cell
Signaling Technology, Inc.
Transfection and drug treatment
The pcDNA3.1 vector (negative control) and the
full-length p53 expression vector were purchased from Shanghai
GeneChem Co., Ltd. Transfections were performed using TurboFect
Transfection Reagent (Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. Briefly, PC3 cells were seeded
(5×105 cells/well) into 6-well plates and cultured
overnight at 37°C. Subsequently, cells were transfected with p53
expression vector (4 µg) or pcDNA3.1 (4 µg). Following transfection
for 4–6 h at 37°C, the PGC-1α activator ZLN005 was added and the
cells were cultured for 24 h at 37°C prior to subsequent
experiments. shRNA sequences targeting human PGC-1α and a
non-target sequence were constructed by GeneChem Co., Ltd.
(Shanghai, China.), as previously described (16). The sequences of PGC-1α shRNA and the
non-target shRNA were 5′-GTTATACCTGTGATGCTTT-3′ and
5′-TTCTCCGAACGTGTCACGT-3′, respectively. Cells were transfected
with shRNAs (4 µg) using the GV248 vector (GeneChem, Inc.),
according to the aforementioned protocol.
Immunofluorescence staining and
confocal laser microscopy
Coverslips were placed in 24-well plates and
8×104 cells were added into each well. The transfection
efficiency of the p53 plasmid and the co-localization of p53 with
PGC-1α were examined using indirect immunofluorescence, as
previously described (17). Stained
cells were observed in five random fields of view using an FV1000
confocal laser microscope (Olympus Corporation; magnification,
×200).
Reverse transcription-quantitative
(RT-q) PCR
Cells were cultured in 6-well plates at a density of
5×105 cells/well. Total RNA was extracted from cells
using TRIzol® (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. Subsequently,
total RNA was reverse transcribed into cDNA using 0.5 µg total RNA
and the SuperScript Pre-amplifcation system (Promega Corporation),
according to the manufacturer's protocol. qPCR was performed using
the CFX96 Touch™ Real-Time PCR detection system (Bio-Rad
Laboratories, Inc.), as previously described (15), and the TransStart Top Green qPCR
SuperMix (cat. no. AQ131; Beijing Transgen Biotech Co., Ltd.), with
a reaction system consisting of 1 µl cDNA, 0.2 µM forward primer,
0.2 µM reverse primer, 10 µl qPCR SuperMix and 8.6 µl nuclease-free
water. The following thermocycling conditions were used for qPCR:
Initial denaturation at 94°C for 30 sec, 40 cycles of 94°C for 5
sec and a final extension at 60°C for 30 sec. The following primer
pairs were used for qPCR: NRF1 forward,
5′-GCTACTTACACCGAGCATAGTA-3′ and reverse,
5′-CTCAAATACATGAGGCCGTTTC-3′; TFAM forward,
5′-TTCCAAGAAGCTAAGGGTGATT-3′ and reverse,
5′-AGAAGATCCTTTCGTCCAACTT-3′; SDHA forward,
5′-GTCCCTCCAATTAAACCAAACG-3′ and reverse,
5′-GTTCCGATGTTCTTATGCTTCC-3′; MFN1 forward,
5′-GTGGCAAACAAAGTTTCATGTG-3′ and reverse,
5′-CACTAAGGCGTTTACTTCATCG-3′; MFN2 forward,
5′-GTGCTTCTCCCTCAACTATGAC-3′ and reverse,
5′-ATCCGAGAGAGAAATGGAACTC-3′; DRP1 forward,
5′-GAGATGGTGTTCAAGAACCAAC-3′ and reverse,
5′-CAATAACCTCACAATCTCGCTG-3′; and β-actin forward,
5′-CCTGGCACCCAGCACAAT-3′ and reverse, 5′-GGGCCGGACTCGTCATAC-3′.
mRNA expression levels were quantified using the 2−ΔΔCq
method (18) and normalized to the
internal reference gene β-actin.
