Introduction
According to the Global Cancer Observatory
statistics, cancer is the leading cause of death in the world, with
9.9 million deaths in 2020; the incidence rate of cancer was 20% in
the Caribbean and South America, with high mortality rates (14%).
Worldwide, gastric cancer (GC) is estimated to be the fifth most
common cancer type in both sexes, ranking sixth for new cases, with
over one million cases per year and third in mortality (1).
In Mexico, according to statistics from the National
Institute of Statistics and Geography, three out of 10
cancer-associated deaths among patients aged 30–59 years were due
to cancer of the digestive system. From 2011 to 2016, four out of
10 and three out of 10 cancer-associated deaths respectively
occurred in females and males aged >60 years and resulted from
tumors in digestive organs (2).
GC refers to any malignancy originating in the
region between the gastroesophageal junction and the pylorus. The
World Health Organization and the Lauren classification system
(3) have classified GC into two
types: Intestinal GC (IGC) and diffuse GC (DGC). Intestinal or
differentiated gastric cancer is characterized by localized and
expansive growth, while DGC has an infiltrating growth pattern, is
an undifferentiated adenocarcinoma and features dispersed cells
with individual or group invasive capacity (4). The development of IGC is preceded by
a precancerous process of several years and stages: Active chronic
gastritis, multifocal atrophic gastritis, complete intestinal
metaplasia, incomplete intestinal metaplasia, dysplasia and
adenocarcinoma (5). GC has a
multifactorial origin: Diet, lifestyle, genetics and socioeconomic
factors, and it has been observed that 80% of cases of IGC are
associated with previous Helicobacter pylori (H. pylori)
infection (6,7). GC is characterized by a complex
etiology with a set of factors, including genetic alterations and
external factors. However, it has been reported that <3% of GC
is due to heredity and includes hereditary DGC, proximal polyposis
of the stomach and hereditary colorectal cancer not associated with
polyposis (6). With respect to
molecular pathogenesis, chromosomal instability (aneuploidy,
chromosomal translocation, amplification, deletions and loss of
heterozygosity), gene fusion and microsatellite instability
(hypermethylation of gene repair promoters) are involved (7).
Copy number alterations (CNA) represent a class of
genetic variation that involve cumulative somatic variations. CNA
are defined as non-inherited genetic alterations that occur in
somatic cells (8). These
unbalanced structural variants usually contain gains or losses.
Their interpretation and the CNA report continue to be a topic of
interest in health and have an important role in GC (9,10).
The majority of gastric adenocarcinomas, similar to
numerous other types of solid tumor, exhibit defects in the
maintenance of genome stability, resulting in DNA CNA that may be
analyzed using comparative genomic hybridization (CGH) (11). This is a widespread and common
phenomenon among humans and several studies have focused on
understanding these genomic alterations that are responsible for
cancer and may be used for its diagnosis and prognosis (12).
At present, there are few published studies
involving genotyping of GC samples using high-density microarrays
(13–15); however, in those altered
chromosomes, gains and losses have a phenotypic impact and
different signaling pathways are involved. The presence of CNA
changes the genetic dose and would modify several molecular
mechanisms, such as epithelial-mesenchymal transition (EMT), which
is the transformation of epithelial cells to mesenchymal cells and
is a critical stage for the transition to metastasis (16). There are currently >1,184 genes
in the EMT Gene Database (dbEMT 2.0), which are involved in other
cancer-related processes, such as proliferative signaling, evasion
of growth suppressors, avoidance of immune destruction,
inactivation of replicative immortality, tumor-promoting
inflammation, induction of angiogenesis, genomic instability,
mutation, resting cell death, deregulation of cellular energetic
activity, invasion and cell plasticity (17). EMT includes activation of
transcription factors, expression of specific cell-surface
proteins, reorganization and expression of cytoskeletal proteins
and the production of extracellular matrix-degrading enzymes
(18). EMT has been associated
with the progression of cancer and increased stemness of tumors
(18,19), and was observed to be involved in
the formation, invasion and metastasis of GC (20,21). In addition, an association has
been established between the presence of CNA and its effect on the
expression level of EMT-associated genes in different cancer cell
lines (22). Studies on CNA
events involving Latin American populations are limited (23,24). In fact, at present, only a small
number of studies have performed GC genotyping using whole-genome
high-density microarrays in Mexican patients with GC (14,15). Therefore, the present study aimed
to determine CNA in DGC and IGC to identify, through bioinformatics
analyses, the main genes and signaling pathways involving
EMT-associated genes.
Materials and methods
Samples
Institutional Review Board approval was obtained for
the present study (approval no. 2008-785-001). The samples were
collected at the Regional General Hospital No. 1 ‘Dr. Carlos
MacGregor Sánchez Navarro’, Specialty Hospital ‘Dr. Bernardo
Sepúlveda’ and Oncology Hospital from IMSS in Mexico City (Mexico).
Clinical data and patient samples were processed following
obtainment of written informed consent. All of the samples were
collected over three years (April 2010 to May 2013) following
standardized endoscopy preservation protocols (25). Histological assessment of the
biopsies was performed by two trained pathologists independently.
They assigned the phenotypic diagnosis of diffuse or intestinal
tumors and non-atrophic gastritis (NAG) samples. Only samples with
the same diagnosis (‘identical results’) by two independent expert
pathologists were included in the analysis.
A total of 21 patients (5 females and 16 males) with
tissue samples that met the criteria for DGC (n=7) and IGC (n=7)
diagnoses, as well as subjects with NAG (n=7) as controls, were
included. In the absence of an established measurement (gold
standard), a value was arbitrarily determined to provide guidance
to investigate relevant alterations. To identify the most relevant
alterations for GC, the present analysis focused on alterations
present in at least three patients (cut-off, ≥3 patients; ≥40%
samples).
DNA extraction
DNA extraction was performed using a commercial kit
(QIAamp® DNA Micro Kit; Qiagen GmbH) according to the
manufacturer's protocol. The extraction was modified to include an
initial incubation at 95°C for 15 min, followed by a 5-min
incubation at room temperature, prior to digestion with proteinase
K (Qiagen GmbH) for three days at 56°C in a water bath, and fresh
enzyme was added at 24 h intervals, as described previously
(26).
DNA quality assessment and
preparation
The extracted DNA was quantified using
spectrophotometry (Nanodrop 2000; Thermo Fisher Scientific, Inc.).
Multiplex PCR was performed to assess the quality of DNA (Multiplex
PCR kit; Qiagen GmbH) with a set of primers to amplify various
regions of the GAPDH gene (27).
Products were visualized using 1% agarose gel electrophoresis
(RedGel® Nucleic acid gel stain; Biotium) and documented
under an ultraviolet light transilluminator system (Syngene).
High-density whole-genome microarray
analysis
The samples were analyzed using the
Affymetrix® CytoScan™ microarray (Affymetrix; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol
and with 250 ng DNA, except for the addition of five PCR cycles to
increase the DNA sample. The PCR products (90 µg) were fragmented
and labeled using additional PCR (https://assets.thermofisher.com/TFS-Assets/LSG/manuals/703038_cytoscan_assay_UG.pdf).
Copy number processing
The raw intensity files (.CEL), retrieved from the
commercial platform, were analyzed using their proprietary
software, Chromosome Analysis Suite (ChAS) v3.2 and NetAff 33
Libraries, based on the construction of the hg19 genome (February
2009) as a reference model.
Data processing was based on the segmentation
algorithm, where the Log2 ratio for each marker was
calculated relative to the reference signal profile. To calculate
the copy number variation (CNV), the data were normalized to
baseline reference intensities using the reference model (provided
by ChAS), including 270 HapMap samples and 96 healthy individuals.
