Introduction
Inflammatory bowel diseases (IBDs) such as
ulcerative colitis and Crohn's disease are refractory disorders
characterized by the recurrence of gastrointestinal inflammation
(1). Several studies have revealed
that intestinal dysbiosis leads to pro-inflammatory immunological
responses and induces gastrointestinal inflammation (2–4).
Intestinal macrophages influence the homeostasis of the
gastrointestinal tract and help maintain a balance between the
commensal intestinal microbiota and the host, but intestinal
dysbiosis promotes macrophage dysfunction. Therefore, disorders of
intestinal macrophages may invoke immune responses against
commensal bacteria, leading to the development of chronic
intestinal inflammation; thus, they play an important role in the
pathogenesis of IBDs (5–7). Macrophages are innate immune cells
involved in homeostasis, immune response, inflammation, and
regeneration and resolution of tissues (8). Macrophage polarization states are
mainly divided into the following two types: M1, pro-inflammatory
or classically activated; and M2, anti-inflammatory or
alternatively activated (9). M2
macrophages are capable of anti-inflammatory responses and tissue
repair. Macrophages dynamically alter their phenotype, switching
from the M1 to M2 phenotype during tissue repair (10). However, little is known about the
factors promoting this conversion during tissue repair. Previous
reports have shown that hepatocyte growth factor (HGF) signaling
affects cellular responses in macrophages (11–13).
HGF was originally purified from the plasma of
patients with fulminant hepatic failure as a protein that
stimulates DNA synthesis in hepatocytes (14). Increased HGF expression is observed
after experimental hepatic, renal, cardiac, or pulmonary injury and
is associated with tissue repair (15). The ligand HGF binds to the
tyrosine-protein kinase Met (c-MET), a single-pass transmembrane,
disulfide-linked a/b heterodimer receptor (16). HGF-MET signaling promotes
angiogenesis, cellular motility, growth, invasion, morphological
differentiation, embryological development, tissue regeneration,
and wound healing in various organs (17). Previous studies have shown that HGF
also ameliorates experimental colitis by accelerating intestinal
epithelial regeneration, promoting cell proliferation, and
preventing apoptosis. HGF aids in intestinal repair by mainly
contributing to intestinal epithelial regeneration (18–20).
However, a recent study has revealed that post-injury intestinal
repair is regulated by the anti-inflammatory processes of
macrophages (21,22); thus, further analyses are required
to clarify the effects of HGF on intestinal macrophages.
In this study, we investigated the effect of HGF on
intestinal immune cells, particularly intestinal macrophages using
a dextran sodium sulfate (DSS)-induced colitis mouse model.
Materials and methods
Mice
Specific-pathogen-free C57BL/6J mice (6–8-week-old)
were sourced from Charles River Laboratories, Japan. The mice were
maintained under standard conditions (24°C, 50–60% humidity, and a
12 h light/dark cycle) with ad libitum access to standard
mouse chow diet (LabDiet Autoclavable Rodent Diet 5010, #0006524,
PMI Nutrition International). We monitored body and stool
conditions and measured the body weight of the mice each day. The
mice were euthanized via cervical dislocation after being
anesthetized with intraperitoneal administration of ketamine (75
mg/kg) and medetomidine (1 mg/kg), and colonic tissues were
collected following euthanasia. Animal experimental procedures were
approved by the Institutional Animal Care and Use Committee of
Kagoshima University (Permit no: MD19057), and the experiments were
performed in accordance with the committee guidelines for animal
experiments.
Induction and assessment of
colitis
The DSS-induced colitis mouse model is an
established experimental model that enables the investigation of
various colitis symptoms, including diarrhea, weight loss, bloody
stool, mucosal ulceration, and shortening of the large intestine.
In this study, acute experimental colitis was induced in mice using
3% DSS (FUJIFILM Wako Pure Chemical Industries, Ltd.; average
molecular weight: 5,000). Control non-colitis mice were
administered distilled water. Colon tissue was collected 5 days
after DSS treatment for pathological and histological analyses.