Western blot analysis
Cells were lysed using RIPA lysis buffer (Takara
Bio, Inc.), sonicated at 20 kHz for 30 sec on ice and incubated on
ice for 45 min. Cell lysates were centrifuged at 3,000 × g for 15
min at 4°C. Total protein was quantified using the Coomassie G250
assay (Beyotime Institute of Biotechnology). Protein samples (20-30
µg) were separated by 12% w/v SDS-PAGE and transferred onto PVDF
membranes. The PVDF membranes were blocked with 5% skim milk powder
for 2 h at room temperature. Subsequently, the membranes were
incubated overnight at 4°C with primary antibodies (primary
antibodies purchased from Santa Cruz Biotechnology, Inc. were
diluted at 1:200; primary antibodies purchased from ProteinTech
Group, Inc. or Cell Signaling Technology, Inc. were diluted at
1:1,000.). Anti-β-actin was use as loading control. Following
primary incubation, the membranes were incubated with
horseradish-peroxidase-conjugated secondary antibodies (1:2,000;
cat.no. SA00001-1 and SA00001-2; ProteinTech Group, Inc.) for 2 h
at room temperature. Protein bands were visualized using Pierce ECL
Western Blotting Substrate (Thermo Fisher Scientific, Inc.) and
imaged using a Syngene BioImaging system (Synoptics). Protein
expression was quantified using ImageJ software (version 1.48;
National Institutes of Health) with β-actin as the loading
control.
Oxygen consumption rate (OCR)
OCR was measured using the MitoXpress®
Xtra-Oxygen Consumption assay (Luxcel Biosciences Ltd.) as
previously reported (19).
Mitochondrial mass
Coverslips were placed in 24-well plates and
8×104 cells were added into each well. Mitochondria were
stained in live cells using MitoTracker™ Red CMXRos (Invitrogen;
Thermo Fisher Scientific, Inc.), as previously described (16).
Cell viability assays
Cells were cultured (1.2×104 cells/well)
in 96-well plates overnight at 37°C and subsequently cultured for
24 h at 37°C after the addition of various concentrations of ZLN005
(5,10,15 or 20 µM). MTT assays were performed as previously
described (19). Briefly, 10 µl MTT
(10 mg/ml) reagent was added to each well and incubated for 4–6 h.
The formazan crystals were dissolved with 150 µl DMSO and the
absorbance was measured at a wavelength of 490 nm.
Annexin V and cell death assay
Cells were cultured in 6-well plates
(5×105 cells/well) and transfection was performed within
24 h. At 4–6 h post-transfection, ZLN005 (15 µM) was added to the
culture. After incubation for 24 h at 37°C, the Annexin V Apoptosis
Detection Kit II (BD Biosciences) was used to measure cellular
apoptosis, according to the manufacturer's protocol. Apoptotic
cells were detected by flow cytometry using a BD Accuri C6
cytometer (BD Biosciences), and data analysis was conducted using
BD Accuri C6 software (version 1.0.264.21; BD Biosciences).
Dataset analysis
All dataset analysis results were obtained from
cBioPortal (http://www.cbioportal.org/), which hosts multiple
cancer genomics studies, including all of the data from The Cancer
Genome Atlas (TCGA). Patient survival was determined by
Kaplan-Meier and log-rank analyses and correlation analysis was
determined by Spearman and Pearson methods.
Statistical analysis
Each experiment was repeated three times. Results
are expressed as the mean ± standard deviation. One-way analysis of
variance was performed to compare results among the groups,
followed by Bonferroni's multiple comparison test to determine
statistical significance. All statistical analyses were performed
using SPSS 24 statistical software (IBM Corp.).
