Introduction
Renal ischemia/reperfusion (I/R) is a leading cause
of acute kidney injury (AKI) that typically occurs during renal
surgery and predisposes individuals to severe dysfunction in
multiple organ systems. I/R-induced cell death and inflammation
within renal tissue is associated with complex pathophysiological
processes and promotes the decline in renal function, thereby
leading to the accumulation of metabolic waste product (1). In addition to ischemia-induced renal
tissue damage, reperfusion has also been found to trigger a complex
pathophysiological cascade that involves excessive release of free
radicals and the accumulation of inflammatory cells, subsequently
promoting inflammation and further aggravating local ischemia and
cellular damage (2).
Subfamilies of the nucleotide-binding domain and
leucine-rich repeat containing family serve a key role in the
development of the inflammatory response (3). The NLR family pyrin domain containing
3 (NLRP3) inflammasome (previously known as NACHT, LRR and PYD
domains-containing protein 3 and cryopirin) is the most
characterized inflammasome to date. An increasing number of studies
have reported an association between NLRP3 and renal I/R. In AKI,
NLRP3 is involved in renal I/R injury (4,5). For
example, knockdown of NLRP3 and its adaptor PYD and CARD domain
containing (ASC) alleviates I/R-induced renal dysfunction and
excessive neutrophil influx into kidney tissue (6). In addition, the knockdown of NLRP3
and/or caspase-1 protects mice from AKI induced by sepsis or
lipopolysaccharide (7,8). These findings suggest that the NLPR3
inflammasome may serve a key role in the pathogenesis of renal
damage.
Nrf2 is an endogenous antioxidant gene, located in
the cytosol, which combines with Kelch-like ECH-associated protein
1 (Keap). Once activated, Nrf2 is released from Keap and
translocates into the nucleus, where it subsequently binds to
antioxidant response elements to initiate the transcription of its
downstream antioxidant and cytoprotective target genes, such as
superoxide dismutase, catalase, glutathione peroxidase,
glutathione-S-transferase isozymes, catalytic and modifier subunits
of γ-glutamyl cysteine ligase and NADP(H): Quinone oxidoreductase
(9). It was previously reported
that Nrf2 is associated with AKI induced by environmental insult,
ischemia or xenobiotics (10).
Apart from its reported protective role in tissue injury, previous
studies have also demonstrated that overexpression of Nrf2
suppresses NLRP3 inflammasome activity and ameliorates tissue
injury (11–13). However, the underlying mechanism of
the Nrf2 signaling pathway and its effect on NLRP3 inflammasome
activation in renal I/R remains unknown.
Hydrogen sulfide (H2S) has gained
recognition for its ability to regulate mammalian homeostasis and
fundamental cell processes, such as autophagy (14). A number of studies have reported
that H2S serves a protective role against apoptosis,
oxidative stress and inflammation in renal injury (15–20).
Nevertheless, the underlying protective mechanism of H2S
in renal injury induced by I/R remains unknown. Therefore, the
present study investigated the expression levels of NLRP3 and its
adapter, ASC, in renal I/R model mice, and subsequently determined
the effect of Nrf2 on NLRP3 expression. The present study also
aimed to determine whether H2S protects kidney tissue
against renal injury, inflammation and apoptosis via Nrf2-mediated
NLRP3 inhibition.
Materials and methods
Establishment of renal I/R model and
treatment groups
A total of 24 male wild-type mice (weight, 20–25 g;
age, 6–8 weeks) were obtained from the Laboratory Animal Center of
the Academy of Military Medical Sciences (Beijing, China). In
addition, nine Nrf2-knockout (KO) male mice (weight, 20–25 g; age,
6–8 weeks) were obtained from Junke Biological Co., Ltd. All animal
protocols were approved by the Animal Care and Use Committee of
General Hospital of Tianjin Medical University (approval no.
IRB2020-DW-04; Tianjin, China), and all efforts were made to
minimize animal suffering and the number of animals used. The mice
were acclimated to the environment for 3 days prior to the
experiment at 22°C with 40–60% humidity, a 12-h light/dark cycle
and free access to water and rodent chow. After mice were
anesthetized with intraperitoneal (i.p.) 50 mg/kg sodium
pentobarbital, the bilateral renal pedicles were ligated for 30 min
using microaneurysm clamps. Subsequently, the microaneurysm clamps
were removed. In the control group, surgery was performed using the
same process; however, renal pedicles were not ligated. Following
surgery, 0.9% sodium chloride solution was used to help mice
recover. Mice were then sacrificed following reperfusion for 24 h.
A total of 24 wild-type mice were randomly divided into the
following six groups: i) Control (Con) group (n=9); ii) I/R group
(n=9); iii) I/R+MCC950 group (n=3); and iv) I/R+NaHS group (n=3). A
total of nine Nrf2-KO mice were randomly divided into the following
three groups: i) Con group (n=3); ii) I/R group (n=3); and iii)
I/R+NaHS group (n=3).
According to previous research and preliminary
experiments (21,22), 20 mg/kg MCC950 (Sigma-Aldrich; Merck
KGaA) was injected (i.p.) daily for 14 days prior to surgery in the
I/R+MCC950 group. A total of 50 µmol/kg sodium hydrosulfide [NaHS;
i.p.; diluted in normal (0.9%) saline; Sigma-Aldrich; Merck KGaA]
was injected prior to surgery in the I/R+NaHS group. Once all
experimental procedures were complete, mice were sacrificed by
cervical dislocation and tissue and blood were collected for
subsequent analysis.
Western blotting
Kidney tissue was collected from I/R model mice
following 24 h reperfusion to analyze protein expression levels.