After intraperitoneal administration of 200 µg (10
mg/kg) HGF or phosphate-buffered saline (PBS), as a vehicle for
HGF, to DSS-induced colitis mice or untreated control mice for 5
day, body weight, colon length, and disease activity index (DAI)
based on clinical scores for weight loss, stool consistency, and
bleeding (each score from 0 to 4, and the parameter values were
summed, maximum score 12) were measured (23), and the colon tissues were extracted
for the assessment of pathological score and gene expression assay.
This colitis model is characterized by the absence of severe
epithelial defects in the colon.
Histological assessment and
evaluation
Colon tissue was fixed in 10% phosphate-buffered
formalin, embedded in paraffin, sectioned, and stained with
hematoxylin and eosin. The stages of colitis were evaluated via
blinded histopathological analysis, according to previously
described morphological criteria, namely epithelial properties,
depth of inflammation, and degree of ulceration (24). Epithelial properties were scored as
follows: 0, intact crypt; 1, loss of the basal one-third of the
crypt; 2, loss of the basal two-thirds of the crypt; 3, loss of the
entire crypt, with intact surface epithelium; and 4, loss of the
entire crypt and surface epithelium (erosion). The depth of
inflammation was scored as follows: 0, no infiltration; 1, crypt
base; 2, mucosa; 3, submucosa; 4, submucosa (extensive). The degree
of ulceration was scored as follows: 0, none; 2, positive; 4,
positive (extensive). A histological score ranging from 0 to 12 was
assigned to each histological specimen.
Isolation of murine colonic lamina
propria mononuclear cells (LPMCs)
LPMCs were isolated from the large intestine of the
mice according to a previous report (23). Briefly, the colon samples were cut
into small pieces in a Ca2+/Mg2+-free 1.5%
Hank's balanced salt solution (1.5% HBSS), washed three times at
37°C with PBS, and incubated with HBSS containing 1 mM
dithiothreitol (#15-508-031, Invitrogen; Thermo Fisher Scientific,
Inc.) and 5 mM EDTA (#15-575-020, Invitrogen) for 30 min at 37°C to
remove the epithelial layer. The samples were then placed in a
digestion solution containing 1.5% HBSS, 1.0-3.0 mg/ml collagenase
A (#11-088-793-001, Roche Diagnostics GmbH), and 0.1 mg/ml DNase I
(#11-284-932-001, Roche Diagnostics GmbH) for 1–2 h at 37°C. The
samples were passed through a 100-µm strainer, transferred to a
50-ml Falcon tube, and centrifuged 200 × g, for 5 min at 4°C. The
supernatant was washed, and the pellets were resuspended in 40%
Percoll and overlaid on a 75% Percoll fraction. Mononuclear cells
were collected at the interphase, washed, and resuspended in
RPMI-1640 medium (#11875-093, Life Science Products Inc.)
containing 10% FBS and 1% penicillin/streptomycin. After isolation,
the LPMCs were seeded in 12-well plates (1×106 cells per
well) and treated with 10 ng/ml human HGF (E3112; EA Pharma Co.,
Ltd.) for 24 h in RPMI-1640 medium containing 10% FBS and 1%
penicillin/streptomycin.
Cell separation of LPMCs by
magnetic-activated cell sorting (MACS)
Colonic LPMCs were separated using MACS. LPMCs with
specific CD (cluster of differentiation) antibodies were
magnetically labeled with their respective magnetic beads (CD11b;
#130-049-601, Miltenyi Biotec). The cell suspension was loaded onto
a MACS LS Column (#130-042-401, Miltenyi Biotec), which was placed
in the magnetic field of a MACS separator (#130-042-301, Miltenyi
Biotec). The MACS separation buffer contained BSA (#A9576, Miltenyi
Biotec) at a final concentration of 0.5%. After the cells were
separated, they were analyzed for gene expression.