The Hidden Markov Model, available in ChAS, was used to determine
the CN state and their breakpoints. The customized high-resolution
condition was used as a filter for the determination of CNV: CN
gains with a 50-marker count and 400 Kb, and CN losses with a
50-marker count and 100 Kb. The median absolute pairwise difference
(MAPD) and the single nucleotide polymorphism quality control (SNP
QC) score were used as the quality control parameters. Only samples
with values of MAPD >0.25 and SNP QC <15 were included in the
further analysis.
Bioinformatics analysis
A Perl script was developed to load the CNV segment
data files generated by ChAS for each sample to compare the files
to generate a list of genes that contained event types (gains or
losses), frequencies of altered regions, including chromosomes and
cytogenetic bands and Online Mendelian Inheritance in Man
information, and to incorporate additional information from
different databases (haploinsufficiency information from the
DECIPHER database of genomic variation, genes reported at dbEMT 2.0
and genes affected in gastric adenocarcinoma from Harmonized Cancer
Datasets; Table SI).
The genes altered in at least three patients
(cut-off, ≥3) with DGC, IGC or NAG were included for analysis and
visualizations were performed using R v4.0.2 and Bioconductor v3.12
packages (Table SII). The
karyotype was created with KaryoploteR and the Bioconductor
software annotation package (BSgenome.Hsapiens.UCSC.hg19 v1.4.0).
The comparison among samples was performed by generating Venn
diagrams with the jvenn server and a heatmap with gplots. Gene
Ontology (GO) analyses were performed with the ClusterProfiler
v3.16.1 packages (org.Hs.eg.db v3.11.4, enrich plot v1.8.1 and
GOplot v1.0.2), with the support of functional enrichment analysis
using the database for annotation, visualization and integrated
discovery (DAVID) v6.8 resource (Table SII). The profile of altered
molecular function (MF) terms in GC was summarized according to the
proportion of CNA-associated genes and the MF GO terms from the
DAVID database, adjusted by the false discovery rate. Dot plots,
heatmaps and chord plots were utilized to visualize the GC CNA
profiles for DGC, IGC and NAG.
To identify the main genes and signaling pathways
involving CNA EMT-associated genes, GC CNA-associated genes
(cut-off, ≥3) were analyzed and compared according to those
previously reported in the dbEMT 2.0, accessed on 12th October,
2020.
Finally, to establish the profile-associated
hallmarks of cancer involving DGC, IGC and NAG EMT-associated
genes, an interaction network was generated using CNA type (gains
and losses) based on genetic and physical interactions and
biological pathways. Furthermore, associations were determined
using the GeneMANIA prediction server and Cytoscape v.3.8.2,
including the manual annotation of their corresponding cancer
hallmarks [adhesion, angiogenesis, inflammation, migration,
metastasis, morphogenesis, proliferation and survival (28)], with punctual scrutiny and
assistance from databases, such as The Human Protein Atlas.
Table SII provides information
on the databases, protocols, software and specific packages
used.
Results
Sample characteristics
Samples from 21 patients with GC from Mexico
(third-generation Mexicans) between 35 and 91 years of age (mean ±
SD, 59.61 ± 15.94 years), without any previous cancer treatment
(naïve) were included in the present study. The samples included
seven cases who had DGC, seven who had IGC and seven who had NAG
(control samples). The raw data were deposited in the NCBI Gene
Expression Omnibus database (ID no. GSE117093). There are seven
adjacent tissue files (.CEL); however, these files were not
included in the data analysis, as certain adjacent tissues were
contaminated with cancer cells or these were not of the quality
required for subsequent analyses.
Table I presents
the ID and the percentage of neoplastic cells for tumor tissues
ranging between 50 and 70%. Blood agar culture indicated that one
patient with IGC and three patients with NAG were positive for
H. pylori (data obtained from our biobank database). The
patient data are also presented in Table I (29–31).
 | Table I.Characteristics of GC and NAG cases
analyzed in the present study (n=21). |
Table I.
Characteristics of GC and NAG cases
analyzed in the present study (n=21).
| ID | Age, years | Sex | Cancer type | % CC | H.
pylori | TNM | Treatment |
|---|
| 3CG-008 | 72 | M | Intestinal | 70 | Positive | IB T1 N1 M0 | Naïve |
| 3CG-126 | 80 | M | Intestinal | 60 | Negative | IIA T4 N0 M0 | Naïve |
| 3CG-128 | 91 | M | Intestinal | 70 | Negative | IIA T3 N2 M0 | Naïve |
| 3CG-046 | 52 | F | Intestinal | 60 | Negative | IV T4 N2 M0 | Naïve |
| 3CG-099 | 59 | M | Intestinal | 50 | Negative | II T3 N0 M0 | Naïve |
| 3CG-146 | 71 | M | Intestinal | 60 | Negative | IIB T3 N2 M0 | Naïve |
| 3CG-104 | 69 | M | Intestinal | 60 | Negative | III A T4 N0 M0 | Naïve |
| 3CG-047 | 58 | M | Diffuse | 70 | Negative | IV T4 N3 M0 | Naïve |
| 3CG-173 | 76 | M | Diffuse | 70 | Negative | III A T2 N3 M0 | Naïve |
| 8CG-004 | 76 | M | Diffuse | 70 | Negative | II T1 N0 M0 | Naïve |
| 1CG-001 | 45 | M | Diffuse | 60 | Negative | IV T4N2M1 | Naïve |
| 3CG-035 | 55 | M | Diffuse | 60 | Negative | IV T4N2M0 | Naïve |
| 3CG-042 | 64 | M | Diffuse | 50 | Negative | IV T4, N2 M0 | Naïve |
| 3CG-064 | 38 | M | Diffuse | 50 | Negative | IV T4 N2 M0 | Naïve |
| 4GB-001 | 64 | M | NAG | 0 | Negative | NA | NA |
| 4GB-031 | 62 | M | NAG | 0 | Negative | NA | NA |
| 4GB-015 | 35 | F | NAG | 0 | Negative | NA | NA |
| 4GB-025 | 39 | M | NAG | 0 | Positive | NA | NA |
| 4GB-033 | 76 | F | NAG | 0 | Positive | NA | NA |
| 4GB-036 | 38 | F | NAG | 0 | Positive | NA | NA |
| 4GB-042 | 77 | F | NAG | 0 | Negative | NA | NA |
Genomic detection of CNA
The total number of CNAs was obtained and they were
classified as either gains or losses for each chromosome in the GC
and NAG samples. From the total CNAs, DGC had more CNAs compared to
IGC (3,505 and 2,781, respectively), while there were 828 events in
the NAG samples. With respect to the tissue, more gains than losses
were observed in both cancer types, DGC (2,310 and 1,195,
respectively) and IGC (1,550 and 1,231, respectively), but the
opposite was observed in NAG (375 and 453, respectively) (Table SIII).
To identify the most relevant CNA in GC and NAG,
alterations occurring in at least three patients (cut-off, ≥3) were
analyzed. This comparison indicated a similar pattern for total
CNA, with more events in DGC (n=710) than in IGC (n=590) or in NAG
(n=332). In addition, more gains than losses were observed in DGC
(516 and 194, respectively), IGC (314 and 276, respectively) and
even in NAG (196 and 136, respectively), which was different when
all of the patients were included. Furthermore, DGC had the highest
number of gains and IGC had the highest number of losses (Table SIII). Table II lists chromosomes and sizes
with gain and loss numbers, representative and summarized.
| Table II.Principal affected chromosomes by CNA
cumulative length in DGC, IGC and NAG. |
Table II.