Results
p53 mutation is associated with
decreased disease-free survival in prostate cancer patients
The TCGA data demonstrated that 12% of prostate
cancer patients harbor p53 mutations (Fig. 1A). Most of these mutations occur in
the p53 DNA-binding domain and severely impair the function of the
p53 protein (Fig. 1B). Survival
analysis demonstrated no significant difference in overall survival
between patients with p53 mutations and p53 non-mutation patients,
but the disease-free survival rate of patients with p53 mutations
was decreased compared to p53 non-mutation patients (Fig. 1C). Unlike p53, the mutation rate of
PGC-1α in prostate cancer patients was only 0.8% and these
mutations had a weak effect on the function of PGC-1α (Fig. S1A); therefore, the finding that
PGC-1α mutation reduced the survival rate requires verification
using a larger sample size (Fig.
S1B).
p53 inhibits the protein expression
and nuclear localization of PGC-1α
The deletion or mutation of p53 may contribute to
the enhancement of PGC-1α expression and function. However, whether
p53 negatively regulates PGC-1α is unclear. To explore the effect
of wild-type p53 on PGC-1α, p53-deleted PC3 prostate cancer cells
were transfected with a construct that overexpresses p53.
Immunofluorescence staining (Fig.
2A) and western blot analysis (Fig.
2B) confirmed that p53 was successfully overexpressed in PC3
cells transfected with the p53 overexpression vector, while no p53
was detected in negative control (NC) cells. Next, the protein
expression and nuclear localization of PGC-1α in response to p53
overexpression was examined. As revealed in Fig. 2C, a decrease in PGC-1α protein
expression was detected in p53-overexpressing cells compared with
the NC cells. Furthermore, immunofluorescence assay demonstrated
that the nuclear staining of PGC-1α observed in NC cells was
decreased upon p53 overexpression (Fig.
2D). These results indicated that p53 inhibited the protein
expression and nuclear localization of PGC-1α. In addition,