Gene expression analysis
Total RNA was extracted from the colon tissues or
LPMCs using TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) was performed using SYBR (Applied Biosystems). After
detection of the threshold cycle for each mRNA in each sample,
relative mRNA concentrations were calculated and normalized to that
of β-actin. PCR conditions included an initial holding period of
95°C for 30 sec, followed by a 2-step PCR program consisting of 40
cycles of 95°C for 5 sec and 60°C for 34 sec. All reactions were
performed in duplicate. The following genes were analyzed (forward,
reverse primers: Takara Bio Inc.): β-actin, inducible nitric oxide
synthase (iNOS), CD86, arginase-1 (Arg-1), CD206, TNF-α, tumor
necrosis factor-α (TNF-α), interleukin (IL)-6, IL-10, and
transforming growth factor-β1 (TGF-β1). The primers used in this
experiment are listed in Table
I.
 | Table I.Primer sequences used in this
study. |
Table I.
Primer sequences used in this
study.
| Gene | Forward, 5′-3′ | Reverse, 5′-3′ |
|---|
| iNOS |
CAAGCTGAACTTGAGCGAGGA |
TTTACTCAGTGCCAGAAGCTGGA |
| CD86 |
ATATGACCGTTGTGTGTGTTCTGGA |
AGGGCCACAGTAACTGAAGCTGTAA |
| Arg-1 |
AGCTCTGGGAATCTGCATGG |
ATGTACACGATGTCTTTGGCAGATA |
| CD206 |
AGCTTCATCTTCGGGCCTTTG |
GGTGACCACTCCTGCTGCTTTAG |
| TNF-α |
TATGGCCCAGACCCTCACA |
GGAGTAGACAAGGTACAACCCATC |
| IL-6 |
CCACTTCACAAGTCGGAGGCTTA |
CCAGTTTGGTAGCATCCATCATTTC |
| L-10 |
GCCAGAGCCACATGCTCCTA |
GATAAGGCTTGGCAACCCAAGTAA |
| TGF-β1 |
GTGTGGAGCAACATGTGGAACTCTA |
CGCTGAATCGAAAGCCCTGTA |
| β-actin |
CATCCGTAAAGACCTCTATGCCAAC |
ATGGAGCCACCGATCCACA |
Flow cytometry
LPMCs were washed with PBS and then blocked with
anti-mouse CD16/32 Antibody (Clone:93, BioLegend) in a
fluorescence-activated cell sorter (FACS) buffer for 10 min. After
Fc receptor blockade, the LPMCs were stained with AF700-conjugated
anti-CD11b (Clone: M1/70, BioLegend), PE-conjugated anti-c-MET
(Clone: eBioclone7, Invitrogen; Thermo Fisher Scientific, Inc.),
APC-A750-conjugated anti-CD86 (Clone: FA-11, Invitrogen; Thermo
Fisher Scientific, Inc.), PC7-conjugated anti-iNOS (Clone: CXNFT,
Invitrogen; Thermo Fisher Scientific, Inc.), eFluor-conjugated
anti-Arg-1 (Clone: A1exF5, Invitrogen; Thermo Fisher Scientific,
Inc.), and APC-conjugated anti-CD206 (Clone: C068C2, BioLegend) for
1 h on ice. Cells were washed and then resuspended in 4%
paraformaldehyde PBS (Fujiwako), 2% bovine calf serum
(Sigma-Aldrich; Merck KGaA), 0.1% sodium azide (Sigma-Aldrich;
Merck KGaA), and 0.1% HEPES. They were then assayed on a CytoFLEX
flow cytometer (Beckman Coulter) and analyzed using FlowJo (Becton
Dickinson & Company).
Colon organ cultures
Segments of the distal colon were removed from each
animal on day 5 after DSS administration, sectioned longitudinally,
and washed in PBS containing penicillin and streptomycin. Segments
of length 1 cm each were then placed in 24-well flat-bottom culture
plates (Asahi Glass) containing 1 ml fresh RPMI-1640 medium
supplemented with penicillin and streptomycin and incubated at 37°C
for 24 h. This experiment was performed on 13 mice from each group.
The culture supernatants were stored at 30°C until further
analysis.
