Introduction
Traumatic brain injury (TBI) is a major public
health problem and a major cause of mortality and disability that
imposes a substantial economic burden worldwide, A previous study
reported that there were ~288,000 TBI-related hospitalizations and
>56,000 TBI-related deaths in the US alone, and the global
economic burden of TBI was estimated at ~$400 billion per annum
(1,2). TBI has a high incidence in low-income
and middle-income countries, as well as developing countries such
as Iran and China (3–5). The incidence of TBI is increasing
rapidly due to the significant increase in road traffic collisions,
including motor vehicle accidents (5). Although an increasing number of
randomized controlled trials including intracranial pressure
monitoring, therapeutic hypothermia, surgical methods and drug
administration have been performed in recent years and the
long-term outcome has substantially improved, a significant benefit
is not observed following drug interventions (4–10).
Hence, studies aiming to further clarify the pathophysiological
mechanisms of TBI and search for new and effective pharmacological
intervention targets are important and necessary. The
pathophysiology of TBI includes several different physiological
changes and mainly involves primary brain injury and secondary
brain injury, which lead to neuronal death, neurological deficits
and mortality following TBI (11).
Primary brain injuries lead to brain tissue disorganization,
intracerebral hemorrhage and blood-brain barrier (BBB) damage,
which is a direct physical injury to brain tissue that is difficult
to prevent and usually cannot be reversed. Secondary brain injury,
including calcium overload, oxidative stress, neuroinflammation,
autophagy, lipid peroxidation and apoptosis, can be reversed
(12,13).
Autophagy is the main cellular lysosomal degradation
mechanism for degrading and recycling intracellular proteins and
organelles under different physiological and pathological
conditions (14). Autophagy has
been reported to have a core role in many central nervous system
diseases, including acute brain injury (12,15,16),
intracerebral hemorrhage (17),
subarachnoid hemorrhage (SAH) (18)
and Huntington's disease (19).
Tang et al (15) reported
significant decreases in neural apoptosis and necrotic cell death
following autophagy was inhibited by fibroblast growth factor-2 and
the autophagy activator rapamycin aggravated brain injury.
According to Fang et al (16), activation of autophagy decreases
mitochondrial apoptosis, improves neurological function, decreases
cerebral edema and alleviates the disruption of the BBB following
TBI in mice. Currently, the neuroprotective effect of the
activation or inhibition of autophagy remains to be elucidated. A
further study of new potential drug targets in the autophagy
pathway is required.
Dexmedetomidine (DEX) is a highly selective
α-2-adrenergic receptor agonist that provides sedation and
analgesic effects with minimal respiratory depression (20). It is widely used in surgical
procedures and to prevent postoperative delirium (12,21).
Recent studies have confirmed that DEX exerts its protective
effects on various organ injuries (12,20).
Based on accumulating evidence, DEX also improves neurological
function and delirium in patients (21) and alleviates EBI in the TBI model
and the effect of DEX is dose-dependent (12,22,23).
Huang and Hao (22) report that DEX
relieves early brain injury (EBI) by inhibiting inflammation and
decreasing neuronal apoptosis, which may depend on the TLR4/NF-κB
signaling pathway. Li et al (24) also report that DEX alleviates EBI by
inhibiting inflammation through the nuclear factor erythroid
2-related factor 2 (Nrf2) signaling pathway. As shown in the study
by Zhao et al (25), DEX
increases autophagy, decreases reactive oxygen species (ROS)
production and apoptosis and then eliminates damaged mitochondria
in a lipopolysaccharide-induced acute kidney injury model through
the PI3K/AKT/mTOR pathway. The AKT/mTOR signaling pathway is
generally acknowledged as the most important regulatory pathway in
autophagy (26,27). ROS regulate the NFE2L2 (nuclear
factor, erythroid derived 2, like 2) pathway, transcriptionally
activates hypoxia-inducible factor (HIF-1) and p53 and then
promotes autophagy (28,29). Our previous studies revealed an
important role for Nrf2 in neuronal death in diseases of the
central nervous system (30,31).
However, researchers have not determined whether DEX attenuates EBI
by regulating autophagy via the ROS/Nrf2 signaling pathway.
The present study constructed a mouse TBI model to
study the effects of DEX on EBI and explored the crosstalk between
autophagy and inflammation. It also explored the mechanism by which
the ROS/Nrf2 signaling pathway may regulate this process.
Materials and methods
Animals
All animal experiments performed in this study
complied with the National Institutes of Health guidelines for the
handling of laboratory animals (32) and were approved by the Ethics
Committee of the Wuxi Medical College of Anhui Medical University
(approval no. YXLL-2020-112). All experiments were conducted on
healthy adult male C57BL/6J mice (age, 8–10 weeks; weight, 22–25 g;
Anhui Medical University, Hefei, China). The mice were housed in
animal care facilities under environmentally controlled temperature
(25±2°C) and humidity (55±5%) with 12-h light/dark cycles, and had
free access to food and water. The 60 mice were divided into four
major groups (15 animals/group): Sham, TBI, TBI + DEX and TBI + DEX
+ Rap.
Animal TBI model
The TBI model was established in strict accordance
with the Feeney weight-drop model of focal injury (33,34).
Briefly, the mice were anesthetized with an intraperitoneal
injection of 1% sodium pentobarbital (40 mg/kg) and then placed in
a brain stereotaxic apparatus. The rectal temperature was
maintained at 37±0.5°C during the operation using a heating pad.
Then, a burr hole was made in the left hemisphere at the following
coordinates: 0.2 mm posterior, 1 mm lateral and 2.2 mm below the
horizontal plane of the bregma. The bone flap was removed to expose
the dura mater. The dura was placed under a weight-drop device with
an impact sensor. A metal (weight 240 g, tip diameter 3 mm) was
dropped from 1 cm above the dura onto the dura mater through a
catheter. Then, the scalp was closed and the mice were removed from
the apparatus. Finally, the hole was covered with medical bone wax.
The animals in the Sham group received similar surgical procedures
but without weight-drop impact. At 72 h following TBI, the mice
were sacrificed with 100 mg/kg sodium pentobarbital via i.p.
injection. Mortality was confirmed by observing respiration and by
using the corneal reflection method.