Briefly, total and nucleic protein were extracted using RIPA buffer
(Thermo Fisher Scientific, Inc.) and protein concentration was
determined using a BCA Protein Assay kit (Thermo Fisher Scientific,
Inc.). A total of 40 µg protein per lane was separated via 10%
SDS-PAGE and transferred onto polyvinylidene difluoride membranes,
then blocked with 5% milk for 2 h at room temperature. The
membranes were subsequently incubated with the following primary
antibodies overnight at 4°C: Anti-NLRP3 (1:500; cat. no. ab214185;
Abcam), anti-ASC (1:200; cat. no. sc-514414; Santa Cruz
Biotechnology, Inc.), anti-caspase-1 (1:200; cat. no. sc-56036;
Santa Cruz Biotechnology, Inc.), anti-IL-1β (1:1,000; cat. no.
ab200478; Abcam), anti-Nrf2 (1:1,000; cat. no. ab137550; Abcam),
anti-heme oxygenase (HO)-1 (1:2,000; cat. no. ab68477; Abcam),
anti-β-actin (1:2,000; cat. no. ab8227; Abcam) and anti-histone H3
(1:2,000; cat. no. ab176842; Abcam). Following primary antibody
incubation, the membranes were incubated with the corresponding
HRP-conjugated secondary antibodies (1:5,000; cat nos. ab6721 and
ab6728; both Abcam) for 1 h at room temperature. Protein bands were
visualized by enhanced chemiluminescence kit (Thermo Fisher
Scientific, Inc.) using a Bio-Imaging system (Bio-Rad Laboratories,
Inc.) and semi-quantified using Quantity One (V4.6) software
(Bio-Rad Laboratories, Inc.). Protein expression levels were
normalized to β-actin for cytoplasmic protein or histone for
nucleic protein.
Reverse transcription-quantitative
(RT-q)PCR
Kidney tissue was collected from I/R model mice
following 24 h reperfusion to analyze mRNA expression levels.
Briefly, total RNA was extracted from tissue using
TRIzol® reagent (cat. no. 15596026; Invitrogen; Thermo
Fisher Scientific, Inc.). Total RNA was reverse transcribed into
cDNA using a RevertAid First Strand cDNA Synthesis kit (cat. no.
K1621; Thermo Scientific™; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. qPCR was subsequently
performed using SYBR PCR Master Mix (Thermo Fisher Scientific,
Inc.) on a 7500 Real-Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The thermocycling conditions were as
follows: Denaturation at 95°C for 5 min; 40 cycles at 95°C for 15
sec; and extension at 60°C for 20 sec. The gene-specific primer
sequences are listed in Table I.
The relative expression levels of the target genes were calculated
using the 2−ΔΔCq method (23) and normalized to GAPDH expression
levels.
 | Table I.Primer sequences for reverse
transcription-quantitative PCR. |
Table I.
Primer sequences for reverse
transcription-quantitative PCR.
| Gene | Primer sequence
(5′→3′) |
|---|
| Nrf2 | Forward:
GTGGTTTAGGGCAGAAGG |
|
| Reverse:
TCTTTCTTACTCTGCCTCTA |
| NLRP3 | Forward:
CCACAGTGTAACTTGCAGAAGC |
|
| Reverse:
GGTGTGTGAAGTTCTGGTTGG |
| GAPDH | Forward:
AAGACCCAGAAATGAAC |
|
| Reverse:
TCTACACGATAACAACCA |
Immunohistochemistry (IHC)
Kidney tissue was collected from I/R model mice
following 24 h reperfusion to determine NLRP3 and Nrf2 expression
levels. Briefly, kidney tissue was fixed at room temperature for 48
h in 10% formalin, embedded in paraffin and cut into 5 µm sections.
The sections were subsequently blocked with 5% milk at 37°C for 30
min and incubated with anti-NLRP3 (1:100; cat. no. ab214185; Abcam)
or anti-Nrf2 (1:200; cat. no. ab137550; Abcam) primary antibodies
overnight. Following primary antibody incubation, the sections were
incubated with HRP-labeled anti-rabbit secondary antibody (1:200;
cat. no. ab214880; Abcam) at 37°C for 30 min, developed using
diaminobenzidine solution for <3 min and counterstained with
0.5% hematoxylin for 3 min at room temperature. A light microscope
was used to visualize IHC staining (magnification, ×200; Biorevo
BZ-9000; Keyence Corporation).
Renal function assay
A total of 2–3 ml blood was collected following
centrifugation at 3,000 × g for 10 min at 4°C from I/R model mice
following 24 h reperfusion. The serum was obtained to analyze renal
function via ELISA to determine levels of creatinine (cat. no.
C011; Nanjing Jiancheng Bioengineering Institute) and blood urea
nitrogen (BUN; cat. no. C013; Nanjing Jiancheng Bioengineering
Institute), according to the manufacturer's instructions. Kidney
injury molecule-1 (KIM-1) levels in serum were measured by ELISA
(cat. no. EK0880; Wuhan Boster Biological Technology Co., Ltd.)
according to the manufacturer's instructions.
Hematoxylin and eosin staining
Kidney tissue was collected from I/R model mice
following 24 h reperfusion to evaluate renal histopathological
changes. Tissue was fixed at room temperature for 48 h in 10%
formalin and then embedded in paraffin. Paraffin-embedded sections
were cut into 5-µm thick sections and stained with 0.5% hematoxylin
for 5 min and eosin for 3 min at room temperature (25°C). The
presence of hemorrhage, tubular cell necrosis, tubular dilatation
and cytoplasmic vacuole formation in the tissue was scored as
follows: 0, normal kidney; 1, minimal damage; 2, moderate damage;
and 3, severe damage (24). Renal
injury scores were calculated in a blinded manner by two
researchers under a light microscope (magnification, ×200; Biorevo
BZ-9000; Keyence Corporation).