The concentrations of TNF-α, IL-6, IL-10, and TGF-β1
in the culture supernatants were measured using a Mouse Quantikine
ELISA Kit (R&D Systems) according to the manufacturer's
instructions and analyzed in duplicate using a microplate reader
(Bio-Rad Laboratories) at 450 nm.
Statistical analysis
Differences between two or three groups were
appropriately analyzed using Kruskal-Wallis test followed by Dunn
test and Mann-Whitney U test (IBM SPSS version 28). P<0.05
indicated statistical significance. The in vitro experiments
were performed at least twice independently.
Results
HGF-altered cytokine expression in
LPMCs
First, we investigated whether LPMCs of mice with
DSS-induced colitis expressed c-MET. The expression of c-Met
of LPMC was higher in mice with DSS-induced colitis than in
non-colitis mice. In addition, the percentage of c-MET-positive
LPMCs was higher in mice with DSS-induced colitis than in
non-colitis mice (Fig. 1A). Next,
we investigated whether HGF affected the cytokine expression from
LPMCs in mice with DSS-induced colitis or non-colitis. The LPMCs
isolated from both mice groups were cultured under both conditions
of presence and absence of HGF for 24 h. The expression of several
cytokines and the genes encoding them was evaluated. A significant
increase was observed in the expression of IL-10, TGF-β,
TNF-α, and IL-6 in LPMCs cultured with HGF compared to
that in the untreated group (P=0.002, 0.004, 0.003, 0.005,
respectively). Furthermore, the expression of IL-10 and IL-6 was
numerically increased in cell culture supernatant with HGF-treated
LPMCs compared to that in the untreated LPMCs (P=0.05), whereas no
difference was observed in TNF-α and TGF-β1 expression in both
groups (Fig. 1B and C).
 | Figure 1.HGF upregulates the expression of
c-MET and anti-inflammatory cytokines. (A) The LPMCs were isolated
from mice with DSS-induced colitis or non-colitis mice.
c-MET mRNA expression in LPMCs was analyzed using RT-qPCR.
Obtained values were normalized to that of β-actin and expressed in
comparison to that of the LPMCs isolated from untreated mice (n=3).
The flow cytometry plots and the percentage of c-MET positive LPMCs
from mice with DSS-induced and non-colitis mice were analyzed using
flow cytometry (n=20). (B) TNF-α, IL-6, TGF-β, and
IL-10 mRNA expression in LPMCs treated with HGF or PBS for
24 h were analyzed using RT-qPCR. The obtained values were
normalized to that of β-actin and expressed in comparison to that
of the LPMCs isolated from untreated mice (n=10-16). (C)
Concentrations of cytokines in the culture supernatant of LPMCs
treated with HGF or PBS for 24 h. TNF-α, IL-6, TGF-β, and IL-10
expression was measured using ELISA (n=5). The results are shown as
mean ± SD. Data were analyzed using (A) Mann-Whitney U test or (B
and C) Kruskal-Wallis followed by Dunn test. *P<0.05. HGF,
hepatocyte growth factor; c-MET, tyrosine-protein kinase Met;
LPMCs, lamina propria mononuclear cells; DSS, dextran sodium
sulfate; RT-qPCR, reverse transcription-quantitative polymerase
chain reaction; IL, interleukin; PBS, phosphate-buffered saline;
TNF-α, tumor necrosis factor-α; Arg-1, arginase-1; TGF-β1,
transforming growth factor-β1; ELISA, enzyme-linked immunosorbent
assay. |
HGF-induced polarization of M1
macrophages to M2-like phenotype
We investigated whether HGF affected the phenotype
of macrophages, which are CD11b cells derived from the LPMCs of
mice with DSS-induced colitis by MACS. No difference was observed
in iNOS mRNA expression between HGF-treated and untreated
CD11b cells, whereas Arg-1 and CD206 expression
numerically increased in the CD11b cells treated with HGF compared
to that in the untreated cells, and CD86 expression
decreased. The levels of M2 markers Arg-1 and CD206 increased in
HGF-treated CD11b cells (Fig. 2A).