Drug administration
DEX was purchased from Jiangsu Hengrui Medicine Co.,
Ltd., dissolved in 0.9% sterile saline and administered i.p. at a
dose of 20 µg/kg 2 h after recovery (12) (Fig.
1A). A dose of 2 mg/kg rapamycin (Selleck Chemicals; dissolved
in 2% DMSO) was administered i.p. 30 min before TBI induction in
the TBI + rapamycin + DEX group.
Modified neurological severity score
(mNSS)
The severity of brain injury was evaluated by
determining neurological function 72 h after TBI using a previously
described neurological grading system (15). The neurological scores of the
animals in each group were evaluated by an independent observer.
The scoring system consisted of motor, sensory, reflex and balance
tests. The neurological scores (mNSS scores) ranged from 0 to 18
points and were calculated by adding the scores together; all mice
in each group underwent a behavioral assessment and a higher score
represented worse neurological function. All mouse behavior scores
were recorded by the same independent observer who was blinded to
the study groups.
Brain water content measurement
The severity of brain edema was evaluated by
measuring the brain water content using the standard wet-dry
method, as previously reported (18,31,35).
The mice were sacrificed 72 h after TBI and the entire brain was
harvested and separated into the ipsilateral and contralateral
cortices, ipsilateral and contralateral basal ganglia and
cerebellum (wet weight). Then, brain specimens from each group were
dehydrated at 105°C for 24 h to acquire the dry weight. The
percentage of brain water content was equal to (wet weight-dry
weight)/wet weight ×100%.
Evans blue extravasation
Evans blue extravasation was performed as previously
described (36). Briefly, mice were
anesthetized by 1% sodium pentobarbital (50 mg/kg) injection 72 h
after TBI. Evans blue dye (2%, 5 ml/kg; Sigma-Aldrich; Merck KGaA)
was injected into the left femoral vein over 2 min and circulated
for 60 min. Then, the mice were sacrificed with 100 mg/kg sodium
pentobarbital via i.p. injection and with phosphate-buffered saline
(PBS) intracardial perfusion. Mortality was identified by observing
respiration and by using the corneal reflection method. The brains
were removed and quickly divided into the left and right cerebral
hemispheres, weighed, homogenized in saline and centrifuged at
15,000 × g for 30 min at room temperature. Subsequently, the
resultant supernatant was added with an equal volume of
trichloroacetic acid, incubated overnight at 4°C and centrifuged at
15,000 × g for 30 min at room temperature. Next, the resultant
supernatant was collected and spectrophotometrically quantified at
610 nm for Evans blue dye.
Cytokine measurements
IL-1β (cat. no. ab197742; Abcam), IL-6 (cat. no.
ab222503; Abcam), TNF-α (cat. no. ab208348; Abcam) and NF-κB (cat.
no. ab176663; Abcam) levels were measured using ELISAs according to
the manufacturer's instructions.
Analysis of ROS
The nonfluorescent diacetylated
2′,7′-dichlorofluorescein (DCF-DA) probe (Sigma-Aldrich; Merck
KGaA), which becomes highly fluorescent upon oxidation, was used to
evaluate intracellular ROS production according to the
manufacturer's instructions.
Analysis of lipid peroxidation
Malondialdehyde (37) levels were detected with a lipid
peroxidation (37) assay kit (Ex/Em
532/553 nm, cat. no. ab118970; Abcam) according to the
manufacturer's instructions.
TdT-mediated dUTP-biotin nick end
labeling (TUNEL) assay
A TUNEL assay was conducted to assess neuronal death
in the hippocampus. The TUNEL reaction mixture (50 µl) was added to
each sample and the slides were incubated in a humidified chamber
for 60 min at 37°C in the dark. The slides were then incubated with
DAPI (0.1 mg/ml) for 5 min at room temperature in the dark to stain
the nuclei, followed by imaging with a fluorescence microscope. The
procedure was performed with a TUNEL staining kit according to the
manufacturer's instructions (cat. no. TUN11684817, Roche
Diagnostics GmbH). A negative control (without the TUNEL reaction
mixture) was used. The apoptotic index (%) was calculated as the
ratio of the number of TUNEL-positive cells/total number of cells
×100. The cell count was confirmed in four randomly selected
high-power fields and the data obtained from each field were
averaged (magnification, ×400).
Western blot analysis
Western blot analyses were performed as previously
described (18). Briefly, cerebral
cortex samples were collected, homogenized and total protein was
extracted using RIPA buffer (CoWin Biosciences). A BCA Protein
Assay kit (Beyotime Institute of Biotechnology) was used to measure
protein concentrations with the bicinchoninic acid method. Total
protein (30 µg) was separated via 12% SDS-PAGE and transferred onto
PVDF membranes. The membranes were blocked with 5% nonfat milk at
room temperature for 1 h. The membranes were then incubated with
the following primary antibodies overnight at 4°C: Rabbit
anti-βactin (1:1,000; rabbit polyclonal; cat. no. ab8227), rabbit
anti-caspase-3 (1:2,000; rabbit polyclonal; cat. no. ab184787),
rabbit anti-Nrf2 (1:1,000; rabbit polyclonal; cat. no. ab31163),
rabbit anti-heme oxygenase (HO)-1 (1:1,000; rabbit polyclonal; cat.
no. ab13243), rabbit anti-Beclin-1 (1 µg/ml, rabbit monoclonal;
cat. no. ab62557) and rabbit anti-LC-3B (1 µg/ml, rabbit
monoclonal; cat. no. ab48394; all from Abcam). After washing the
membranes with TBST (0.5% Tween-20) three times at room temperature
for 20 min, HRP-conjugated goat anti-rabbit IgG or goat anti-mouse
IgG secondary antibodies (1:2,000; cat. no. 7074s; Cell Signaling
Technology, Inc.) were applied and the membranes were incubated
with the secondary antibodies at room temperature for 1.5 h. The
protein bands were detected using a Bio-Rad imaging system (Bio-Rad
Laboratories, Inc.) and quantified with ImageJ software (version
1.52; National Institutes of Health).
Statistical analysis
The data are reported as the means ± standard error
of mean. SPSS v14.0 (SPSS, Inc.) and GraphPad Prism 6 (GraphPad
Software, Inc.) software were used for the statistical analyses.