correlation analysis demonstrated that p53 and PGC-1α tended to be
negatively associated, which is consistent with the results of the
present study, although the P-value was not significant (Fig. S1C).
Inhibition of PGC-1α is involved in
p53-induced mitochondrial dysfunction
Lack of PGC-1α has been reported to be a major cause
of mitochondrial dysfunction (20).
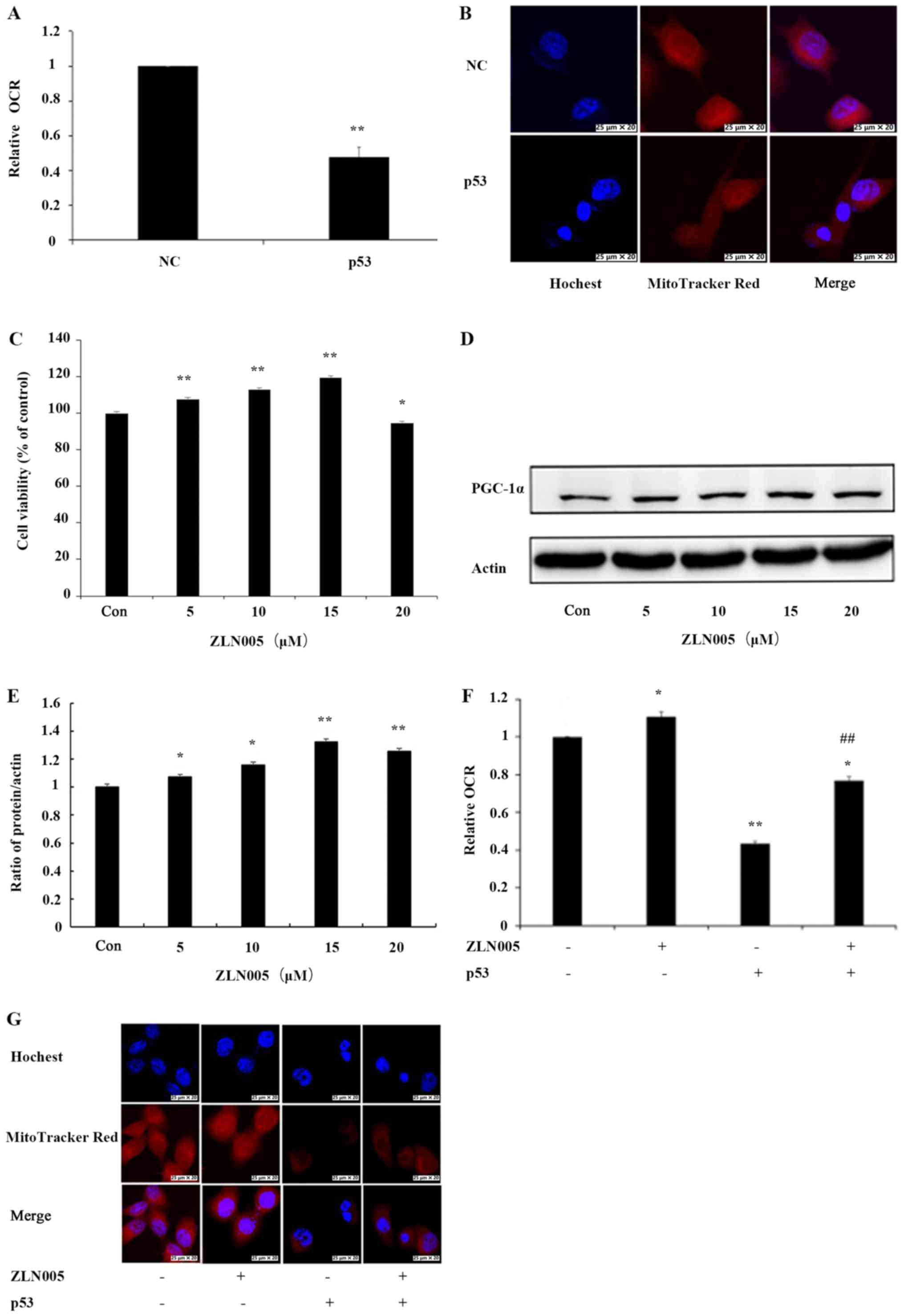
OCR and mitochondrial mass were examined to determine the effect of
p53 on mitochondrial function. The effect of p53 on OCR in PC3
cells was examined and there was a decrease in OCR by 52% in PC3
cells transfected with the p53 overexpression vector compared with
NC cells (Fig. 3A). To evaluate the
effect of p53 on mitochondrial mass, PC3 cells were stained with
MitoTracker Red, a fluorescence dye that stains mitochondria in a
mass-dependent fashion. A marked decrease in mitochondrial mass in
p53-overexpressing PC3 cells compared with NC cells was observed
(Fig. 3B). These results confirmed
that p53 induced mitochondrial dysfunction in PC3 cells.
To examine whether p53-induced mitochondrial
dysfunction involves inhibition of PGC-1α, the mitochondrial
function in PC3 cells after activation of PGC-1α by ZLN005 was
examined. First, PC3 cells were treated with various concentrations
of ZLN005. MTT assays indicated that treatment of PC3 cells with
ZLN005 at concentrations of ≤15 µM resulted in increased cell
survival (Fig. 3C). Next, PGC-1α
expression was examined by western blotting. The results indicated
that the levels of the PGC-1α protein were significantly increased
as the concentration of ZLN005 increased (Fig. 3D and E). A concentration of 15 µM,
which demonstrated beneficial effects on cell survival, was
selected for subsequent experiments. p53-overexpressing cells were
treated with ZLN005 and the effects on OCR and mitochondrial mass
were examined. The OCR was increased by 30% in PC3 cells with p53
overexpression treated with ZLN005 compared with PC3 cells
overexpressing p53 alone (Fig. 3F).