Principal affected chromosomes by CNA
cumulative length in DGC, IGC and NAG.
|
Type/chromosome | Gains | Losses | Length, Mb-cl |
|---|
| DGC |
|
|
|
| 1 | 327 | - | 117.9 |
| 4 | - | 155 | 40.8 |
| 5 | - | 148 | 74.23 |
| IGC |
|
|
|
| 1 | - | 148 | 33.78 |
| 8 | 365 | - | 139.8 |
| X | - | 66 | 167.1 |
| NAG |
|
|
|
| 6 | - | 28 | 0.40 |
| 7 | 21 | - | 0.20 |
| 14 | 10 | - | 3.02 |
| 17 | - | 15 | 1.86 |
| X | - | 87 | 0.47 |
| X | 207 | - | 1.20 |
To visualize the distribution of DGC and IGC
chromosome gains and losses, the identified CNA present in a
karyogram (cut-off, ≥3) was plotted, which displayed alterations
according to the coordinates of the Human genome hg19 (Fig. 1). The top five altered cytobands
are provided in Tables III and
SIV.
| Table III.Top five altered cytobands in DGC,
IGC and NAG. |
Table III.
Top five altered cytobands in DGC,
IGC and NAG.
| Type/cytoband | Gains | Length, Mb-cl | Number of
patients |
|---|
| DGC |
|
|
|
|
Xq28 | 37 | 11.5 | 6 |
|
8q24.22 | 25 | 15.8 | 5 |
|
1q32.1 | 26 | 12.16 | 5 |
|
8q24.3 | 25 | 17.46 | 6 |
|
1q23.3 | 22 | 15.26 | 6 |
| IGC |
|
|
|
|
8q24.3 | 24 | 12.662 | 4 |
|
13q34 | 18 | 4.9018 | 4 |
|
8q24.21 | 18 | 6.2619 | 4 |
|
8q24.22 | 16 | 6.4856 | 4 |
|
8q12.1 | 16 | 5.8523 | 4 |
| NAG |
|
|
|
|
Xq26.2 | 24 | 34.026 | 6 |
|
Xq21.2 | 22 | 33.586 | 7 |
|
Xp22.33 | 19 | 167.504 | 7 |
|
Xq23 | 16 | 63.79 | 6 |
|
Xq26.3 | 13 | 217.774 | 7 |
Of note, in DGC and IGC, the most frequent CNA
lengths were between 100 and 200 Kb, while lengths of 1–50 Kb were
more common in NAG, with respect to gains and losses (Table SV).
GC genes associated with CNA
Overall, 2,441 CNA-associated genes were identified
in DGC, IGC and NAG. GC had 2,420 affected genes (99%), while only
108 genes (4%) were affected in NAG; of these alterations, certain
candidates were shared between GC and NAG. There were 1,317 unique
CNA-associated genes in DGC, 596 in IGC and 21 in NAG. Furthermore,
both cancer types shared 420 genes, while 60 genes were shared
between GC and NAG. In addition, 19 genes in NAG were shared with
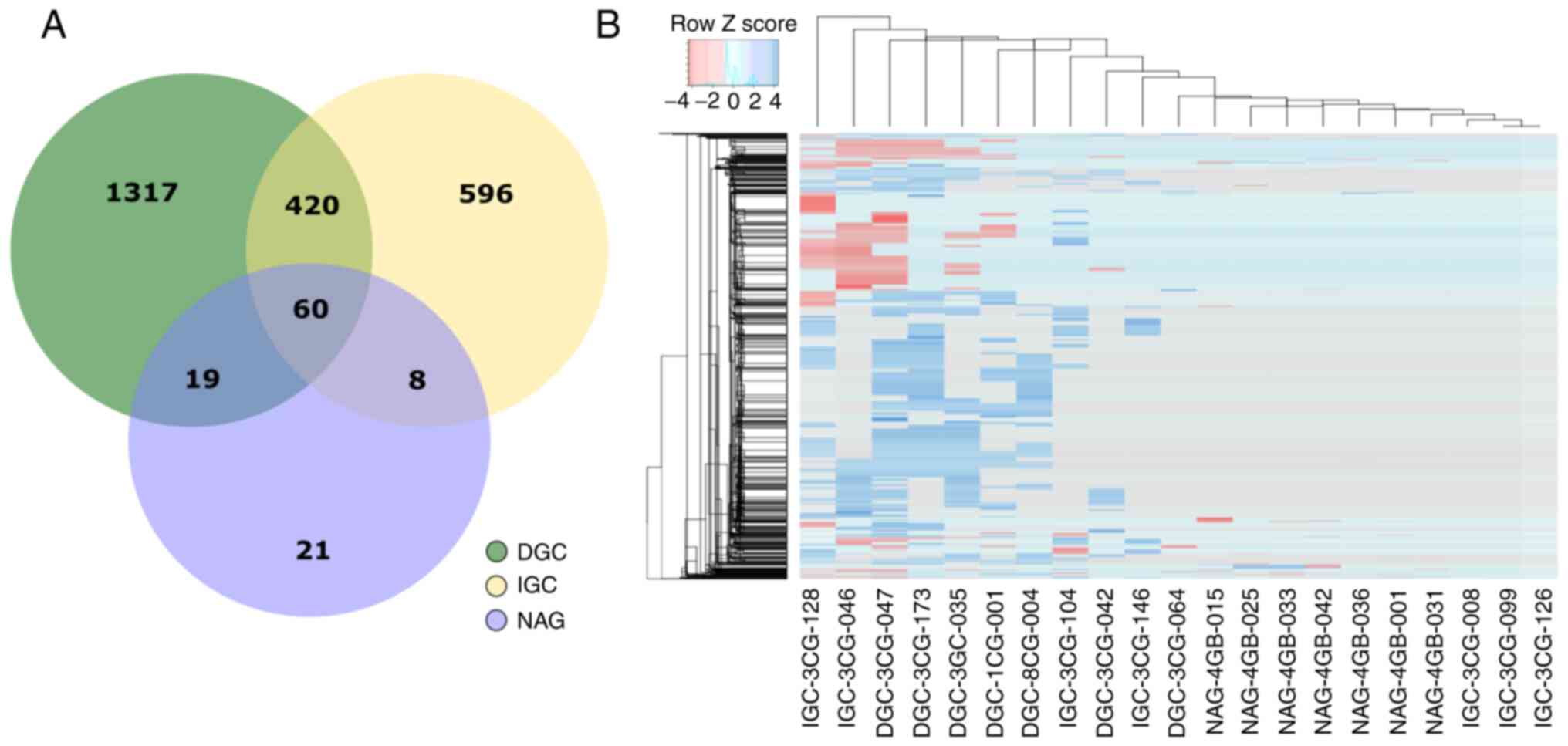
DGC and eight genes with IGC (Fig.
2A; Table SVI).
To identify the possible emerging patterns among the
samples, hierarchical clustering heatmaps were generated (Fig. 2B). The results provided the
molecular signature and hierarchical clustering of samples
according to the 2,441 genes. The emerging pattern of altered genes
affected by CNA distinguishes DGC and IGC from NAG.