Student's t-test (unpaired) was used if two groups were compared
and one-way analysis of variance (ANOVA) followed by Bonferroni's
post hoc test was used if two independent variables were compared.
For nonnormally distributed data and/or data with a nonhomogeneous
variance, the Kruskal-Wallis test followed by Dunn's post hoc test
was used. P<0.05 was considered to indicate a statistically
significant difference.
Results
DEX alleviates neurological deficits
and brain edema following TBI
The modified neurological severity score (mNSS) was
calculated to evaluate neurological deficits and the brain water
content determined using the wet-dry method at 72 h after TBI to
evaluate brain damage and clarify the neuroprotective effect of DEX
on TBI. TBI significantly increased the neurological scores and DEX
administration significantly improved neurological function
(Fig. 1B). Similar results were
obtained for brain water content, which were increased
significantly after TBI and was alleviated by DEX treatment
(P<0.05; Fig. 1C).
DEX alleviates neuronal apoptosis and
BBB permeability following TBI
Neuronal apoptosis and BBB permeability are the main
factors that lead to EBI following TBI. Therefore, a TUNEL assay
was used to evaluate the level of cell death in TBI mice treated
with and without DEX at 72 h after model construction. The
expression levels of apoptosis-related proteins were detected using
Western blotting. Evans blue extravasation was analyzed to clarify
BBB permeability. The results revealed more hippocampal neuronal
death following TBI and DEX decreased neuronal apoptosis (Fig. 2A). The western blot results also
indicated that DEX reduced the expression levels of the
apoptosis-related protein caspase-3 (Fig. 2B-D). Compared with the sham and TBI
groups, DEX significantly decreased the extent of damage to the
blood-brain barrier (Fig. 2E).
Based on these results, DEX exerted neuroprotective effects
following TBI.
DEX alleviates neuroinflammation
following TBI
As previous studies have identified a vital role for
neuroinflammation in EBI following TBI, increased neuroinflammation
aggravates EBI (12,20,22,24).
The inflammatory complex induces the secretion of pro-inflammatory
cytokines, including IL-1β, IL-6 and TNF-α and the subsequent
activation of pro-inflammatory signaling through NF-κB to initiate
pyroptosis. Therefore, the hippocampal levels of IL-1β, IL-6, TNF-α
and NF-κB were measured using ELISAs. The levels of the
pro-inflammatory cytokines were increased significantly following
TBI, while their levels decreased significantly following DEX
treatment (Fig. 3A-D). Hence, these
results suggested that DEX exhibited potent anti-inflammatory
activity against TBI-induced neuroinflammation.
DEX inhibits TBI-induced autophagy
activation in the hippocampus
To clarify whether autophagy serves an important
role in TBI and the regulatory effect of DEX on autophagy, the
levels of the autophagy-related proteins Beclin-1 and LC3 in each
group were detected using western blotting (Fig. 4A). The levels of both Beclin-1 and
LC3 were increased following TBI but decreased significantly
following DEX treatment (Fig. 4B).
LC3 is a biomarker for autophagy activation and LC3 was chosen to
mark autophagic cells. Immunofluorescence staining showed few
LC3-positive neurons in the hippocampus of the Sham group, but were
widespread in the hippocampus following TBI induction. However, the
number of LC3-positive neurons decreased following DEX
administration (Fig. 4C).
Rapamycin stimulates autophagy and
reverses the neuroprotective effect of DEX
Rapamycin is a specific activator of autophagy
(18). Mice were pretreated with
rapamycin before the induction of TBI to investigate the
relationship between autophagy and the neuroprotective effect of
DEX. Pretreatment with rapamycin dramatically enhanced neurological
deficits (Fig 5A), aggravated brain
edema (Fig 5B) and BBB permeability
(Fig 5C) and reversed the
neuroprotective effect of DEX. Additionally, the TUNEL assay also
showed that rapamycin also significantly increased neuronal
apoptosis in the injured hippocampus compared with the TBI + DEX
group (Fig 5D). Levels of
autophagy-related proteins and apoptosis-related proteins were
detected using western blotting (Fig.
5E). DEX significantly decreased the levels of caspase 3,
Beclin-1 and LC3 (Fig. 5F) but
these changes were partially blocked by rapamycin administration.
Thus, rapamycin activated autophagy, abolished the anti-autophagy
effects of DEX and reversed the neuroprotective effects of DEX on
TBI.