ELISA analysis of TNF-α, IL-1β and
IL-6 levels
Blood was collected (1 ml) from I/R model mice
following 24 h reperfusion. The serum was obtained by
centrifugation at 3,000 × g for 10 min at 4°C. The serum levels of
TNF-α (cat. no. MTA00b), IL-1β (cat. no. MLB00C) and IL-6 (cat. no.
M6000B) were analyzed using commercial ELISA kits (R&D Systems,
Inc.), according to the manufacturer's protocol on a microplate
reader (cat. no. CA94089; Molecular Devices, LLC).
TUNEL staining
Kidney tissue was collected from I/R model mice
following 24 h reperfusion to evaluate cell apoptosis using TUNEL
staining. Briefly, tissue was fixed at room temperature for 48 h in
10% formalin, embedded in paraffin and cut into 5-µm thick
sections. Sections were incubated with TUNEL reagent in a
humidified chamber at 37°C for 60 min in the dark (Roche
Diagnostics) and stained with DAPI for 5 min at room temperature.
Then, 10 high-power microscope fields were randomly picked and
observed in each section with a fluorescence microscope
(magnification, ×10).
Statistical analysis
Data are presented as the mean ± SD of three
experiments and were analyzed by GraphPad Prism 5 (GraphPad
Software, Inc.). Statistical differences between two groups were
analyzed using a paired Student's t-test. Statistical differences
between more than three groups were analyzed using one-way ANOVA
followed by post hoc Tukey's multiple comparisons test. All data
were normally distributed and had an equal variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
MCC950 treatment prevents I/R-induced
NLRP3 signaling activation following renal injury in mice
The NLRP3 inflammasome pathway is activated and
serves a crucial role in kidney injury (25,26). A
previous study also reported that NLRP3 signaling is activated in
renal tissue of mice at 24 h post-reperfusion (4). Therefore, 24 h post-reperfusion was
selected as the present assessment time point. NLRP3 protein
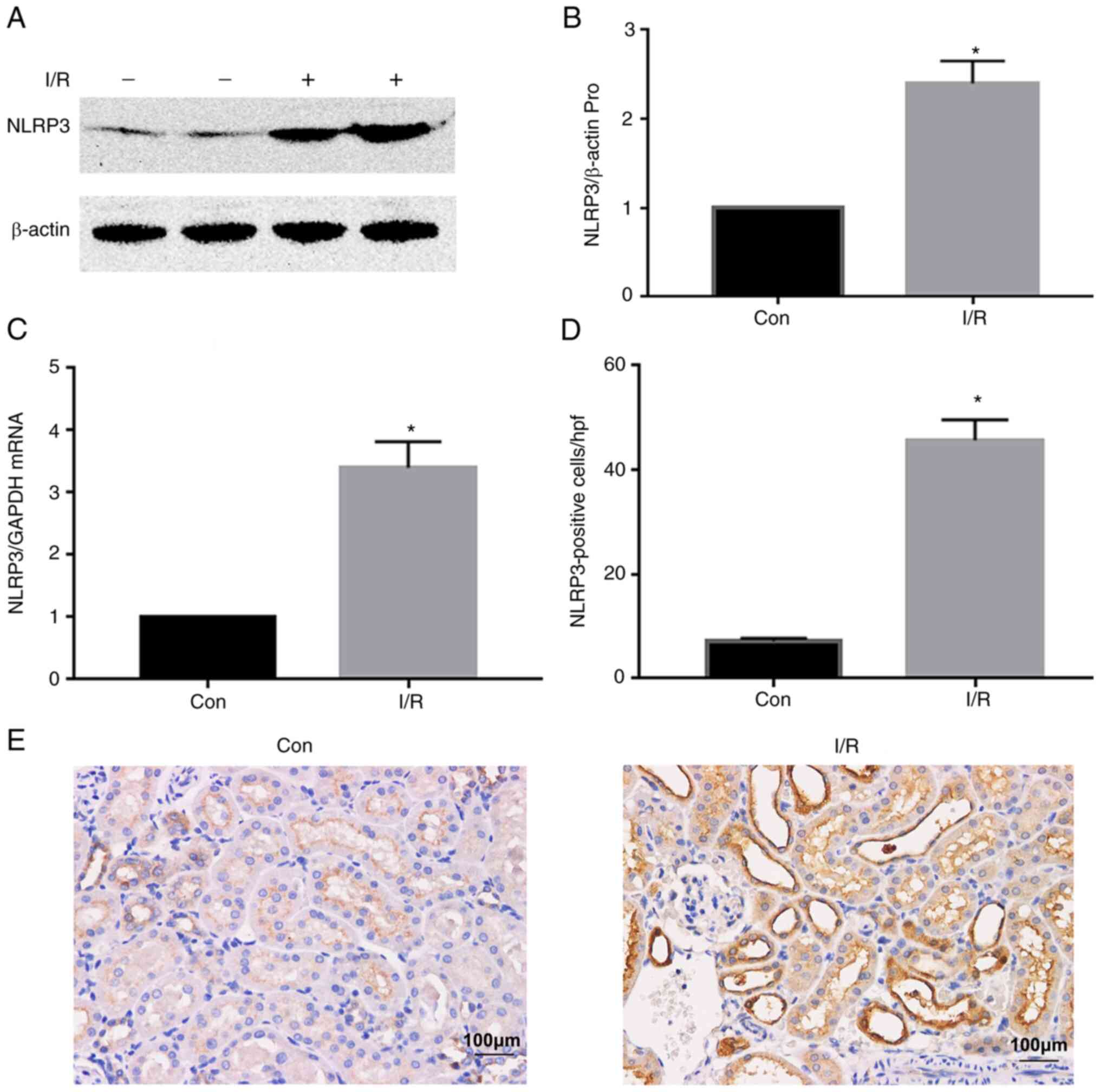
expression levels were upregulated by renal I/R injury in the I/R
group compared with the control (Con) group (Fig. 1A and B). A similar trend was
observed in NLRP3 mRNA expression levels (Fig. 1C). Furthermore, the number of
NLRP3-positive cells increased in the I/R group compared with the
Con group, as determined by IHC staining (Fig. 1D and E).