GO analysis of GC
The functional profile for GC was generated through
enrichment and GO analysis of the 2,420 GC-altered genes; 1,317
genes were only altered in DGC and 596 only altered in IGC. To
identify the principal MFs altered in GC, these CNA-associated
genes were categorized, independently of the GC type, into two
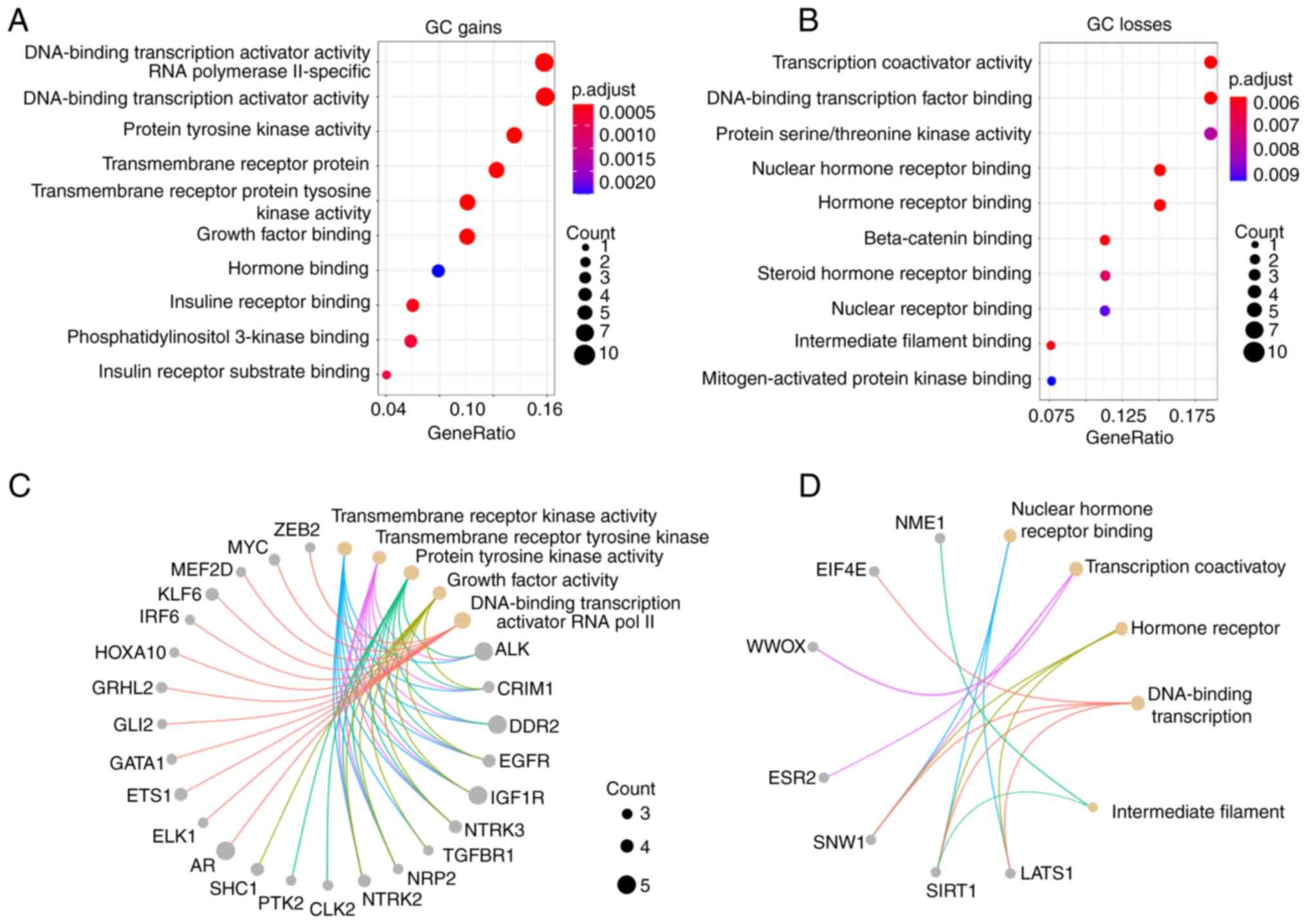
groups: Gains and losses (Fig. 3A and
B). The top 10 MFs associated with CNA gains or losses revealed
that transcription activator, tyrosine kinase activity, growth
factors and hormone binding, as well as intracellular signal
transduction genes were enriched in GC. Gene losses mainly involved
transcription coactivator and serine/threonine kinase activity, as
well as several receptors binding to hormone, steroid hormone,
nuclear receptor, β-catenin, intermediate filament and
mitogen-activated protein kinase binding genes. In addition, the
principal CNA-associated genes affecting the MF by GC type (DGC and
IGC) were identified (Figs. 3C and
D and S1).
CNA-EMT genes in DGC and IGC
To identify the main genes and signaling pathways
involving the CNA-EMT genes in GC and NAG, GC CNA-associated genes
were compared against a comprehensive and annotated database of EMT
genes (dbEMT 2.0). A total of 551 CNA-EMT genes were found in DGC,
619 in IGC and 28 in NAG. Using the cut-off ≥3, 112 genes in DGC,
66 in IGC and 5 in NAG were obtained. The complete data of
EMT-associated genes for DGC, IGC and NAG, with chromosome and
cytoband locations, CNA type (gain or loss), and the P-values are
provided in Table SI.
GO analysis of the EMT-associated
genes
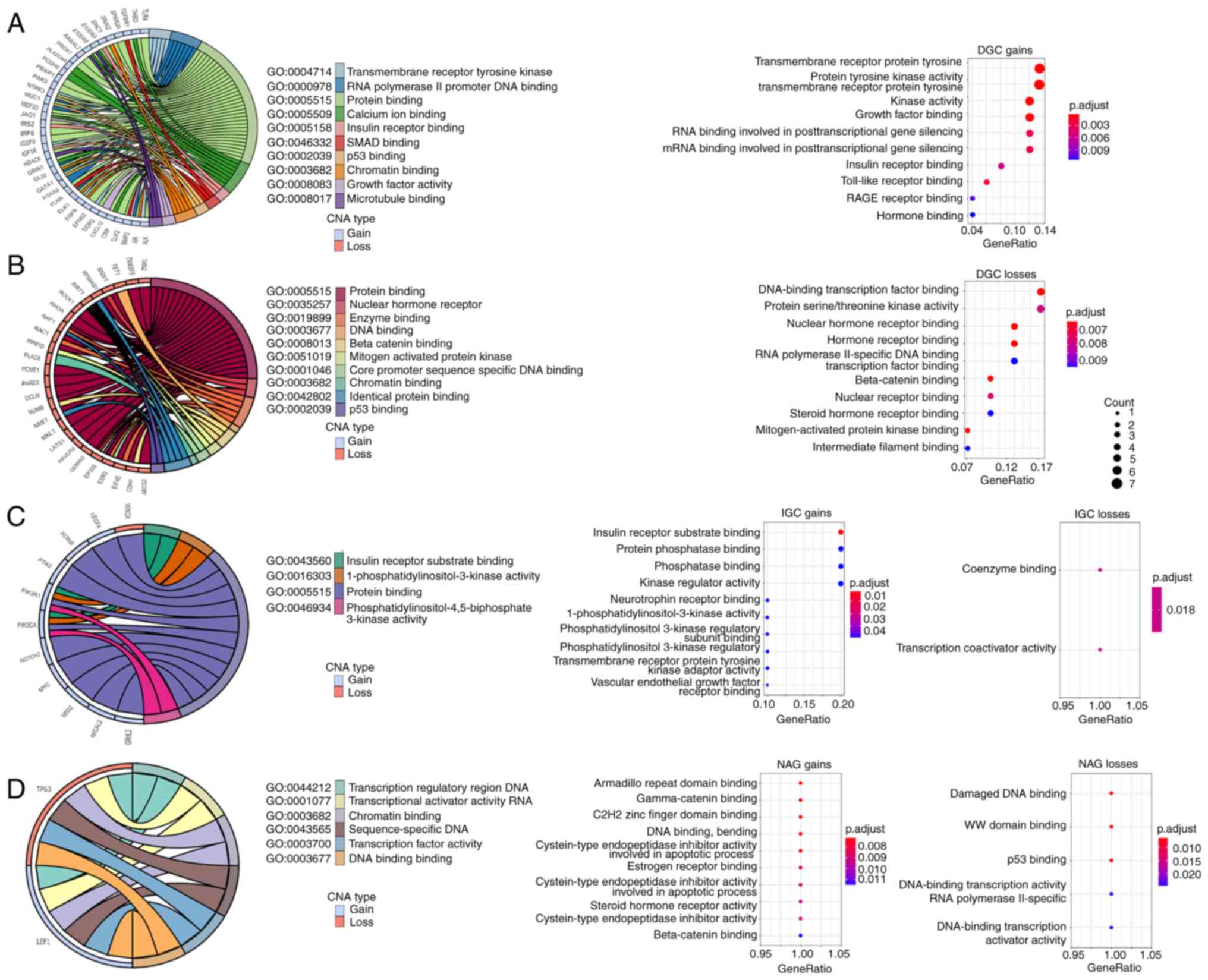
GO enrichment analysis was performed to determine
the MF of the main CNA-EMT genes affected in DGC, IGC and NAG
(Fig. 4). The results indicated
that gains in the CNA-EMT genes in DGC were associated with
transmembrane receptor tyrosine kinase, DNA and RNA binding and
receptor binding for insulin, growth factors, Toll-like receptors,
hormone, as well as SMAD, p53, chromatin, calcium ion binding and
microtubule binding (Fig. 4A).