Similarly, the mitochondrial mass of PC3 cells with p53
overexpression treated with ZLN005 was also increased compared to
cells with p53 overexpression alone (Fig. 3G). These results indicated that
inhibition of PGC-1α may be involved in p53-induced mitochondrial
dysfunction.
p53 decreases the expression of genes
and proteins associated to mitochondrial biogenesis and
fission/fusion by inhibiting PGC-1α
The results of the present study confirmed that
activation of PGC-1α by ZLN005, a PGC-1α activator, ameliorated the
mitochondrial dysfunction induced by p53. Since PGC-1α regulates
mitochondrial biogenesis and fission/fusion, the gene expression
levels of NRF1, TFAM, SDHA, MFN1/2 and DRP1 were next examined. The
results demonstrated that p53 inhibited the expression of NRF1,
TFAM, SDHA, MFN1/2 and DRP1 genes, while ZLN005 partially reversed
the inhibitory effect of p53 on these genes (Fig. 4A and B). The expression levels of
the corresponding proteins were also examined by western blotting.
As revealed in Fig. 4C-F, p53
inhibited the expression of NRF1, TFAM, SDHA, Mfn1/2 and DRP1,
while ZLN005 partially reversed the inhibitory effect of p53 on the
expression of these proteins. These results indicated that p53
decreased the expression of genes and proteins associated to
mitochondrial biogenesis and fission/fusion by inhibiting
PGC-1α.
 | Figure 4.p53 reduces the expression of genes
and proteins associated to mitochondrial biogenesis and
fission/fusion by inhibiting PGC-1α. The expression levels of (A)
mitochondrial biogenesis and (B) fission/fusion genes were detected
by RT-qPCR. Data are presented as the mean ± standard deviation
(n=3). *P<0.05 vs. the control; #P<0.05 vs. p53.
The expression levels of proteins involved in (C) mitochondrial
biogenesis and (D) fission/fusion were analyzed by western
blotting. Quantitation of (E) NRF1, TFAM and SDHA, and (F) Mfn1,
Mfn2 and DRP1 were assessed using ImageJ software. Data are
presented as the mean ± standard deviation (n=3). *P<0.05 and
**P<0.01 vs. the control; #P<0.05 vs. p53,
##P<0.01 vs. p53. PGC-1α, peroxisome
proliferator-activated receptor γ coactivator-1α; RT-qPCR, reverse
transcription-quantitative PCR; Con, control; NRF1, nuclear
respiratory factor 1; TFAM, transcription factor A, mitochondrial;
SDHA, succinate dehydrogenase complex flavoprotein subunit A; Mfn,
mitofusin; DRP1, dynamin-related protein 1. |
p53/PGC-1α-mediated mitochondrial
dysfunction promotes apoptosis of PC3 prostate cancer cells
Peng et al (15) revealed that changes in mitochondrial
biogenesis and fission/fusion not only affect mitochondrial
function but also determine cell survival and death. To investigate
the effect of the p53/PGC-1α pathway on PC3 prostate cancer cells,
the rate of apoptosis of PC3 prostate cancer cells was examined.
Flow cytometric analysis revealed that the rate of apoptosis was
decreased in p53-overexpressing cells treated with ZLN005 compared
with cells only expressing p53 (Fig. 5A
and B). Western blot analysis demonstrated that the expression
of cleaved caspase-3 and Bak were slightly decreased in
p53-overexpressing cells treated with ZLN005 compared with p53
overexpression alone (Fig. 5C and
D). These results indicated that the p53/PGC-1α pathway
promoted apoptosis and the apoptosis induced by the p53/PGC-1α
pathway may be associated with mitochondrial dysfunction. These
findings identified PGC-1α as an essential target of p53-induced
apoptosis in prostate cancer cells, indicating that targeting
PGC-1α may serve as a new therapeutic strategy for prostate cancer.
Consistent with this possibility, western blot analysis
demonstrated that knockout of PGC-1α promoted the expression of
cleaved caspase-3 in PC3 and DU145 prostate cancer cells and
decreased the expression of Bcl-2 (Fig. S1D).