In order to determine the underlying mechanism of
NLRP3 inflammasome activation and the effect on the downstream
signaling pathway following renal I/R injury, mice were treated
with the NLRP3 inhibitor MCC950. Western blotting results
demonstrated that I/R upregulated protein expression levels of
NLRP3 and its adaptor ASC and promoted maturation from
pro-caspase-1 to caspase-1 and pro-IL-1β to and IL-1β in the I/R
group compared with the Con group (Fig.
2A-E). These results indicated that renal I/R may induce
activation of the NLRP3 inflammasome pathway and treatment with
MCC950 may downregulate NLRP3, ASC, caspase-1 and IL-1β expression
levels following renal I/R injury.
Effect of the NLRP3 inflammasome on
renal injury, inflammation and apoptosis in renal I/R model
mice
In order to determine the effect of NLRP3 on renal
injury, I/R model mice were treated with the NLRP3 inhibitor
MCC950. Renal I/R model mice exhibited significantly increased
levels of BUN, serum creatinine and KIM-1, which is a biomarker of
renal injury (Fig. 3A-C),
indicating successful induction of renal I/R injury. Histological
examination of renal tissue from I/R model mice revealed increased
histological score, which was evidenced by severe tubular
epithelial swelling, tubular dilation and interstitial edema,
vacuolar degeneration and loss of the brush border, which suggested
that renal I/R may promoted significant renal tissue damage and
increased histological score (Fig. 3D
and E). Inhibition of NLRP3 by MCC950 treatment significantly
decreased I/R-induced increases in BUN, creatinine and KIM-1 levels
and decreased histological score, with renal tissue exhibiting
fewer severely injured tubules (Fig.
3A-E).
 | Figure 3.MCC950 alleviates renal function and
injury and decreases cytokine levels and apoptosis following renal
I/R. Mice underwent surgery to induce renal I/R via clamping of the
bilateral renal pedicles. Mice were injected with 20 mg/kg MCC950
intraperitoneally daily for 14 days prior to surgery. Following 24
h reperfusion, renal tissue and blood were collected to analyze (A)
Cr, (B) BUN and (C) KIM-1 levels. (D) Histopathological changes
were assessed and (E) scored. Scale bar, 20 µm. (F) TNF-α, (G)
IL-1β and (H) IL-6 expression levels and (I and J) apoptosis were
assessed. Scale bar, 100 µm. Data are presented as the mean ± SD
(n=3). *P<0.05 vs. Con; #P<0.05 vs. I/R. I/R,
ischemia/reperfusion; Cr, creatinine; BUN, blood urea nitrogen;
KIM-1, kidney injury molecule-1; Con, control. |
In addition, levels of inflammatory factors and
apoptotic cells following MCC950 administration were analyzed in
renal I/R model mice. Indicators of inflammation, including TNF-α,
IL-1β and IL-6, were significantly upregulated in the I/R group
compared with the Con group (Fig.
3F-H). Moreover, compared with the Con group, the percentage of
apoptotic cells was significantly increased at 24 h
post-reperfusion in the I/R group (Fig.
3I and J). Compared with the I/R group, the secretory levels of
cytokines, TNF-α, IL-1β and IL-6, and the percentage of apoptotic
cells were decreased by MCC950 treatment in the I/R+MCC950 group
(Fig. 3F-J). These results
indicated that inhibition of the NLRP3 inflammasome may prevent
release of inflammatory factors and apoptosis of renal tissue
induced by I/R.
Nrf2 is essential for renal
I/R-induced regulation of NLRP3 inflammasome activity
A previous study revealed that Nrf2 is activated and
expression levels of Nrf2 target genes are upregulated in kidney
tissue following renal I/R injury (27). Similar findings were obtained in the
present research. Nucleic, total protein and mRNA expression levels
of Nrf2 were analyzed at 24 h post renal I/R injury. The results
demonstrated that, compared with the Con group, the nucleic, total
protein and mRNA expression levels of Nrf2 were upregulated in the
I/R group (Fig. 4A-D). In addition,
IHC analysis found that expression levels of Nrf2 in renal tissue
were increased, which was demonstrated by increased number of
Nrf2-positive cells observed in the I/R group (Fig. 4E and F). These data suggested that
activation of the Nrf2 signaling pathway may initiate a protective
response in renal I/R model mice.