Losses in CNA-EMT genes included associations with DNA and
chromatin binding, nuclear hormone receptor binding, β-catenin,
steroid hormone, mitogen-activated protein binding, intermediate
filament binding, p53 binding and RNA polymerase II-specific DNA
binding (Fig. 4B). Furthermore,
gains in CNA-EMT genes in IGC were associated with insulin receptor
substrate and phosphatase binding, kinase regulation, neurotrophin
receptor binding, 1-phosphatidylinositol-3-kinase activity,
transmembrane receptor protein tyrosine kinase adaptor activity and
VEGF receptor binding, while losses in CNA-EMT genes in IGC were
only associated with coenzyme binding and transcription coactivator
activity (Fig. 4C). On the other
hand, the main MF for gains in the CNA-EMT genes in NAG included
transcription regulatory region DNA, transcriptional activator
activity RNA, armadillo repeat and C2H2 zinc finger domain binding,
γ- and β-catenin binding, as well as cysteine-type endopeptidase
inhibitor activity involved in apoptotic process and estrogen
receptor, as well as steroid hormone receptor activity, while
losses in CNA-EMT genes in NAG were associated with damaged DNA, WW
domain, p53 binding and DNA-binding transcription activator
activity (Fig. 4D).
CNA-EMT genes associated with the
hallmarks of cancer
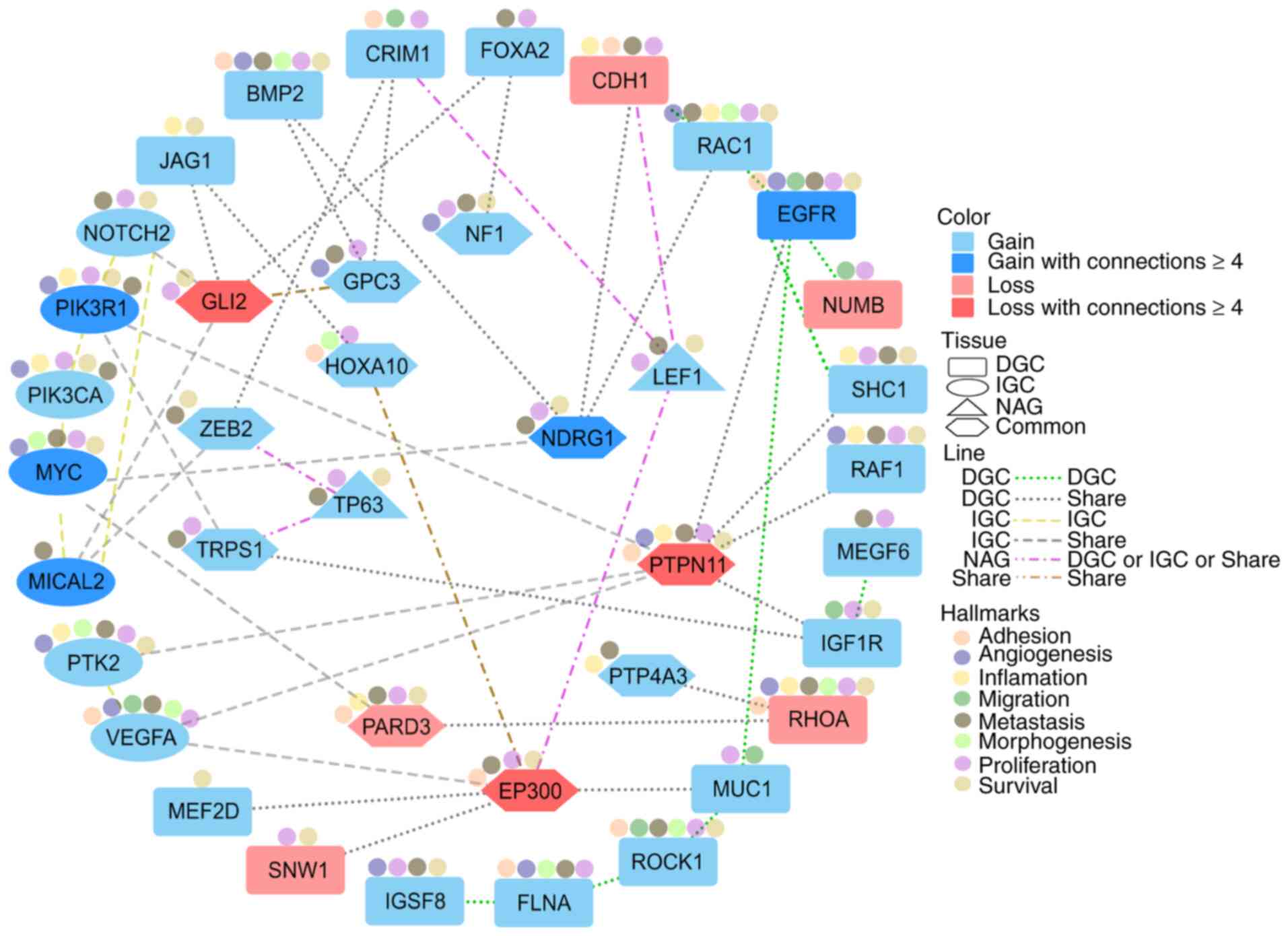
Based on the main molecular profile of altered
CNA-EMT genes in GC and NAG, the functional network between 39
previously selected unique CNA-EMT genes (19 genes for DGC, 7 for
IGC, 11 common to GC and two for NAG; cut-off, ≥3 patients) was
generated. Gained genes, with the highest degree and at least four
interactions per gene, were EGFR, MICAL2, MYC, NDRG and PIK3R1,
while lost genes included GLI2, EP300 and PTPN11. The principal
functions associated with these CNA-EMT genes have been previously
associated with several hallmarks of cancer: Adhesion,
angiogenesis, inflammation, migration, metastasis, morphogenesis,
proliferation and survival (Fig.
5).
Discussion
To the best of our knowledge, the present study was
the first whole-genome high-density array study on GC in Mexican
patients with DGC and IGC, as well as NAG as non-cancerous
controls. Using this experimental strategy, it was possible to
generate a karyogram and obtain molecular signatures for DGC and
IGC, and their association with CNA-EMT genes, independent of age,
sex, percentage of cancer cells, presence/absence of H.
pylori infection, TNM and treatment (naïve samples in the
present study). In addition, the genomic analysis was focused on
the molecular profile of GC, particularly involving alterations of
EMT-associated genes, given their role in cancer progression, as
epithelial cell transformation to mesenchymal cells is fundamental
to metastasis (32) and
chemoresistance (33,34). The results of the present study
are consistent with those previously reported in the literature
(detailed above), which provides validity and robustness to the
results and enables the reporting of novel data or data not yet
investigated to identify potential diagnostic, prognostic and
treatment response markers.
Globally, the alteration profile in GC was dominated
by gains. This phenomenon, where gains are more abundant than
losses, has been previously reported in different tumor cell lines,
including gastric cancer cell lines (35). Chromosomal gains in cancer may
result in increased gene functions, providing cancer cells with a
competitive advantage for the development of metastasis (36), while chromosomal losses may
involve the downregulation of tumor suppressor genes (37), disrupting homeostasis and
accelerating cancer progression. The most affected CNA chromosomes
for DGC were 1, 4 and 5; for IGC 1, 8 and X, and for NAG 6, 7, 14,
17 and X. The altered cytobands associated with GC observed in the
present study are in agreement with previous studies. For instance,
8q24 has been associated with the development of different types of
tumor (38). The highest
frequencies of gains in advanced GC were found at 8q24.21 (65%) and
8q24.3 (60%), and the pattern of CNA in advanced GC was different
from that in early GC. This increase in CNA numbers is associated
with disease progression from early to advanced GC (39). The 8q24 cytoband has also been
reported in Latin American countries, such as Brazil (40) and Venezuela (41), as well as in Asian countries,
including Korea (42).