Moreover, we analyzed M1 and M2 protein expression of CD11b cells
treated with HGF using flow cytometry (Fig. 2B). In HGF-treated cells, iNOS and
CD86-positive cell percentages were higher, whereas Arg-1 and
CD206-positive cell percentages were lower than in untreated cells.
These results suggest that HGF could induce transformation from the
M1 phenotype to an M2-like phenotype in CD11b cells.
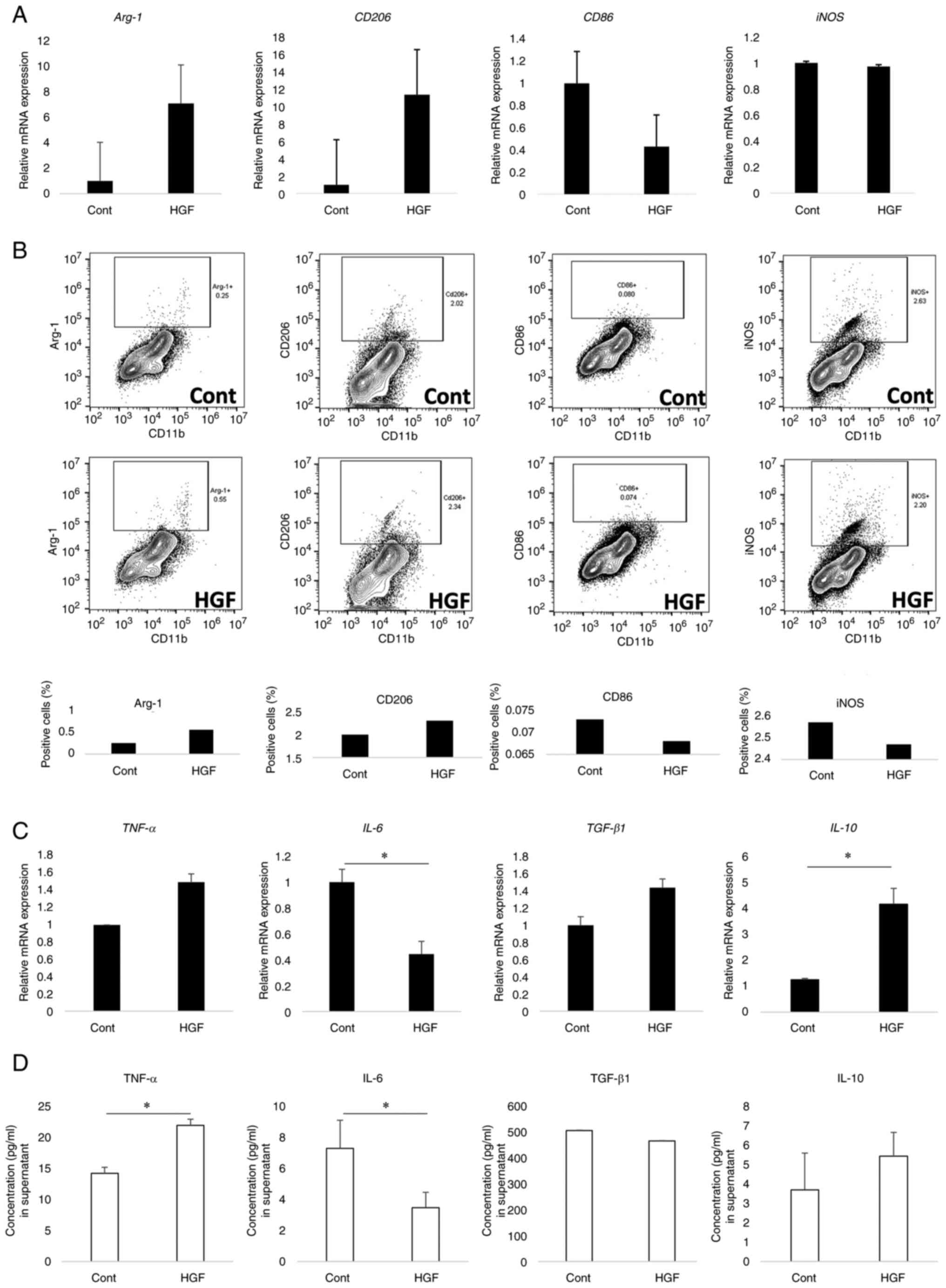 | Figure 2.HGF shifts M1 phenotype to M2-like
phenotype in macrophages. (A) Effects on expression of M1 and M2
markers by HGF treatment. iNOS and CD86 mRNA as M1 markers, Arg-1
and CD206 as M2 markers were analyzed using RT-qPCR. The values
were normalized to that of β-actin and expressed in relation to
CD11b-positive cells without HGF treatment. n=4. (B) Effects of HGF
treatment on protein expression of M1 and M2 markers. iNOS, CD86,
Arg-1, and CD206 were compared between HGF-treated and untreated
CD11b-positive cells using flow cytometry. n=20. (C) Effects of HGF
treatment on gene expression (n=8-11) and (D) secretion of
cytokines (n=4) in CD11b-positive cells. TNF-α, IL-6, TGF-β, and
IL-10 gene and protein expressions were measured using RT-qPCR and
ELISA, respectively. The results are expressed as mean ± SD. Data
were analyzed by Mann-Whitney U test. *P<0.05. HGF, hepatocyte
growth factor; iNOS, inducible nitric oxide synthase; CD, cluster
of differentiation; DSS, dextran sodium sulfate; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; IL,
interleukin; TNF-α, tumor necrosis factor-α; Arg-1, arginase-1;
TGF-β1, transforming growth factor-β1; ELISA, enzyme-linked
immunosorbent assay. |
Furthermore, IL-10 expression was
significantly increased in CD11b cells treated with HGF compared to
that in the untreated group (P=0.038), whereas IL-6
expression decreased (P<0.001). We observed a significant
increase in TGF-β expression in CD11b cells treated with HGF
compared to that in the untreated group. However, no difference was