In order to verify the effect of Nrf2 on the NLRP3
inflammasome pathway in renal I/R, Nrf2-KO mice were used in
subsequent experiments. Protein expression levels of Nrf2 and its
target gene, HO-1, were significantly downregulated following renal
I/R in KO mice compared with wild-type mice (Fig. 5A-C). Conversely, compared with the
I/R wild-type group, the expression levels of NLRP3 and its
adaptor, ASC, caspase-1 and IL-1β were upregulated in the I/R KO
group (Fig. 5A and D-G). These data
indicated that renal I/R may induce activation of the NLRP3
inflammasome in Nrf2-KO mice.
 | Figure 5.Nrf2 regulates the inhibition of
NLRP3 inflammasome activity. Wild-type and Nrf2-KO mice underwent
surgery to induce renal I/R via clamping of the bilateral renal
pedicles. (A) At 24 h post-reperfusion, renal tissue was collected
for western blot analysis of (B) Nrf2, (C) HO-1, (D) NLRP3, (E)
ASC, (F) caspase-1 and (G) IL-1β protein expression levels. Data
are presented as the mean ± SD (n=3). *P<0.05 vs. Con wild-type
mice; #P<0.05 vs. I/R wild-type mice. NLRP3, NLR
family pyrin domain containing 3; ASC, PYD and CARD domain
containing; HO-1, heme oxygenase 1; KO, knockout; Con, control;
I/R, ischemia/reperfusion. |
NaHS alleviates I/R-induced
upregulation of NLRP3 protein expression levels in wild-type, but
not in Nrf2-KO, mice
NaHS has been demonstrated to exert effects on Nrf2
expression and NLRP3 inflammasome activation in different disease
models (28–30). The present study investigated
whether the effect of NaHS on activation of the NLRP3 pathway
occurs via the Nrf2 signaling pathway by analyzing expression
levels of Nrf2 and NLRP3 inflammasome-associated proteins in
wild-type and KO mice. In wild-type mice, compared with the Con
group, the expression levels of Nrf2, NLRP3, caspase-1 and IL-1β
were upregulated by renal I/R injury. In addition, NaHS treatment
further upregulated Nrf2 expression and downregulated NLRP3,
caspase-1 and IL-1β expression levels in the I/R+NaHS group
compared with the I/R group (Fig.
6A-E). In Nrf2-KO mice, no statistically significant
differences were observed in Nrf2, NLRP3, caspase-1 and IL-1β
expression levels between the I/R and I/R+NaHS groups (Fig. 6A-E). In the I/R+NaHS group, Nrf2
expression levels were significantly downregulated, whereas NLRP3,
caspase-1 and IL-1β expression levels were significantly
upregulated in Nrf2-KO mice compared with wild-type mice. These
results suggested that NaHS may decrease NLRP3 inflammasome
activation via the Nrf2 signaling pathway in renal I/R injury.
NaHS alleviates renal injury,
inflammation and apoptosis via the Nrf2 signaling pathway in renal
I/R model mice
NaHS serves an important role in renal damage in
AKI, I/R and other disease models (31). NaHS alleviated renal I/R-induced
histopathological damage, which was demonstrated by decreased
tubular epithelial swelling, tubular dilation and interstitial
edema and kidney histological scores in the I/R+NaHS group compared
with the I/R group in wild-type mice (Fig. 7A and B). Indicators of renal
function, including creatinine, BUN and KIM-1, were also decreased
by NaHS treatment in the I/R+NaHS group compared with the I/R group
in wild-type mice (Fig. 7C-E).
However, in Nrf2-KO mice, NaHS was unable to decrease the increased
kidney histological score and levels of renal function indicators
induced by renal I/R.
 | Figure 7.NaHS alleviates renal dysfunction,
excessive release of cytokines and cell apoptosis induced by renal
I/R via the Nrf2 signaling pathway. Wild-type and Nrf2-KO mice
underwent surgery to induce renal I/R via clamping of the bilateral
renal pedicles. Mice were treated with 50 µmol/kg intraperitoneal
NaHS prior to renal ischemia. (A) At 24 h post-reperfusion, blood
and renal tissue were collected to measure (B) histopathological
changes and (C) Cr, (D) BUN and (E) KIM-1 levels. Scale bar, 20 µm.
(F and G) Apoptosis and expression levels of (H) TNF-α, (I) IL-1β
and (J) IL-6 were assessed. Scale bar, 100 µm. Data are presented
as the mean ± SD (n=3). *P<0.05 vs. Con wild-type mice;
#P<0.05 vs. I/R wild-type mice;
&P<0.05 vs. I/R+NaHS wild-type mice;
$P<0.05 vs. Con KO mice. NaHS, sodium hydrosulfide;
I/R, ischemia/reperfusion; KO, knockout; BUN, blood urea nitrogen;
KIM-1, kidney injury molecule-1; Con, control; Cr, creatinine. |
Furthermore, the percentage of apoptotic cells and
release of cytokines, TNF-α, IL-1β and IL-6, were all decreased
following NaHS treatment in the I/R+NaHS group compared with the
I/R group in wild-type mice (Fig.
7F-J). However, NaHS was unable to exert a protective effect
against apoptosis and excessive release of inflammatory factors
following reperfusion of renal tissue in the I/R+NaHS group of
Nrf2-KO mice compared with I/R+NaHS wild-type mice (Fig. 7F-J). These results suggested that
NaHS may alleviate renal injury, inflammation and apoptosis in
renal I/R wild-type mice, but not in Nrf2-KO mice.
Discussion
Inflammation is a key pathogenic processes of AKI
induced by I/R (4). The NLRP3
inflammasome and Nrf2 signaling pathway participate in the
regulation of inflammation in kidney injury (32). The present study found that AKI
induced by I/R activated the NLRP3 inflammasome and its adaptor,
ASC, promoted the maturation of pro-caspase-1 and pro-IL-1β and
accelerated the excessive release of cytokines and apoptosis, which
culminated in severe renal dysfunction. Inhibition of the NLRP3
inflammasome by MCC950 treatment improved renal function, levels of
inflammation and apoptosis in renal tissue. In addition to
activating NLRP3, renal I/R also activated the Nrf2 signaling
pathway. However, the absence of Nrf2 in Nrf2-KO mice led to
further upregulation of the NLRP3 inflammasome and its adaptor.