Of note, the most frequent CNA length in GC was
100–200 Kb, in both DGC and IGC compared with 1–50 Kb in NAG. The
biological implications of this difference in length in GC compared
with non-cancerous tissues, such as NAG, is yet to be determined.
Furthermore, it is important to highlight that a resolution of
100–200 Kb versus Mb is an advantage of molecular resolution
approaches over classical cytogenetics (CGH and fluorescence in
situ hybridization) to discover ‘small’ potentially important
alterations in cancer samples.
The cumulative length averages (Megabases, Mb) of
these alterations were 183.44 Mb for DGC, 113.56 Mb for IGC and
1.19 Mb for NAG. These lengths, whether gained or lost, describe
the magnitude of global alterations per tissue; however, their
relevance lies in the MFs, biological processes and interaction
networks in which they participate.
In the present study, a molecular profile that
distinguishes GC from NAG was identified based on 2,441 genes
affected by CNA. They are associated with GC, as well as the
differences and similarities among histological subtypes
(undifferentiated DGC and well-differentiated IGC) compared with
that in non-cancerous tissue, such as NAG (43). Of note, 60 affected genes shared
between GC and NAG were identified; 19 genes were shared
exclusively with DGC, while only eight were shared with IGC. This
emerging pattern of shared altered genes between cancerous and
non-cancerous tissues should be further studied to identify
possible CNA-dependent oncogenic pathways and progression
trajectories from NAG to either GC subtype, particularly in
conjunction with environmental factors, such as H. pylori
infection, diet and lifestyle, that may be associated with the
spread patterns affecting patient survival (43).
In the heatmap, a separation between NAG and GC was
observed, exhibiting clusters based on the molecular profiles of
CNA-associated genes. There is a greater heterogeneity among the
IGC samples in clusters but there were more genes affected in DGC.
The front-line tool for IGC distinction has been based on different
criteria, such as the Lauren histopathological classification
system. However, due to challenges, including disagreements in the
correct assignment, diagnosis and treatment, new criteria have been
proposed, such as molecular characterization according to The
Cancer Genome Atlas (TCGA) Research Network, which divides GC into
four subtypes (44). The results
of the present study agree with the requirement for new proposals
for the classification of GC, which includes defined subgroups,
with the integration of several genomic and genetic parameters
where CNA are present.
In the present study, the MF profile of GC
CNA-associated genes was analyzed and determined. With respect to
gains, there were increased alterations involving transcription,
signaling, tyrosine kinases, growth factors, hormones and insulin,
while with respect to losses, molecules involved in transcription,
serine/threonine and MAP kinases, steroid hormones, β-catenin
binding and filament binding were decreased. These gene sets are
important in GC biology. There were 13 CNA genes in IGC (including
CDH1, LAST1, ROCK1 and WWOX) and 49 CNA genes in DGC (including
CRIM1, EGFR, MIR9-1, MUC1, MYC, NDRG1, SCRIB, SNAI2, VEGF and
ZEB2); therefore, these genes were further analyzed with the
intention of comparing the results of the present study with those
of others and organizing the data in a biologically coherent
context. For instance, CDH1 codes for E-cadherin and, from a
simplified viewpoint, E-cadherin maintains the epithelial
phenotype; if CDH1 is lost, this promotes the mesenchymal
phenotype, i.e., it favors loss of adhesion and metastasis
(32).
In the present study, an enrichment analysis of
unique CNA-associated genes for all tissues was performed and
several shared MFs, such as protein binding, were obtained. Several
gained-genes that encode for RNA-binding proteins (45) have diverse targets and participate
in tumor progression by regulating homeostasis and changing
expression patterns. Chromatin binding is another altered function
in GC that participates in regulating eukaryotic gene expression,
methylation profile modulation, and genome stability maintenance
(46). EMT is a process that
involves changes in histone modification, DNA methylation and
chromatin accessibility. These changes may be promoted through
transcription, allowing the cell to have an identity or to have a
mesenchymal-epithelial transition-EMT conversion (47). The kinase function in DGC and IGC
gains have recently been considered key regulators in the
development of cancer (48).
Numerous kinases were associated with the initiation and
progression of carcinogenesis and are one of the main therapeutic
targets for the development of inhibitors in the clinical field.
Kinases are able to promote EMT and enhance invasion, migration and
evasion of apoptosis (49). In
the present study, PIK3R1 and PIK3CA were associated with IGC. The
PI3K pathway is a key regulatory hub for cell growth, survival and
metabolism (50). Activation of
PI3K is a frequent hallmark of cancer, highlighted by the
prevalence of somatic mutations in genes encoding key components of
this pathway (51). These enzymes
are responsible for transferring a phosphate group; however, the
reverse process is performed by phosphatases, which are also
affected in IGC. PIK3R1 is a gene frequently affected by mutations
or copy numbers in various types of cancer, according to the TCGA
project. These genes converge with the PI3K/AKT/mTOR pathway, which
is involved in the regulation of several processes (51).
To date, the differences between DGC and IGC have
been insufficiently investigated and understood; differences in
etiology, location, incidence and genetic profiles have been
observed (52). In the present
study, a CNA-EMT network for GC was generated with relevant genes
according to different criteria: Frequency among patients, genetic
connections, reported pathways and experimental associations with
several databases [dbEMT 2.0, The Human Protein Atlas, COSMIC
(53)] and Cancer Hallmark Genes
(54). Shared and exclusively
altered genes were observed for each tissue type. The common
CNA-EMT genes between DGC and IGC include GLI2, which has been
associated with proliferation (55); EP300, with multiple functions as
an inhibitor of antitumor immune response via metabolic modulation
(56); PTPN11, associated with GC
progression; and NDRG1, associated with metastasis and poor
prognosis in GC (57). Another
relevant gene in DGC is EGFR, which, due to its association with
CNV GC, is now the target for the development of anti-GC therapies
(58). IGC-associated EMT genes
include MICAL2, MYC and PIK3R1. MICAL2, a destabilizing F-actin in
cytoskeletal dynamics, has been associated with poor prognosis in
GC (59). MYC gains have also
been reported in several GC studies, as expected for a common
oncogenic gene (60) associated
with proliferation, differentiation and apoptosis (61). PIK3R1 participates in the PI3K/AKT
signaling pathway, with roles in apoptosis and cell survival, as
well as chemotherapy resistance in GC (62).
A large amount of data remains to be analyzed,
including loss of heterozygosity, mosaicism and other gene sets
that participate in different hallmarks of cancer. Another
limitation of the present study was the absence of a transcriptomic
analysis to validate the GC EMT signature, particularly for DGC and
IGC. Yet, the concordance of CNA with expression alterations in
EMT-associated genes is plausible, as previously observed for
multiple types of cancer from TCGA (35). In addition, further inclusion of
precancerous stages would allow further analysis of the ‘profile’
of IGC progression. The results of the present genomic approach
coincide with those already reported in the literature, which
provides validity and solidity to the results. After all, this
strategy allowed us to report novel or thus far scarce data, or
those not previously investigated, to identify differential GC CNA,
identify associations with relevant MFs associated with the
hallmarks of cancer and predict the EMT signature for DGC and IGC.
It may be hypothesized that these networks will potentially provide
treatment targets, as well as diagnostic and prognostic markers. In
addition, the use of NAG as a non-malignant control allowed for
investigation of the molecular and cellular events of GC and the
identification of potential biomarkers for the ‘early’ stages of
GC.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Ms. Irma P.