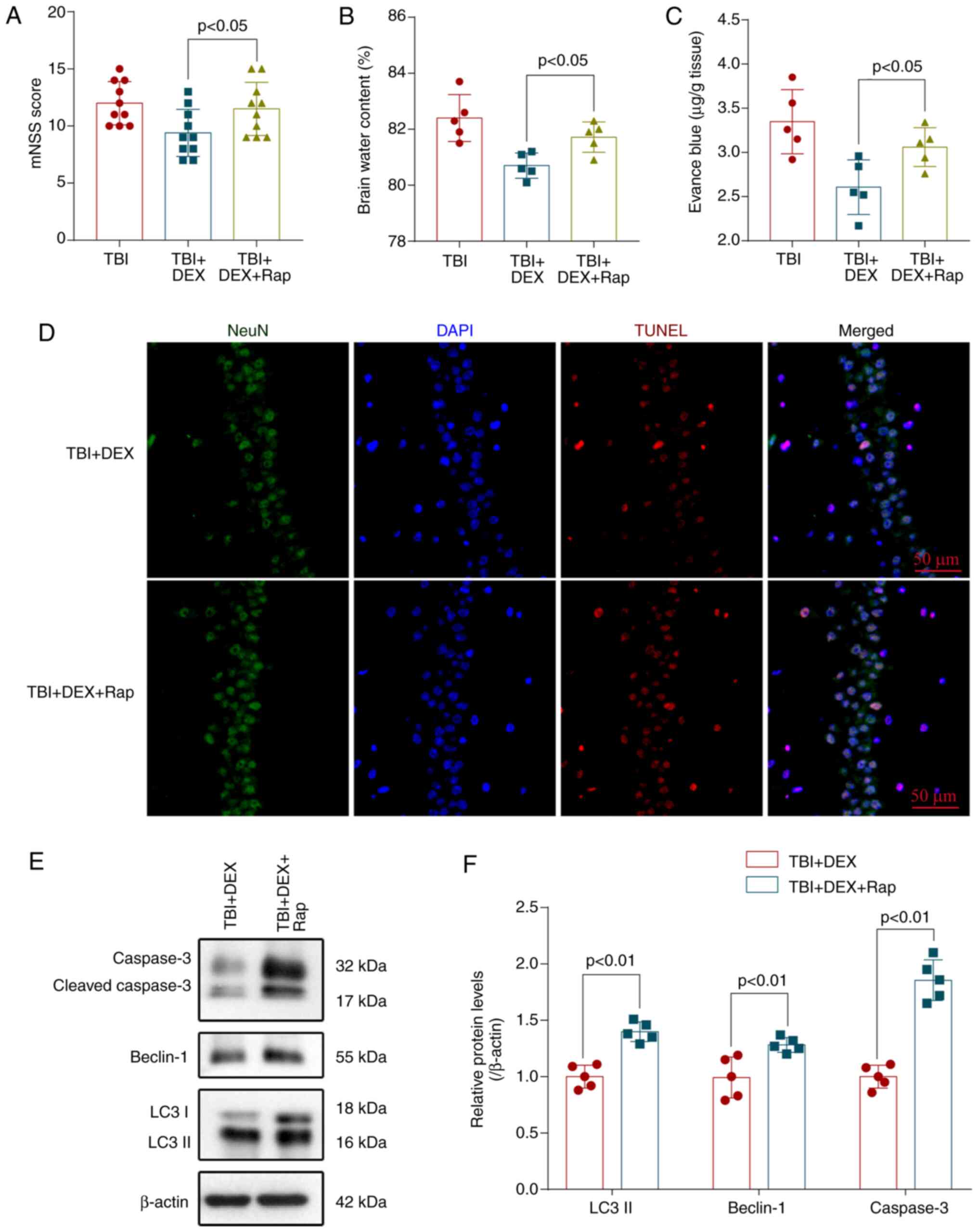 | Figure 5.Rap stimulates autophagy and reverses
the neuroprotective effect of DEX. Pretreatment with rapamycin
significantly increased the (A) mNSS, (B) The brain water content
and (C) BBB permeability compared with the TBI + DEX group (n=10,
P<0.05). (D) Rapamycin increased neuronal apoptosis in the
injured hippocampus compared with the TBI + DEX group
(magnification, ×400). (E) Levels of LC3, Beclin-1 and caspase-3 in
the brain cortex of mice with TBI were determined using western
blotting. (F) Quantification of LC3, Beclin-1 and caspase-3 levels
in the brain cortex relative to β-actin, the loading control. Rap
increases the LC3, Beclin-1 and caspase-3 levels in DEX-treated
mice following TBI (n=5, ANOVA; means ± standard error of mean).
Rap, rapamycin; DEX, dexmedetomidine; mNSS, modified neurological
severity score; TBI, traumatic brain injury; BBB, blood-brain
barrier; DAPI, 4′6-diamidino-2-phenylindole. |
DEX regulates autophagy by modulating
the ROS/Nrf2 signaling pathway following TBI
AKT/mTOR is a core signaling pathway of cell
autophagy. According to previous studies, activation of the
AKT/mTOR signaling pathway is partially dependent on ROS production
(38,39). The present study explored whether
autophagy inhibition by ROS occurred through the ROS/Nrf2 signaling
pathway following DEX treatment. ROS levels were detected with a
DCF-DA probe and the degree of membrane lipid peroxidation
evaluated by measuring the MDA content. The levels of ROS and MDA
were significantly increased following TBI but decreased following
DEX treatment (Fig. 6A and B). The
levels of the Nrf2 and HO-1 protein were detected by performing
western blotting to investigate neuronal autophagy (Fig. 6C). The levels of Nrf2 and HO-1 were
decreased significantly in the TBI group and increased following
DEX administration (Fig. 6D and E).
Thus, these results showed that DEX may inhibit TBI-induced
autophagy by regulating the ROS/Nrf2 signaling pathway.
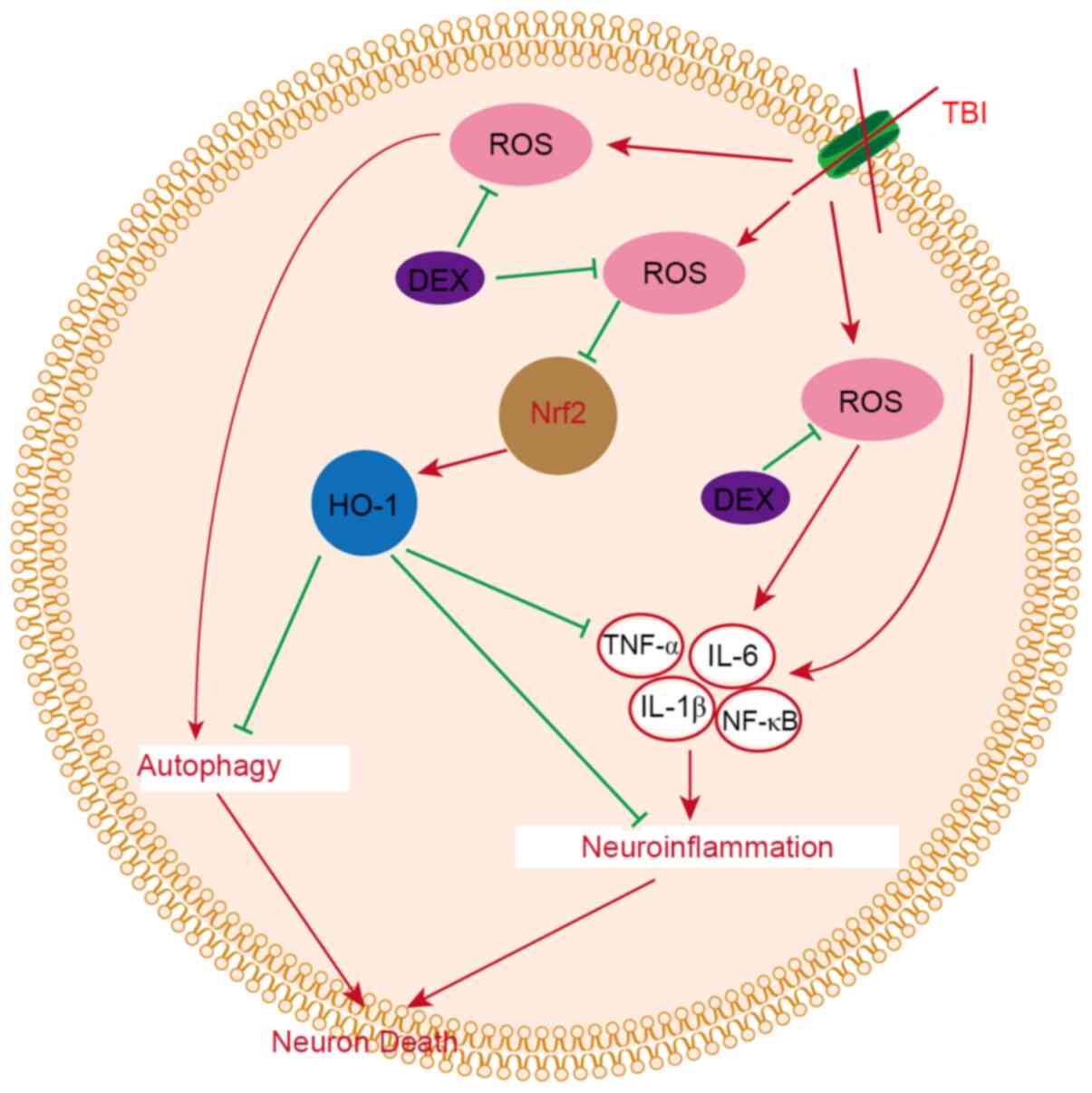
Discussion
The present study evaluated the therapeutic
potential of DEX to alleviate EBI in a mouse model of TBI. It was
demonstrated that DEX is a neuroprotective agent that attenuated
EBI following TBI. It was found that DEX i) improved neurological
dysfunction following TBI, ii) alleviated brain damage in a mouse
TBI model, iii) relieved neuroinflammation following TBI and then
decreased inflammatory damage in the brain and iv) prevented
autophagy following TBI and alleviated neuronal death, and v) the
anti-neuroinflammatory and anti-autophagy effects of DEX may be
related to the ROS/Nrf2 pathway (Fig.
7).
The present study, to avoid bias of biological sex
and gonadal hormone status, used healthy adult male C57BL/6J mice
not females. The main reason was that studies indicate that
biological sex and sex hormone levels may affect the outcome
following TBI (40,41). A recent study also recommends that
studies should include information on gonadal hormone status and
need to include biological sex and gonadal hormone status in the
design and analysis following TBI (42).
DEX, a highly selective α-2-adrenergic receptor
agonist with anxiolytic, sedative and analgesic properties, has
been widely used as an adjuvant during general anesthesia and
anti-delirium with minimal respiratory depression (12,21).
Clinical studies have shown that the administration of low-dose DEX
to critically ill adults reduces the incidence of delirium during