NaHS treatment was found to alleviate NLRP3 inflammasome activity
via the Nrf2 signaling pathway. Moreover, renal injury,
inflammation and apoptosis were decreased by NaHS treatment via
Nrf2-mediated inhibition of NLRP3 inflammasome activation.
Renal I/R injury is an important clinical problem
and primary cause of AKI, which leads to increased risk of
developing chronic kidney disease (33). Inflammation is an important
pathological feature of ischemic injury and occurs as a consequence
of immune cell activation via recognition of pathogen- and
damage-associated molecular patterns (34). Moreover, it was previously reported
that the NLRP3 inflammasome serves a role in a range of kidney
diseases by regulating inflammation, pyroptosis, apoptosis and
fibrosis (35). The NLRP3
inflammasome is a protein complex comprising NLRP3, its adapter
protein, ASC, and caspase-1. Caspase-1 cleaves pro-IL-1β and
pro-IL-18 into mature, activated secretory forms. Renal injury
induced by different pathologies, including ischemia (33), activates the inflammasome complex,
upregulates NLRP3 expression and promotes the subsequent maturation
of pro-IL-1β (36). NLRP3 KO using
small interfering RNA in mice or renal tubular epithelial cells
exerts a protective effect against renal tissue injury induced by
ischemia or cell injury in the absence of glucose (37). The present results indicated that
renal I/R induced activation of the NLRP3 inflammasome, while
inhibition of NLRP3 using MCC950 significantly downregulated NLRP3
and ASC expression levels and the maturation of pro-caspase-1 and
pro-IL-1β in the renal I/R injury model. In addition, NLRP3
inhibition by MCC950 ameliorated renal dysfunction, and decreased
histopathological injury and score, excessive release of cytokines
and the number of apoptotic cells. These data suggested that NLRP3
activation may serve a key role in renal injury and inhibition of
NLRP3 may exert a protective effect against tissue injury and
dysfunction induced by renal I/R.
Previous studies have reported that Nrf2
participates in the inflammatory response (10–12).
In addition, Nrf2 serves a key anti-inflammatory, anti-oxidative or
anti-apoptotic role in ischemic conditions. Nrf2/antioxidant
response element signaling pathway activation attenuates
inflammation and apoptosis in renal tissue of
cyclophosphamide-induced mice (38). Another study demonstrated that Nrf2
activation by sulforaphane inhibits NF-κB signaling pathway
activity, thereby relieving inflammation in dystrophic muscle
tissue (39). Furthermore, the
knockdown of Nrf2 promotes NLRP3 inflammasome activation and leads
to IL-1β activation in an ischemia model (11). The present data supported the
findings that renal I/R may induce the Nrf2 signaling pathway and
expression of its downstream target genes, such as HO-1. Nrf2-KO
mice further promoted activation of the NLRP3 inflammasome, which
indicated that NLRP3, ASC, caspase-1 and IL-1β expression levels
were increased in the renal I/R model. These results suggested that
Nrf2 exerted an inhibitory effect on NLRP3 inflammasome activation
in renal I/R injury.
Previous research has reported that the kidney
produces H2S (40).
Furthermore, H2S increases renal blood flow and promotes
the clearance function of the kidney, as demonstrated by elevated
glomerular filtration rate (41).
Moreover, H2S not only alleviates inflammation and
oxidative stress, but also serves a crucial role in regulating
endothelial dysfunction and hypertension (28). H2S participates in the
regulation of renal-associated disease, such as I/R injury and
obstructive and diabetic nephropathy (42), however, the underlying mechanisms
remain poorly understood. NaHS has been used as a H2S
exogenous donor in research (43).
In the present study, the delivery of H2S was in the
form of NaHS. The present results demonstrated that NaHS
ameliorated tissue injury, kidney dysfunction, excessive release of
cytokines and apoptosis induced by renal I/R. It was previously
reported that NaHS prevents microglial activation and inflammation
induced by nerve injury via regulating the Nrf2 signaling pathway
(44). Consistent with the
hypothesis that NaHS regulates inflammation via the Nrf2 signaling
pathway, the present results demonstrated that NaHS upregulated
Nrf2 expression levels and inhibited expression levels of NLRP3 in
renal I/R model mice. Furthermore, Nrf2 KO abolished the regulatory
effects of NaHS on Nrf2 and the NLRP3 inflammasome. The present
study also investigated the role of Nrf2 on renal injury and
function following NaHS treatment; inhibition of Nrf2 not only
partially reversed the protective effect of NaHS on kidney injury
and dysfunction, but also reversed the inhibitory effect of NaHS on
the inflammatory response and apoptosis following renal I/R. These
results suggested that Nrf2 may be required for the protective
effect of NaHS on AKI induced by I/R, and NaHS may exert its
protective role against tissue injury via Nrf2-mediated NLPR3
inflammasome inhibition.