Ramos-Vega, Infectious and Parasitic Diseases Medical Research Unit
(UIMEIP), High Specialty Medical Unit (UMAE)-Pediatrics Hospital
‘Dr. Silvestre Frenk Freund’, XXI Century National Medical Center,
IMSS; Mr. Brian-Alexander Cruz-Ramírez and Ms. Alejandra
García-Bejarano, Oncological Diseases Medical Research Unit
(UIMEO), UMAE-Oncology Hospital, XXI Century National Medical
Center, IMSS for their technical assistance.
Funding
The present study was supported by the Fondo de Investigación en
Salud-Instituto Mexicano del Seguro Social (grant nos.
FIS/IMSS/PROT/G16/1573 and FIS/IMSS/PROT/PRIO/13/027).
Availability of data and materials
All datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The data have been deposited in the Gene Expression
Omnibus database (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession no.
GSE117093. Pre-print: doi: https://doi.org/10.1101/2021.11.22.469612.
Authors' contributions
VLS, HAVS and JDME performed the molecular
experiments, participated in data analysis to provide the results
and prepared, wrote and discussed the manuscript. JT, MCP and PPS
were responsible for clinical aspects and recruited the patients,
discussed the data and revised the manuscript. MERT contributed to
the design of the study, supervised the study and critically
reviewed, revised and wrote the manuscript. JT and MERT were
responsible for acquiring financial support. VLS and MERT confirm
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Institutional Review Board approval was obtained for
the study (approval no. 2008-785-001). Clinical data and patient
samples were processed following written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
INEGI-Social Communication, . Statistics
on World Cancer Day (February 4)-National Data. INEGI. 2018.
|
|
3
|
Lauren P: The two histological main types
of gastric carcinoma: Diffuse and so-called intestinal-type
carcinoma: An attempt at a histo-clinical classification. Acta
Pathol Microbiol Scand. 64:31–49. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Espejo Romero H and Navarrete Siancas J:
Classification of stomach adenocarcinomas. Rev Gastroenterol Peru.
23:199–212. 2003.(In Spanish). PubMed/NCBI
|
|
5
|
Piazuelo MB, Epplein M and Correa P:
Gastric cancer: An infectious disease. Infect Dis Clin North Am.
24853–869. (VII)2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nagini S: Carcinoma of the stomach: A
review of epidemiology, pathogenesis, molecular genetics and
chemoprevention. World J Gastrointest Oncol. 4:156–169. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McLean MH and El-Omar EM: Genetics of
gastric cancer. Nat Rev Gastroenterol Hepatol. 11:664–674. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mikhail FM, Biegel JA, Cooley LD, Dubuc
AM, Hirsch B, Horner VL, Newman S, Shao L, Wolff DJ and Raca G:
Technical laboratory standards for interpretation and reporting of
acquired copy-number abnormalities and copy-neutral loss of
heterozygosity in neoplastic disorders: A joint consensus
recommendation from the American College of Medical Genetics and
Genomics (ACMG) and the Cancer Genomics Consortium (CGC). Genet
Med. 21:1903–1916. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wallander K, Eisfeldt J, Lindblad M,
Nilsson D, Billiau K, Foroughi H, Nordenskjöld M, Liedén A and Tham
E: Cell-free tumour DNA analysis detects copy number alterations in
gastro-oesophageal cancer patients. PLoS One. 16:e02454882021.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Han B, Ren D, Mao B, Song X, Yang W, Zhang
H and Gao F: Tumor copy number alteration (CNA) burden as a
prognostic factor for overall survival in Chinese gastric cancers.
J Clin Oncol. 37 (Suppl 15):e155552019. View Article : Google Scholar
|
|
11
|
Milne AN: Early-onset gastric cancer:
Learning lessons from the young. World J Gastrointest Oncol.
2:59–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Iafrate AJ, Feuk L, Rivera MN, Listewnik
ML, Donahoe PK, Qi Y, Scherer SW and Lee C: Detection of
large-scale variation in the human genome. Nat Genet. 36:949–951.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Seabra AD, Araújo TM, Mello Junior FA, Di
Felipe Ávila Alcântara D, De Barros AP, De Assumpção PP, Montenegro
RC, Guimarães AC, Demachki S, Burbano RM and Khayat AS:
High-density array comparative genomic hybridization detects novel
copy number alterations in gastric adenocarcinoma. Anticancer Res.
34:6405–6415. 2014.PubMed/NCBI
|
|
14
|
Morales-Guerrero SE, Rivas-Ortiz CI, Ponce
de León-Rosales S, Gamboa-Domínguez A, Rangel-Escareño C,
Uscanga-Domínguez LF, Aguilar-Gutiérrez GR,
Kershenobich-Stalnikowitz D, Castillo-Rojas G and López-Vidal Y:
Translation of gastric disease progression at gene level
expression. J Cancer. 11:520–532. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nadauld LD, Garcia S, Natsoulis G, Bell
JM, Miotke L, Hopmans ES, Xu H, Pai RK, Palm C, Regan JF, et al:
Metastatic tumor evolution and organoid modeling implicate TGFBR2
as a cancer driver in diffuse gastric cancer. Genome Biol.
15:4282014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vasaikar SV, Deshmukh AP, den Hollander P,
Addanki S, Kuburich NA, Kudaravalli S, Joseph R, Chang JT,
Soundararajan R and Mani SA: EMTome: A resource for pan-cancer
analysis of epithelial-mesenchymal transition genes and signatures.
Br J Cancer. 124:259–269. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Varga J and Greten FR: Cell plasticity in
epithelial homeostasis and tumorigenesis. Nat Cell Biol.
19:1133–1141. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Christofori G: New signals from the
invasive front. Nature. 441:444–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Murai T, Yamada S, Fuchs BC, Fujii T,
Nakayama G, Sugimoto H, Koike M, Fujiwara M, Tanabe KK and Kodera
Y: Epithelial-to-mesenchymal transition predicts prognosis in
clinical gastric cancer: EMT in clinical gastric cancer. J Surg
Oncol. 109:684–689. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xia P and Xu XY: Epithelial-mesenchymal
transition and gastric cancer stem cell. Tumour Biol.
39:10104283176983732017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang G and Anastassiou D: Pan-cancer
driver copy number alterations identified by joint expression/CNA
data analysis. Sci Rep. 10:171992020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Owen GI, Pinto MP, Retamal IN, Fernádez
MF, Cisternas B, Mondaca S, Sanchez C, Galindo H, Nervi B, Ibañez
C, et al: Chilean gastric cancer task force: A study protocol to
obtain a clinical and molecular classification of a cohort of
gastric cancer patients. Medicine (Baltimore). 97:e04192018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Araújo TM, Seabra AD, Lima EM, Assumpção
PP, Montenegro RC, Demachki S, Burbano RM and Khayat AS: Recurrent
amplification of RTEL1 and ABCA13 and its synergistic effect
associated with clinicopathological data of gastric adenocarcinoma.
Mol Cytogenet. 9:522016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eltoum I, Fredenburgh J, Myers RB and
Grizzle WE: Introduction to the theory and practice of fixation of
tissues. J Histotechnol. 24:173–190. 2001. View Article : Google Scholar
|
|
26
|
Fischer I, Cunliffe C, Bollo RJ, Weiner
HL, Devinsky O, Ruiz-Tachiquin ME, Venuto T, Pearlman A, Chiriboga
L, Schneider RJ, et al: Glioma-like proliferation within tissues
excised as tubers in patients with tuberous sclerosis complex. Acta
Neuropathol. 116:67–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Utrera-Barillas D, Valdez-Salazar HA,
Gómez-Rangel D, Alvarado-Cabrero I, Aguilera P, Gómez-Delgado A and
Ruiz-Tachiquin ME: Is human cytomegalovirus associated with breast
cancer progression? Infect Agent Cancer. 8:122013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jeon J and Cheong JH: Clinical
implementation of precision medicine in gastric cancer. J Gastric
Cancer. 19:235–253. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zubarayev M, Min EK and Son T: Clinical
and molecular prognostic markers of survival after surgery for
gastric cancer: Tumor-node-metastasis staging system and beyond.