stays in intensive care units and reduces cognitive decline for up
to one postoperative month in elderly patients undergoing scheduled
laparotomy (21,43,44),
but the specific neuroprotective mechanism remains to be
elucidated. Chen et al (45)
aimed to further clarify the mechanisms and report that DEX
protects against MTX-induced cognitive impairment by inhibiting
neuronal toxicity and inflammation. In hypoxia-activated BV2
microglia and neonatal rats subjected to hypoxia, DEX decreases
hippocampal synaptic loss and neuronal damage and the potential
mechanisms may be that DEX prevents hypoxia-induced microglial NOX2
activation and then inhibits oxidative stress and the
neuroinflammatory response (46).
Mei et al (47) also report
that DEX alleviates BBB disruption, learning and memory
impairments, systemic inflammation and neuroinflammation in a cecal
ligation and puncture (CLP)-induced septic model. Hence, the
anti-inflammation and anti-neuroinflammation effects serve an
important role in neuroprotection following DEX treatment. As shown
in the present study, DEX alleviated neuroinflammation in mouse TBI
models. Additionally, DEX directly regulates several cell death
models (22,25,48–51).
According to Gao et al (49), DEX induces the expression of the
neuroglobin protein in hippocampal tissues and then inhibits
neuronal apoptosis through the mitochondrial pathway. Sun et
al (50) indicate that DEX may
preserve brain function and ultimately improve the outcome of
sepsis by decreasing pyroptosis and subsequently protecting
neurons. In Shen et al (51), DEX postconditioning also improves
cardiac outcomes and neurological function following cardiac arrest
and resuscitation in swine, partly by suppressing cell necroptosis.
Zhao et al (25) report that
DEX alleviates acute kidney injury by increasing autophagy through
the modulation of the PI3K/AKT/mTOR pathway. At present, the
neuroprotective effect of DEX-regulated autophagy activation or
inhibition remains to be elucidated.
Autophagy regulates the turnover of cellular
constituents to ensure the removal and recycling of toxins and is
important in cell homeostasis (52). The role of autophagy has been
confirmed in many central nervous system diseases, including acute
brain injury (12,15,16),
intracerebral hemorrhage (17), SAH
(18) and Huntington's disease
(19). Autophagy transports
materials in cells to lysosomes for degradation through different
pathways involved in the regulation of cell survival and death
mechanisms following TBI (53).
Therefore, autophagy serves a very important role in neuronal
injury and repair following TBI. Xue et al (54) report that DEX improves long-term
learning cognitive function by inhibiting neuronal autophagy in a
neonatal rat model of hypoxic-ischemic brain injury. As shown in Li
et al (12), DEX reverses
upregulated circLrp1b and Dram2 expression and downregulates
miR-27a-3p expression to subsequently inhibit excessive autophagy
and improve the TBI-induced neurological impairment. Based on the
findings reported by Zhao et al (25), DEX promotes cell autophagy and
subsequent LPS-induced acute kidney injury. Yang et al
(20) also report that the
protective effect of DEX is based on the activation of autophagy
through the α2-adrenoreceptor/AMPK/mTOR pathway. In the present
study, autophagy was excessively activated following TBI and then
led to neurological impairments, BBB disruption, brain edema and
neuronal apoptosis, which were reversed by the DEX treatment.