observed in TNF-α expression between the HGF-treated and
untreated CD11b cells. Moreover, the secretion of TNF-α was
significantly increased in the cell culture supernatant of CD11b
cells treated with HGF (P=0.029), and the secretion of IL-10 was
numerically increased in the cell culture supernatant of CD11b
cells treated with HGF, whereas the secretion of IL-6 was
significantly decreased in the cells treated with HGF compared to
that in the untreated cells (P=0.029). In addition, no difference
was observed in TGF-β concentration between the two groups
(Fig. 2C and D).
HGF ameliorate DSS-induced
colitis
The DSS-induced colitis mouse model was evaluated on
day 5 after DSS administration. The body weight of the mice in the
vehicle group was significantly decreased compared to that in the
HGF-treated group, and the DAI in the vehicle group was
significantly increased compared to that in the HGF-treated group
(P<0.05, Fig. 3A and B).
Although no significant differences were observed in colon length
and pathological score between the vehicle and HGF-treated groups,
the DAI and colon length in the HGF-treated group were lower and
longer than those in the vehicle group, respectively (Fig. 3C-E).
 | Figure 3.Effects of HGF in mice with
DSS-induced colitis. (A) Final body weight relative to that on Day
0 (%). (B) Change in DAI scores. #P<0.05 (HGF vs. PBS
group). (C) Histopathology of the distal large intestine.
Magnification, ×10, ×100. colon tissue stained with hematoxylin and
eosin (D) Histological score. (E) Colon lengths (mm). (F) Cytokine
gene expression in CD11b-positive cells of colonic lamina propria
mononuclear cells (n=8). (G) Expression of cytokines from the
transverse colons obtained from PBS-treated and HGF-treated mice
(n=6). Colons were cultured for 24 h in serum-free medium, and
cytokine levels in the culture supernatants were measured using
ELISA. Data were analyzed by (D, F and G) Mann-Whitney U test and
(A, B and E) Kruskal-Wallis test followed by Dunn test *P<0.05.