Discussion
The p53 mutation data in prostate cancer from TCGA
demonstrated that more than 85% of p53 mutations occur in the p53
DNA binding domain. These mutations not only severely damage the
function of the p53 protein, but also reduced the disease-free
survival of prostate cancer patients. The present study explored
the functional association between p53 and PGC-1α by overexpressing
p53 in p53-deficient prostate cancer PC3 cells and revealed that
p53 inhibited the protein expression and nuclear localization of
PGC-1α. The inhibition of PGC-1α by p53 decreased the expression of
genes and proteins associated to mitochondrial biogenesis and
fission/fusion and led to mitochondrial dysfunction and apoptosis.
These results revealed that PGC-1α was an essential target of
p53-induced apoptosis in prostate cancer cells and indicated that
targeting PGC-1α may provide a new therapeutic strategy for
prostate cancer.
A study reported that overexpression of p53
negatively affects the normal mitochondrial homeostasis in HepG2
cells, but the precise mechanism of mitochondrial dysfunction has
not been investigated (21).
Another study demonstrated that PGC-1α promotes prostate cancer
cell growth by activating the AR. However, whether the survival and
death of prostate cancer cells are determined by mitochondrial
function regulated by PGC-1α remains unclear (5). The present study demonstrated that
inhibition of PGC-1α by p53 induced mitochondrial dysfunction and
apoptosis of PC3 cells, indicating an essential involvement of
PGC-1α in p53-mediated control of mitochondrial dysfunction and
apoptosis. The results not only revealed that inhibition of PGC-1α
by p53 is associated with mitochondrial dysfunction but also
confirmed the importance of PGC-1α in cell survival.
The present study focused on the regulation of
PGC-1α by p53. Previous studies have demonstrated that p53
transcriptionally inhibits and promotes PGC-1α in other non-tumor
cells (22,23). For example, p53 binds to the
repressive-954 and −564 regions of the mouse PPARGC1A promoter and
inhibits the expression of PGC-1α (22,23),
However, upon antioxidant glutathione shortage, p53 is released
from the two repressive regions and binds to the −2317 region,
which is positively associated to increased PGC-1α expression
(22). The various activities of
p53 on PGC-1α expression may be associated with redox modification
of critical p53 amino acids that affect its DNA binding activity
(22). The present study revealed
that p53 induced a decrease of PGC-1α protein expression and
nuclear localization in PC3 tumor cells. However, Aquilano et
al (22) demonstrated that p53
binds to the −1237 region in the human PPARGC1A promoter in SH-SY5Y
cells to enhance PGC-1α expression. Collectively, these results
indicate that p53 exhibits various effects on PGC-1α expression in
tumor cells. There are at least two possible reasons for these
contradictory activities of p53 on PGC-1α expression in tumor
cells. First, post-transcriptional modification of p53 may result
in the binding of p53 to different regions of the PGC-1α promoter.
It was hypothesized that the p53-binding region in the human
PPARGC1A promoter may be the functional homolog of the −954 and
−564 regions and not the −1237 region in the mouse PPARGC1A
promoter. Second, there may be protein-protein interactions between
p53 and PGC-1α. Based on the results of the present study,
inhibition of PGC-1α by p53 at the transcriptional level does not
fully explain the decreased expression of PGC-1α in the nucleus.
Protein-protein interactions between p53 and PGC-1α may explain
PGC-1α reduction in the nucleus and this possibility will be the
focus of a future study.
The present study identified that activation of
PGC-1α by ZLN005 resulted in amelioration of the mitochondrial
dysfunction and apoptosis induced by p53. Activation of PGC-1α by
ZLN005 regulated mitochondrial biogenesis and fission/fusion, which
is probably involved in the maintenance of mitochondrial function
and promotion of cell growth and survival. Although previous
studies have demonstrated the effects of ZLN005 in increasing the
expression of PGC-1α, further experiments to evaluate the effect of
PGC-1α overexpression are required, because ZLN005 also
transcriptionally promotes genes encoding the deacetylase SIRT1 and
antioxidant enzymes SOD1 and HO-1 (24–26).