The mechanisms and molecules regulating the
autophagy pathway are complex and involve mTOR-dependent and
nondependent pathways. The upstream regulatory pathway of mTOR
includes AMPK and the PI3K/AKT pathway (13,20).
As shown in the present study, DEX decreased levels of ROS
production, subsequently inhibiting the activation of autophagy. In
the nucleus, ROS regulate the Nrf2 pathway, transcriptionally
activate hypoxia-inducible factor (HIF-1) and p53 and then promote
autophagy (28,29). Wang et al (55) report that rehmapicrogenin improves
adriamycin-induced nephropathy in vivo and in vitro
and the mechanisms may be to reduce ROS accumulation and then alter
the expression levels of Nrf2. Nrf2 regulates the expression of
>250 genes through a specific binding site. The majority of
these genes regulate oxidative stress and cell apoptosis, autophagy
and ferroptosis (30). TBI leads to
intracellular ROS accumulation and decreases the expression levels
of Nrf2 and HO-1 and the Nrf2/HO-1 signaling pathway also directly
regulates autophagy (56). In the
present study, DEX decreased ROS levels and then downregulated Nrf2
and HO-1 expression in the experimental TBI model. DEX ameliorated
EBI following TBI by inhibiting neuroinflammation and autophagy via
the Nrf2/HO-1 pathway. The specific mechanism remains unclear and
other potential molecular mechanisms may serve important roles.
Therefore, further research is needed to explore these mechanisms.
In addition, the experiments was performed in mice and debate
persists regarding whether the treatment is effective in humans. In
the future, the clinical effect of DEX will be explored on patients
with TBI.
In summary, the present study provided evidence that
autophagy, which is mediated by ROS and Nrf2, was an important
cellular regulatory mechanism and contributed to EBI following TBI.
The present study reported the DEX-mediated regulation of autophagy
by the ROS/Nrf2 pathway and provided a new way to explore the
biological effects and mechanisms underlying the anti-autophagy,
anti-inflammatory and neuroprotective properties of DEX.
Acknowledgements
Not applicable.
Funding
The present study was supported. by the National
Natural Science Foundation of China (grant. no. 81871589) and the
Wuxi Municipal Bureau on Science and Technology (grant no.
N20201008).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XF and WM performed the experiments and wrote the
manuscript. XF, JZ, WJ and YW assisted in performing the
experiments and prepared all the figures. YW and WM designed the
study and revised the manuscript. XF, YW and WM confirm the
authenticity of all the raw data All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
All animal experiments performed in this study
complied with the National Institutes of Health guidelines for the
handling of laboratory animals and were approved by the Ethics
Committee of the Wuxi Medical College of Anhui Medical University
(approval no. YXLL-2020-112).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rowell SE, Meier EN, McKnight B, Kannas D,
May S, Sheehan K, Bulger EM, Idris AH, Christenson J, Morrison LJ,
et al: Effect of out-of-Hospital tranexamic acid vs placebo on
6-month functional neurologic outcomes in patients with moderate or
severe traumatic brain Injury. JAMA. 324:961–974. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shankar JJ and Vandorpe R: CT perfusion
for confirmation of brain death. AJNR Am J Neuroradiol.
34:1175–1179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang JY, Gao GY, Feng JF, Mao Q, Chen LG,
Yang XF, Liu JF, Wang YH, Qiu BH and Huang XJ: Traumatic brain
injury in China. Lancet Neurol. 18:286–295. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen J, Li M, Chen L, Chen W, Zhang C,
Feng Y, Wang Y and Chen Q: The effect of controlled decompression
for severe traumatic brain injury: A randomized, controlled trial.
Front Neurol. 11:1072020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen JH, Li PP, Yang LK, Chen L, Zhu J, Hu
X and Wang YH: Value of ventricular intracranial pressure
monitoring for traumatic bifrontal contusions. World Neurosurg.
113:e690–e701. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nichol A, French C, Little L, Haddad S,
Presneill J, Arabi Y, Bailey M, Cooper DJ, Duranteau J, Huet O, et
al: Erythropoietin in traumatic brain injury (EPO-TBI): A
double-blind randomised controlled trial. Lancet. 386:2499–2506.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hutchinson PJ, Kolias AG, Timofeev IS,
Corteen EA, Czosnyka M, Timothy J, Anderson I, Bulters DO, Belli A,
Eynon CA, et al: Trial of decompressive craniectomy for traumatic
intracranial hypertension. N Engl J Med. 375:1119–1130. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cooper DJ, Nichol AD, Bailey M, Bernard S,
Cameron PA, Pili-Floury S, Forbes A, Gantner D, Higgins AM, Huet O,
et al: Effect of early sustained prophylactic hypothermia on
neurologic outcomes among patients with severe traumatic brain
injury: The POLAR randomized clinical trial. JAMA. 320:2211–2220.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wright DW, Yeatts SD, Silbergleit R,
Palesch YY, Hertzberg VS, Frankel M, Goldstein FC, Caveney AF,
Howlett-Smith H, Bengelink EM, et al: Very early administration of
progesterone for acute traumatic brain injury. N Engl J Med.
371:2457–2466. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Robertson CS, Hannay HJ, Yamal JM,
Gopinath S, Goodman JC, Tilley BC; Epo Severe TBI Trial
Investigators, ; Baldwin A, Rivera Lara L, Saucedo-Crespo H, et al:
Effect of erythropoietin and transfusion threshold on neurological
recovery after traumatic brain injury: A randomized clinical trial.