Transl Gastroenterol Hepatol. 4:592019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brierley JD, Gospodarowicz MK and
Wittekind C: TNM Classification of Malignant Tumours: Edition 8.
John Wiley & Sons; New Jersey: pp. 1–16. 2016, PubMed/NCBI
|
|
32
|
Bure IV, Nemtsova MV and Zaletaev DV:
Roles of E-cadherin and noncoding RNAs in the
epithelial-mesenchymal transition and progression in gastric
cancer. Int J Mol Sci. 20:28702019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marin JJG, Perez-Silva L, Macias RIR,
Asensio M, Peleteiro-Vigil A, Sanchez-Martin A, Cives-Losada C,
Sanchon-Sanchez P, Sanchez De Blas B, Herraez E, et al: Molecular
bases of mechanisms accounting for drug resistance in gastric
adenocarcinoma. Cancers (Basel). 12:21162020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
De Las Rivas J, Brozovic A, Izraely S,
Casas-Pais A, Witz IP and Figueroa A: Cancer drug resistance
induced by EMT: Novel therapeutic strategies. Arch Toxicol.
95:2279–2297. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao M, Liu Y and Qu H: Expression of
epithelial-mesenchymal transition-related genes increases with copy
number in multiple cancer types. Oncotarget. 7:24688–24699. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wee Y, Wang T, Liu Y, Li X and Zhao M: A
pan-cancer study of copy number gain and up-regulation in human
oncogenes. Life Sci. 211:206–214. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhao M and Zhao Z: Concordance of copy
number loss and down-regulation of tumor suppressor genes: A
pan-cancer study. BMC Genomics. 17 Suppl 7(Suppl 7):5322016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tong Y, Tang Y, Li S, Zhao F, Ying J, Qu
Y, Niu X and Mu D: Cumulative evidence of relationships between
multiple variants in 8q24 region and cancer incidence. Medicine
(Baltimore). 99:e207162020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Arakawa N, Sugai T, Habano W, Eizuka M,
Sugimoto R, Akasaka R, Toya Y, Yamamoto E, Koeda K, Sasaki A, et
al: Genome-wide analysis of DNA copy number alterations in early
and advanced gastric cancers. Mol Carcinog. 56:527–537. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Anauate AC, Leal MF, Wisnieski F, Santos
LC, Gigek CO, Chen ES, Calcagno DQ, Assumpção PP, Demachki S,
Arasaki CH, et al: Analysis of 8q24.21 miRNA cluster expression and
copy number variation in gastric cancer. Future Med Chem.
11:947–958. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Labrador L, Torres K, Camargo M, Santiago
L, Valderrama E and Chiurillo MA: Association of common variants on
chromosome 8q24 with gastric cancer in Venezuelan patients. Gene.
566:120–124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jin DH, Park SE, Lee J, Kim KM, Kim S, Kim
DH and Park J: Copy number gains at 8q24 and 20q11-q13 in gastric
cancer are more common in intestinal-type than diffuse-type. PLoS
One. 10:e01376572015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Korivi BR, Faria S, Aly A, Sun J, Patnana
M, Jensen CT, Wagner-Bartak N and Bhosale PR: Intestinal and
diffuse gastric cancer: A retrospective study comparing primary
sites. Clin Imaging. 56:33–40. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cancer Genome Atlas Research Network, .
Comprehensive molecular characterization of gastric adenocarcinoma.
Nature. 513:202–209. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qin H, Ni H, Liu Y, Yuan Y, Xi T, Li X and
Zheng L: RNA-binding proteins in tumor progression. J Hematol
Oncol. 13:902020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Morgan MA and Shilatifard A: Chromatin
signatures of cancer. Genes Dev. 29:238–249. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bhullar KS, Lagarón NO, McGowan EM, Parmar
I, Jha A, Hubbard BP and Rupasinghe HPV: Kinase-targeted cancer
therapies: Progress, challenges and future directions. Mol Cancer.
17:482018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Olea-Flores M, Zuñiga-Eulogio MD,
Mendoza-Catalán MA, Rodríguez-Ruiz HA, Castañeda-Saucedo E,
Ortuño-Pineda C, Padilla-Benavides T and Navarro-Tito N:
Extracellular-signal regulated kinase: A central molecule driving
epithelial-mesenchymal transition in cancer. Int J Mol Sci.
20:28852019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Castel P, Toska E, Engelman JA and
Scaltriti M: The present and future of PI3K inhibitors for cancer
therapy. Nat Cancer. 2:587–597. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang Y, Kwok-Shing Ng P, Kucherlapati M,
Chen F, Liu Y, Tsang YH, de Velasco G, Jeong KJ, Akbani R,
Hadjipanayis A, et al: A pan-cancer proteogenomic atlas of
PI3K/AKT/mTOR pathway alterations. Cancer Cell. 31:820–832.e3.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Assumpção PP, Barra WF, Ishak G, Coelho
LGV, Coimbra FJF, Freitas HC, Dias-Neto E, Camargo MC and Szklo M:
The diffuse-type gastric cancer epidemiology enigma. BMC
Gastroenterol. 20:2232020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sondka Z, Bamford S, Cole CG, Ward SA,
Dunham I and Forbes SA: The COSMIC cancer gene census: Describing
genetic dysfunction across all human cancers. Nat Rev Cancer.
18:696–705. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang D, Huo D, Xie H, Wu L, Zhang J, Liu
L, Jin Q and Chen X: CHG: A systematically integrated database of
cancer Hallmark genes. Front Genet. 11:292020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wan J, Zhou J, Zhao H, Wang M, Wei Z, Gao
H, Wang Y and Cui H: Sonic hedgehog pathway contributes to gastric
cancer cell growth and proliferation. Biores Open Access. 3:53–59.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Krupar R, Watermann C, Idel C, Ribbat-Idel
J, Offermann A, Pasternack H, Kirfel J, Sikora AG and Perner S: In
silico analysis reveals EP300 as a panCancer inhibitor of
anti-tumor immune response via metabolic modulation. Sci Rep.
10:93892020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Dong X, Hong Y, Sun H, Chen C, Zhao X and
Sun B: NDRG1 suppresses vasculogenic mimicry and tumor
aggressiveness in gastric carcinoma. Oncol Lett. 18:3003–3016.
2019.PubMed/NCBI
|
|
58
|
Liang L, Fang JY and Xu J: Gastric cancer
and gene copy number variation: Emerging cancer drivers for
targeted therapy. Oncogene. 35:1475–1482. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mariotti S, Barravecchia I, Vindigni C,
Pucci A, Balsamo M, Libro R, Senchenko V, Dmitriev A, Jacchetti E,
Cecchini M, et al: MICAL2 is a novel human cancer gene controlling
mesenchymal to epithelial transition involved in cancer growth and
invasion. Oncotarget. 7:1808–1825. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Herold S, Herkert B and Eilers M:
Facilitating replication under stress: An oncogenic function of
MYC? Nat Rev Cancer. 9:441–444. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhang L, Hou Y, Ashktorab H, Gao L, Xu Y,
Wu K, Zhai J and Zhang L: The impact of C-MYC gene expression on
gastric cancer cell. Mol Cell Biochem. 344:125–135. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Huang X, Li Z, Zhang Q, Wang W, Li B, Wang
L, Xu Z, Zeng A, Zhang X, Zhang X, et al: Circular RNA AKT3
upregulates PIK3R1 to enhance cisplatin resistance in gastric
cancer via miR-198 suppression. Mol Cancer. 18:712019. View Article : Google Scholar : PubMed/NCBI
|