Whether activation of these genes is involved in improving
mitochondrial function and apoptosis is unclear.
In conclusion, the findings of the present study
demonstrated that p53 decreased the expression of mitochondrial
biogenesis and fission-/fusion-associated genes by inhibiting
PGC-1α, leading to mitochondrial dysfunction and ultimately
apoptosis. The results revealed a pro-cancer effect from PGC-1α and
indicated that PGC-1α may be a new therapeutic target for PC3
prostate cancer cells. However, the precise regulatory mechanism
linking p53 and PGC-1α remains to be elucidated and requires
further investigation.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81772794, 81501982,
81672948 and 81572927), Jilin Provincial Research Foundation for
the Development of Science and Technology Projects (grant nos.
20170623021TC and 20160414005GH), Jilin Provincial Industrial
Innovation Project (grant no. 2018C052-7) and Jilin University
Bethune Plan B Projects (grant nos. 2015222 and 2018A02), Jilin
Province Health and Health Technology Innovation Project (grant no.
2018J061) and the Fundamental Research Funds for the Jilin
Universities.
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LS, YL, JS, JL and YNL designed the study. JL and LC
collected and analyzed the data. JL, YX, RG and BY performed the
experiments. JL drafted the manuscript. YNL, JS and YL reviewed the
manuscript. All authors reviewed the manuscript and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ji K, Wang B, Shao YT, Zhang L, Liu YN,
Shao C, Li XJ, Li X, Hu JD, Zhao XJ, et al: Synergistic suppression
of prostatic cancer cells by coexpression of both murine double
minute 2 small interfering RNA and wild-type p53 gene in vitro and
in vivo. J Pharmacol Exp Ther. 338:173–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chappell WH, Lehmann BD, Terrian DM,
Abrams SL, Steelman LS and McCubrey JA: p53 expression controls
prostate cancer sensitivity to chemotherapy and the MDM2 inhibitor
Nutlin-3. Cell Cycle. 11:4579–4588. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shiota M, Yokomizo A, Tada Y, Inokuchi J,
Tatsugami K, Kuroiwa K, Uchiumi T, Fujimoto N, Seki N and Naito S:
Peroxisome proliferator-activated receptor gamma coactivator-1alpha
interacts with the androgen receptor (AR) and promotes prostate
cancer cell growth by activating the AR. Mol Endocrinol.
24:114–127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ogasawara MA, Liu J, Pelicano H, Hammoudi
N, Croce CM, Keating MJ and Huang P: Alterations of mitochondrial
biogenesis in chronic lymphocytic leukemia cells with loss of p53.
Mitochondrion. 31:33–39. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Basu S, Gnanapradeepan K, Barnoud T, Kung
CP, Tavecchio M, Scott J, Watters A, Chen Q, Kossenkov AV and
Murphy ME: Mutant p53 controls tumor metabolism and metastasis by
regulating PGC-1α. Genes Dev. 32:230–243. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Villena JA: New insights into PGC-1
coactivators: Redefining their role in the regulation of
mitochondrial function and beyond. FEBS J. 282:647–672. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Piantadosi CA and Suliman HB:
Transcriptional regulation of SDHa flavoprotein by nuclear
respiratory factor-1 prevents pseudo-hypoxia in aerobic cardiac
cells. J Biol Chem. 283:10967–10977. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li PA, Hou X and Hao S: Mitochondrial
biogenesis in neurodegeneration. J Neurosci Res. 95:2025–2029.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martin OJ, Lai L, Soundarapandian MM,
Leone TC, Zorzano A, Keller MP, Attie AD, Muoio DM and Kelly DP: A
role for peroxisome proliferator-activated receptor γ coactivator-1
in the control of mitochondrial dynamics during postnatal cardiac
growth. Circ Res. 114:626–636. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cartoni R, Léger B, Hock MB, Praz M,
Crettenand A, Pich S, Ziltener JL, Luthi F, Dériaz O, Zorzano A, et
al: Mitofusins 1/2 and ERRalpha expression are increased in human
skeletal muscle after physical exercise. J Physiol. 567:349–358.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guo K, Lu J, Huang Y, Wu M, Zhang L, Yu H,
Zhang M, Bao Y, He JC, Chen H and Jia W: Protective role of PGC-1α
in diabetic nephropathy is associated with the inhibition of ROS
through mitochondrial dynamic remodeling. PLoS One.