JAMA. 312:36–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y, Wang L, Hu T, Wang F, Han Z, Yin
Z, Ge X, Xie K and Lei P: Hydrogen improves cell viability partly
through inhibition of autophagy and activation of PI3K/Akt/GSK3β
signal pathway in a microvascular endothelial cell model of
traumatic brain injury. Neurol Res. 42:487–496. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H, Lu C, Yao W, Xu L, Zhou J and Zheng
B: Dexmedetomidine inhibits inflammatory response and autophagy
through the circLrp1b/miR-27a-3p/Dram2 pathway in a rat model of
traumatic brain injury. Aging (Albany NY). 12:21687–21705. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Zhao M, Shang L, Zhang Y, Huang C,
He Z, Luo M, Wu B, Song P, Wang M and Duan F: Homer1a protects
against neuronal injury via PI3K/AKT/mTOR signaling pathway. Int J
Neurosci. 130:621–630. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ceccariglia S, Cargnoni A, Silini AR and
Parolini O: Autophagy: A potential key contributor to the
therapeutic action of mesenchymal stem cells. Autophagy. 16:28–37.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang C, Shan Y, Hu Y, Fang Z, Tong Y, Chen
M, Wei X, Fu X and Xu X: FGF2 attenuates neural cell death via
suppressing autophagy after rat mild traumatic brain injury. Stem
Cells Int. 2017:29231822017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fang J, Zhu Y, Wang H, Cao B, Fei M, Niu
W, Zhou Y, Wang X, Li X and Zhou M: Baicalin protects mice brain
from apoptosis in traumatic brain injury model through activation
of autophagy. Front Neurosci. 12:10062018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao M, Gao J, Cui C, Zhang Y, Jiang X and
Cui J: Inhibition of PTEN ameliorates secondary hippocampal injury
and cognitive deficits after intracerebral hemorrhage: Involvement
of AKT/FoxO3a/ATG-mediated autophagy. Oxid Med Cell Longev.
2021:54726052021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen JH, Wu T, Xia WY, Shi ZH, Zhang CL,
Chen L, Chen QX and Wang YH: An early neuroprotective effect of
atorvastatin against subarachnoid hemorrhage. Neural Regen Res.
15:1947–1954. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aron R, Pellegrini P, Green EW, Maddison
DC, Opoku-Nsiah K, Oliveira AO, Wong JS, Daub AC, Giorgini F,
Muchowski P and Finkbeiner S: Deubiquitinase Usp12 functions
noncatalytically to induce autophagy and confer neuroprotection in
models of Huntington's disease. Nat Commun. 9:31912018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang T, Feng X, Zhao Y, Zhang H, Cui H,
Wei M, Yang H and Fan H: Dexmedetomidine enhances autophagy via
α2-AR/AMPK/mTOR pathway to inhibit the activation of NLRP3
inflammasome and subsequently alleviates lipopolysaccharide-induced
acute kidney injury. Front Pharmacol. 11:7902020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Subramaniam B, Shankar P, Shaefi S,
Mueller A, O'Gara B, Banner-Goodspeed V, Gallagher J, Gasangwa D,
Patxot M, Packiasabapathy S, et al: Effect of intravenous
acetaminophen vs placebo combined with propofol or dexmedetomidine
on postoperative delirium among older patients following cardiac
surgery: The DEXACET randomized clinical trial. JAMA. 321:686–696.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang GR and Hao FG: Dexmedetomidine
inhibits inflammation to alleviate early neuronal injury via
TLR4/NF-κB pathway in rats with traumatic brain injury. Crit Rev
Eukaryot Gene Expr. 31:41–47. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bilodeau V, Saavedra-Mitjans M, Frenette
AJ, Burry L, Albert M, Bernard F and Williamson DR: Safety of
dexmedetomidine for the control of agitation in critically ill
traumatic brain injury patients: A descriptive study. J Clin Pharm
Ther. 46:1020–1026. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li F, Wang X, Zhang Z, Zhang X and Gao P:
Dexmedetomidine attenuates neuroinflammatory-induced apoptosis
after traumatic brain injury via Nrf2 signaling pathway. Ann Clin
Transl Neurol. 6:1825–1835. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao Y, Feng X, Li B, Sha J, Wang C, Yang
T, Cui H and Fan H: Dexmedetomidine protects against
lipopolysaccharide-induced acute kidney injury by enhancing
autophagy through inhibition of the PI3K/AKT/mTOR pathway. Front
Pharmacol. 11:1282020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bu LL, Liu YQ, Shen Y, Fan Y, Yu WB, Jiang
DL, Tang YL, Yang YJ, Wu P, Zuo CT, et al: Neuroprotection of
exendin-4 by enhanced autophagy in a parkinsonian rat model of
α-synucleinopathy. Neurotherapeutics. Mar 15–2021.(Epub ahead of
print). View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dou R, Qian J, Wu W, Zhang Y, Yuan Y, Guo
M, Wei R, Yang S, Jurczyszyn A, Janz S, et al: Suppression of
steroid 5α-reductase type I promotes cellular apoptosis and
autophagy via PI3K/Akt/mTOR pathway in multiple myeloma. Cell Death
Dis. 12:2062021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li J, Tian M, Hua T, Wang H, Yang M, Li W,
Zhang X and Yuan H: Combination of autophagy and NFE2L2/NRF2
activation as a treatment approach for neuropathic pain. Autophagy.
April 9–2021.(Epub ahead of print). View Article : Google Scholar
|
|
29
|
Shi J, Yu T, Song K, Du S, He S, Hu X, Li
X, Li H, Dong S, Zhang Y, et al: Dexmedetomidine ameliorates
endotoxin-induced acute lung injury in vivo and in vitro by
preserving mitochondrial dynamic equilibrium through the
HIF-1a/HO-1 signaling pathway. Redox Biol. 41:1019542021.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen J, Wang Y, Wu J, Yang J, Li M and
Chen Q: The potential value of targeting ferroptosis in early brain
injury after acute CNS disease. Front Mol Neurosci. 13:1102020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen J, Zhang C, Yan T, Yang L, Wang Y,
Shi Z, Li M and Chen Q: Atorvastatin ameliorates early brain injury
after subarachnoid hemorrhage via inhibition of pyroptosis and
neuroinflammation. J Cell Physiol. Mar 3–2021.(Epub ahead of
print). View Article : Google Scholar
|
|
32
|
National Research Council (US) Institute
for Laboratory Animal Research, . Guide for the Care and Use of
Laboratory Animals. National Academies Press (US); Washington, DC:
1996
|
|
33
|
Flierl MA, Stahel PF, Beauchamp KM, Morgan
SJ, Smith WR and Shohami E: Mouse closed head injury model induced
by a weight-drop device. Nat Protoc. 4:1328–1337. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tian J, Yang L, Wang P, Yang L and Fan Z:
Exogenous CGRP regulates apoptosis and autophagy to alleviate
traumatic brain injury through Akt/mTOR signalling pathway.