In conclusion, I/R-induced renal injury activated
the NLRP3 inflammasome and Nrf2 signaling pathway, and inhibition
of the NLRP3 inflammasome protected against renal injury. Nrf2
negatively regulated NLRP3 inflammasome activation and NaHS
alleviated kidney injury and dysfunction, apoptosis and the
inflammatory response via Nrf2-mediated NLRP3 inflammasome
inhibition. These results provide novel insight into a potential
future target for the treatment of renal ischemic injury.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ML and HC designed the research and wrote the
manuscript. YS and HC constructed the animal model. YW performed
sample collection and experiments. YS and ML analyzed the data. YS,
ML and HC confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All animal protocols were approved by the Animal
Care and Use Committee of General Hospital of Tianjin Medical
University (approval no. IRB2020-DW-04; Tianjin, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Park J, Lee EG, Yi HJ, Kim NH, Rhee SG and
Woo HA: Ablation of peroxiredoxin V exacerbates
Ischemia/Reperfusion-Induced kidney injury in mice. Antioxidants
(Basel). 9:7692020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ghasemzadeh P, Rezayat SM, Adeli S and
Rahbar-Roshandel N: Protective effect of 25Mg-porphyrin-fullerene
nanoparticles on oxygen-glucose deprivation/reperfusion injury in
PC12 cells. Acta Med Iran. 54:478–484. 2016.PubMed/NCBI
|
|
3
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tang TT, Lv LL, Pan MM, Wen Y, Wang B, Li
ZL, Wu M, Wang FM, Crowley SD and Liu BC: Hydroxychloroquine
attenuates renal ischemia/reperfusion injury by inhibiting
cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis.
9:3512018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nazir S, Gadi I, Al-Dabet MM, Elwakiel A,
Kohli S, Ghosh S, Manoharan J, Ranjan S, Bock F, Braun-Dullaeus RC,
et al: Cytoprotective activated protein C averts Nlrp3
inflammasome-induced ischemia-reperfusion injury via mTORC1
inhibition. Blood. 130:2664–2677. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iyer SS, Pulskens WP, Sadler JJ, Butter
LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA,
Leemans JC and Sutterwala FS: Necrotic cells trigger a sterile
inflammatory response through the Nlrp3 inflammasome. Proc Natl
Acad Sci USA. 106:20388–20393. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cao Y, Fei D, Chen M, Sun M, Xu J, Kang K,
Jiang L and Zhao M: Role of the nucleotide-binding domain-like
receptor protein 3 inflammasome in acute kidney injury. FEBS J.
282:3799–3807. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang W, Faubel S, Ljubanovic D, Mitra A,
Falk SA, Kim J, Tao Y, Soloviev A, Reznikov LL, Dinarello CA, et
al: Endotoxemic acute renal failure is attenuated in
caspase-1-deficient mice. Am J Physiol Renal Physiol.
288:F997–F1004. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu Q, Gao Y and Ci X: Role of Nrf2 and
its activators in respiratory diseases. Oxid Med Cell Longev.
2019:70905342019.PubMed/NCBI
|
|
10
|
Shelton LM, Park BK and Copple IM: Role of
Nrf2 in protection against acute kidney injury. Kidney Int.
84:1090–1095. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu X, Zhang L, Ye X, Hao Q, Zhang T, Cui G
and Yu M: Nrf2/ARE pathway inhibits ROS-induced NLRP3 inflammasome
activation in BV2 cells after cerebral ischemia reperfusion.
Inflamm Res. 67:57–65. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hou Y, Wang Y, He Q, Li L, Xie H, Zhao Y
and Zhao J: Nrf2 inhibits NLRP3 inflammasome activation through
regulating Trx1/TXNIP complex in cerebral ischemia reperfusion
injury. Behav Brain Res. 336:32–39. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang H, Lv H, Li H, Ci X and Peng L:
Oridonin protects LPS-induced acute lung injury by modulating
Nrf2-mediated oxidative stress and Nrf2-independent NLRP3 and NF-κB
pathways. Cell Commun Signal. 17:622019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu D, Wang H, Teng T, Duan S, Ji A and Li
Y: Hydrogen sulfide and autophagy: A double edged sword. Pharmacol
Res. 131:120–127. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu LF, Lu M, Wu ZY, Wong PT and Bian JS:
Hydrogen sulfide inhibits rotenone-induced apoptosis via
preservation of mitochondrial function. Mol Pharmacol. 75:27–34.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jha S, Calvert JW, Duranski MR,
Ramachandran A and Lefer DJ: Hydrogen sulfide attenuates hepatic
ischemia-reperfusion injury: Role of antioxidant and antiapoptotic
signaling. Am J Physiol Heart Circ Physiol. 295:H801–H806. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mariggio MA, Minunno V, Riccardi S,
Santacroce R, De Rinaldis P and Fumarulo R: Sulfide enhancement of
PMN apoptosis. Immunopharmacol Immunotoxicol. 20:399–408. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sivarajah A, Collino M, Yasin M, Benetti
E, Gallicchio M, Mazzon E, Cuzzocrea S, Fantozzi R and Thiemermann
C: Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide
in a rat model of regional myocardial I/R. Shock. 31:267–274. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Whiteman M, Armstrong JS, Chu SH, Jia-Ling
S, Wong BS, Cheung NS, Halliwell B and Moore PK: The novel
neuromodulator hydrogen sulfide: An endogenous peroxynitrite
‘scavenger’? J Neurochem. 90:765–768. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zanardo RC, Brancaleone V, Distrutti E,
Fiorucci S, Cirino G and Wallace JL: Hydrogen sulfide is an
endogenous modulator of leukocyte-mediated inflammation. FASEB J.
20:2118–2120. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bai YD, Yang YR, Mu XP, Lin G, Wang YP,
Jin S, Chen Y, Wang MJ and Zhu YC: Hydrogen sulfide alleviates
acute myocardial ischemia injury by modulating autophagy and
inflammation response under oxidative stress. Oxid Med Cell Longev.