10:e01251762015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dabrowska A, Venero JL, Iwasawa R, Hankir
MK, Rahman S, Boobis A and Hajji N: PGC-1α controls mitochondrial
biogenesis and dynamics in lead-induced neurotoxicity. Aging
(Albany NY). 7:629–647. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Peng K, Yang L, Wang J, Ye F, Dan G, Zhao
Y, Cai Y, Cui Z, Ao L, Liu J, et al: The interaction of
mitochondrial biogenesis and fission/fusion mediated by PGC-1α
regulates rotenone-induced dopaminergic neurotoxicity. Mol
Neurobiol. 54:3783–3797. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shen L, Sun B, Sheng J, Yu S, Li Y, Xu H,
Su J and Sun L: PGC1α promotes cisplatin resistance in human
ovarian carcinoma cells through upregulation of mitochondrial
biogenesis. Int J Oncol. 53:404–416. 2018.PubMed/NCBI
|
|
17
|
Xu L, Xie Q, Qi L, Wang C, Xu N, Liu W, Yu
Y, Li S and Xu Y: Bcl-2 overexpression reduces cisplatin
cytotoxicity by decreasing ER-mitochondrial Ca2+ signaling in SKOV3
cells. Oncol Rep. 39:985–992. 2018.PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu Y, Gao WN, Xue YN, Zhang LC, Zhang JJ,
Lu SY, Yan XY, Yu HM, Su J and Sun LK: SIRT3 aggravates
metformin-induced energy stress and apoptosis in ovarian cancer
cells. Exp Cell Res. 367:137–149. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen X, Zhong J, Dong D, Liu G and Yang P:
Endoplasmic reticulum stress-induced CHOP inhibits PGC-1α and
causes mitochondrial dysfunction in diabetic embryopathy. Toxicol
Sci. 158:275–285. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koczor CA, White RC, Zhao P, Zhu L, Fields
E and Lewis W: p53 and mitochondrial DNA: Their role in
mitochondrial homeostasis and toxicity of antiretrovirals. Am J
Pathol. 180:2276–2283. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aquilano K, Baldelli S, Pagliei B, Cannata
SM, Rotilio G and Ciriolo MR: p53 orchestrates the PGC-1α-mediated
antioxidant response upon mild redox and metabolic imbalance.
Antioxid Redox Signal. 18:386–399. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sahin E, Colla S, Liesa M, Moslehi J,
Müller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, et al:
Telomere dysfunction induces metabolic and mitochondrial
compromise. Nature. 470:359–365. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang LN, Zhou HY, Fu YY, Li YY, Wu F, Gu
M, Wu LY, Xia CM, Dong TC, Li JY, et al: Novel small-molecule
PGC-1α transcriptional regulator with beneficial effects on
diabetic db/db mice. Diabetes. 62:1297–1307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li W, Li X, Wang B, Chen Y, Xiao A, Zeng
D, Ou D, Yan S, Li W and Zheng Q: ZLN005 protects cardiomyocytes
against high glucose-induced cytotoxicity by promoting SIRT1
expression and autophagy. Exp Cell Res. 345:25–36. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu Y, Kabba JA, Ruan W, Wang Y, Zhao S,
Song X, Zhang L, Li J and Pang T: The PGC-1α activator ZLN005
ameliorates ischemia-induced neuronal injury in vitro and in vivo.
Cell Mol Neurobiol. 38:929–939. 2018. View Article : Google Scholar : PubMed/NCBI
|