Neurochem Res. 45:2926–2938. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen J, Xuan Y, Chen Y, Wu T, Chen L, Guan
H, Yang S, He J, Shi D and Wang Y: Netrin-1 alleviates subarachnoid
haemorrhage-induced brain injury via the PPAR gamma/NF-KB
signalling pathway. J Cell Mol Med. 23:2256–2262. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li G, Dong Y, Liu D, Zou Z, Hao G, Gao X,
Pan P and Liang G: NEK7 coordinates rapid neuroinflammation after
subarachnoid hemorrhage in mice. Front Neurol. 11:5512020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Das S, Chattopadhyay D, Chatterjee SK,
Mondal SA, Majumdar SS, Mukhopadhyay S, Saha N, Velayutham R,
Bhattacharya S and Mukherjee S: Increase in PPARγ inhibitory
phosphorylation by Fetuin-A through the activation of Ras-MEK-ERK
pathway causes insulin resistance. Biochim Biophys Acta Mol Basis
Dis. 1867:1660502021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao S, Cheng L, Shi Y, Li J, Yun Q and
Yang H: MIEF2 reprograms lipid metabolism to drive progression of
ovarian cancer through ROS/AKT/mTOR signaling pathway. Cell Death
Dis. 12:182021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dando I, Pacchiana R, Pozza ED, Cataldo I,
Bruno S, Conti P, Cordani M, Grimaldi A, Butera G, Caraglia M, et
al: UCP2 inhibition induces ROS/Akt/mTOR axis: Role of GAPDH
nuclear translocation in genipin/everolimus anticancer synergism.
Free Radic Biol Med. 113:176–189. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dick RW: Is there a gender difference in
concussion incidence and outcomes? Br J Sports Med. 43 (Suppl
1):i46–i50. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gupte R, Brooks W, Vukas R, Pierce J and
Harris J: Sex differences in traumatic brain injury: What We Know
and What We Should Know. J Neurotrauma. 36:3063–3091. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Biegon A: Considering biological sex in
traumatic brain injury. Front Neurol. 12:5763662021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kawazoe Y, Miyamoto K, Morimoto T,
Yamamoto T, Fuke A, Hashimoto A, Koami H, Beppu S, Katayama Y, Itoh
M, et al: Effect of dexmedetomidine on mortality and
ventilator-free days in patients requiring mechanical ventilation
with sepsis: A randomized clinical trial. JAMA. 317:1321–1328.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Skrobik Y, Duprey MS, Hill NS and Devlin
JW: Low-Dose nocturnal dexmedetomidine prevents ICU delirium. A
Randomized, Placebo-Controlled Trial. Am J Respir Crit Care Med.
197:1147–1156. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen J, Wang J, Li C, Ding H, Ye J and Xia
Z: Dexmedetomidine reverses MTX-induced neurotoxicity and
inflammation in hippocampal HT22 cell lines via NCOA4-mediated
ferritinophagy. Aging (Albany NY). 13:6182–6193. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen X, Chen D, Li Q, Wu S, Pan J, Liao Y,
Zheng X and Zeng W: Dexmedetomidine alleviates hypoxia-induced
synaptic loss and cognitive impairment via inhibition of microglial
NOX2 activation in the hippocampus of neonatal rats. Oxid Med Cell
Longev. 2021:66431712021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mei B, Li J and Zuo Z: Dexmedetomidine
attenuates sepsis-associated inflammation and encephalopathy via
central α2A adrenoceptor. Brain Behav Immun. 91:296–314. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang YF, Wang H, Song N, Jiang YH, Zhang
J, Meng XW, Feng XM, Liu H, Peng K and Ji FH: Dexmedetomidine
attenuates ischemia/reperfusion-induced myocardial inflammation and
apoptosis through inhibiting endoplasmic reticulum stress
signaling. J Inflamm Res. 14:1217–1233. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gao Y, Zhang Y, Dong Y, Wu X and Liu H:
Dexmedetomidine mediates neuroglobin Up-regulation and alleviates
the hypoxia/reoxygenation injury by inhibiting neuronal apoptosis
in developing rats. Front Pharmacol. 11:5555322020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sun YB, Zhao H, Mu DL, Zhang W, Cui J, Wu
L, Alam A, Wang DX and Ma D: Dexmedetomidine inhibits astrocyte
pyroptosis and subsequently protects the brain in in vitro and in
vivo models of sepsis. Cell Death Dis. 10:1672019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shen R, Pan D, Wang Z, Jin X, Li Z and
Wang H: The effects of dexmedetomidine post-conditioning on cardiac
and neurological outcomes after cardiac arrest and resuscitation in
swine. Shock. 55:388–395. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nixon RA: The role of autophagy in
neurodegenerative disease. Nat Med. 19:983–997. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zeng Z, Zhang Y, Jiang W, He L and Qu H:
Modulation of autophagy in traumatic brain injury. J Cell Physiol.
235:1973–1985. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Xue H, Wu Z, Xu Y, Gao Q, Zhang Y, Li C
and Zhao P: Dexmedetomidine post-conditioning ameliorates long-term
neurological outcomes after neonatal hypoxic ischemia: The role of
autophagy. Life Sci. 270:1189802021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang M, Ke Y, Li Y, Shan Z, Mi W, Cao Y,
Feng W and Zheng X: The nephroprotective effects and mechanisms of
rehmapicrogenin include ROS inhibition via an oestrogen-like
pathway both in vivo and in vitro. Biomed Pharmacother.
138:1113052021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cui G, Li Z, Cao F, Li P, Jin M, Hou S,
Yang X, Mu Y, Peng C, Shao H and Du Z: Activation of Nrf2/HO-1
signaling pathway attenuates ROS-mediated autophagy induced by
silica nanoparticles in H9c2 cells. Environ Toxicol. 36:1389–1401.
2021. View Article : Google Scholar : PubMed/NCBI
|