2018:34028092018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li S, Lin Q, Shao X, Mou S, Gu L, Wang L,
Zhang Z, Shen J, Zhou Y, Qi C, et al: NLRP3 inflammasome inhibition
attenuates cisplatin-induced renal fibrosis by decreasing oxidative
stress and inflammation. Exp Cell Res. 383:1114882019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shingu C, Koga H, Hagiwara S, Matsumoto S,
Goto K, Yokoi I and Noguchi T: Hydrogen-rich saline solution
attenuates renal ischemia-reperfusion injury. J Anesth. 24:569–574.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vilaysane A, Chun J, Seamone ME, Wang W,
Chin R, Hirota S, Li Y, Clark SA, Tschopp J, Trpkov K, et al: The
NLRP3 inflammasome promotes renal inflammation and contributes to
CKD. J Am Soc Nephrol. 21:1732–1744. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yuan X, Wang X, Chen C, Zhou J and Han M:
Bone mesenchymal stem cells ameliorate ischemia/reperfusion-induced
damage in renal epithelial cells via microRNA-223. Stem Cell Res
Ther. 8:1462017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kalbolandi SM, Gorji AV, Babaahmadi-Rezaei
H and Mansouri E: Luteolin confers renoprotection against
ischemia-reperfusion injury via involving Nrf2 pathway and
regulating miR320. Mol Biol Rep. 46:4039–4047. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li J, Teng X, Jin S, Dong J, Guo Q, Tian D
and Wu Y: Hydrogen sulfide improves endothelial dysfunction by
inhibiting the vicious cycle of NLRP3 inflammasome and oxidative
stress in spontaneously hypertensive rats. J Hypertens.
37:1633–1643. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li F, Zhang P, Zhang M, Liang L, Sun X, Li
M, Tang Y, Bao A, Gong J, Zhang J, et al: Hydrogen sulfide prevents
and partially reverses ozone-induced features of lung inflammation
and emphysema in mice. Am J Respir Cell Mol Biol. 55:72–81. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xie L, Gu Y, Wen M, Zhao S, Wang W, Ma Y,
Meng G, Han Y, Wang Y, Liu G, et al: Hydrogen sulfide induces Keap1
S-sulfhydration and suppresses diabetes-accelerated atherosclerosis
via Nrf2 activation. Diabetes. 65:3171–3184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ling Q, Yu X, Wang T, Wang SG, Ye ZQ and
Liu JH: Roles of the exogenous H2S-Mediated SR-A signaling pathway
in renal ischemia/reperfusion injury in regulating endoplasmic
reticulum stress-induced autophagy in a rat model. Cell Physiol
Biochem. 41:2461–2474. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yifan Z, Benxiang N, Zheng X, Luwei X,
Liuhua Z, Yuzheng G and Ruipeng J: Ceftriaxone calcium crystals
induce acute kidney injury by NLRP3-mediated inflammation and
oxidative stress injury. Oxid Med Cell Longev. 2020:64284982020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Asvapromtada S, Sonoda H, Kinouchi M,
Oshikawa S, Takahashi S, Hoshino Y, Sinlapadeelerdkul T,
Yokota-Ikeda N, Matsuzaki T and Ikeda M: Characterization of
urinary exosomal release of aquaporin-1 and −2 after renal
ischemia-reperfusion in rats. Am J Physiol Renal Physiol.
314:F584–F601. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu J, Han M, Li J, Yang X, Zhen X, Schlaak
JF, Yang D and Lu M: Pattern recognition receptors and liver
failure. Crit Rev Immunol. 39:289–311. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fan J, Xie K, Wang L, Zheng N and Yu X:
Roles of inflammasomes in inflammatory kidney diseases. Mediators
Inflamm. 2019:29230722019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bakker PJ, Butter LM, Claessen N, Teske
GJ, Sutterwala FS, Florquin S and Leemans JC: A tissue-specific
role for Nlrp3 in tubular epithelial repair after renal
ischemia/reperfusion. Am J Pathol. 184:2013–2022. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shen J, Wang L, Jiang N, Mou S, Zhang M,
Gu L, Shao X, Wang Q, Qi C, Li S, et al: NLRP3 inflammasome
mediates contrast media-induced acute kidney injury by regulating
cell apoptosis. Sci Rep. 6:346822016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
ALHaithloul H, Alotaibi MF, Bin-Jumah M,
Elgebaly H and Mahmoud AM: Olea europaea leaf extract up-regulates
Nrf2/ARE/HO-1 signaling and attenuates cyclophosphamide-induced
oxidative stress, inflammation and apoptosis in rat kidney. Biomed
Pharmacother. 111:676–685. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun CC, Li SJ, Yang CL, Xue RL, Xi YY,
Wang L, Zhao QL and Li DJ: Sulforaphane attenuates muscle
inflammation in dystrophin-deficient mdx mice via NF-E2-related
Factor 2 (Nrf2)-mediated Inhibition of NF-κB signaling pathway. J
Biol Chem. 290:17784–17795. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stipanuk MH and Beck PW: Characterization
of the enzymic capacity for cysteine desulphhydration in liver and
kidney of the rat. Biochem J. 206:267–277. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xia M, Chen L, Muh RW, Li PL and Li N:
Production and actions of hydrogen sulfide, a novel gaseous
bioactive substance, in the kidneys. J Pharmacol Exp Ther.
329:1056–1062. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cao X and Bian JS: The role of hydrogen
sulfide in renal system. Front Pharmacol. 7:3852016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen HG, Han HZ, Li Y, Yu YH and Xie KL:
Hydrogen alleviated organ injury and dysfunction in sepsis: The
role of cross-talk between autophagy and endoplasmic reticulum
stress: Experimental research. Int Immunopharmacol. 78:1060492020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen H, Xie K, Chen Y, Wang Y, Wang Y,
Lian N, Zhang K and Yu Y: Nrf2/HO-1 signaling pathway participated
in the protection of hydrogen sulfide on neuropathic pain in rats.
Int Immunopharmacol. 75:1057462019. View Article : Google Scholar : PubMed/NCBI
|