HGF, hepatocyte growth factor; DAI, disease activity index; DSS,
dextran sulfate sodium; IL, interleukin; TNF-α, tumor necrosis
factor-α; CD, cluster of differentiation; PBS, phosphate-buffered
saline; ELISA, enzyme-linked immunosorbent assay. |
Moreover, gene expression in CD11b cells separated
from LPMCs of HGF-treated and vehicle groups was evaluated by
RT-qPCR. IL-6 expression was significantly decreased in the
HGF-treated group compared to that in the vehicle group (P=0.029),
whereas the expression levels of TNF-α, IL-10, and
TGF-β1 were not significantly different between the vehicle
and HGF-treated groups (Fig. 3F).
IL-6 secretion numerically decreased in the supernatant from the
tissue culture of the HGF-treated group compared to that of the
supernatant from the vehicle group; however, no significant
difference was observed in cytokine secretion between the two
groups (Fig. 3G).
Discussion
In this study, we demonstrated that HGF signaling
induces the polarization of M1 to M2-like macrophages in the LPMCs
of DSS-induced colitis mice. HGF-stimulated macrophages exhibited
increased IL-10 production and decreased IL-6 production.
HGF, which was originally purified from the plasma
of patients with fulminant hepatic failure, is the primary agent
that promotes hepatocyte proliferation. HGF also functions as a
pleiotropic factor, as it acts as a mitogen, morphogen, and motogen
in multiple subsets of various epithelial cells (14,17).
After HGF binds to the c-Met receptor, a signaling cascade is
activated that increases several biological actions
(proliferation/differentiation, survival, and motogenesis). HGF
modulates a major agent that not only promotes the proliferation of
hepatocytes but also of intestinal epithelial cells. Recombinant
HGF and HGF gene therapy attenuates acute colitis, and the
underlying mechanism could be the regeneration and repair of
injured epithelial cells (16,25).
In contrast, HGF also influences immune cells, such as macrophages,
by altering macrophage polarization (26,27).
Therefore, it is highly likely that the therapeutic effect of HGF
uncovered in this study is not the promotion of epithelial
regeneration but the therapeutic response resulting from its effect
on immune cells (28–30).
Macrophages are one of the most abundant leukocytes
in the intestinal mucosa and are essential for maintaining
intestinal homeostasis. They are implicated in the pathogenesis of
various disorders, such as IBD, offering the proteins on their
surface that act as potential targets for novel therapies for IBD.
The intestinal macrophage pool requires continual renewal from the
circulating blood monocytes, unlike most other tissue macrophages,
which appear to be derived from primitive precursors that
subsequently self-renew (31). As
many microbes always inhabit the intestinal tract, the macrophages
must be regulated by some mechanism to prevent an excessive immune
response against these organisms. However, an excessive immune
response and abnormal differentiation of macrophages may be induced
by intestinal microbes in IBD patients (32). The pro-inflammatory M1 (classically
activated) macrophages produce cytokines, such as IL-6, IL-1, and
TNFα; meanwhile, M2 (alternatively activated) macrophages produce
IL-10 and TGFβ, which are thought to be associated with tissue
repair. Therefore, the regulation of macrophage polarization is one
of the most important mechanisms for maintaining immune homeostasis
(33). In this study, the
DSS-induced colitis mouse model was used as the IBD model.
DSS-induced colitis occurs owing to the loss of epithelial barrier
function and entry of intestinal microbes into the lamina propria.
This results in an increase in the secretion of pro-inflammatory
cytokines and chemokines, and M1 macrophages migrate into the
lamina propria (34–36). c-MET is expressed by activated
macrophages (11,12,26).
In addition, we reported that HGF induced a
transformation from M1 to M2-like in macrophages extracted from
bone marrow (26). M2 macrophages
were generally negative for M1 markers (iNOS and CD86). However,
the macrophages in our previous report primarily expressed M2
markers, but some M1 markers were expressed a little. Therefore, we
defined the HGF-treated macrophages as M2-like. In this study, M1
macrophages also switched to M2-like macrophages, which showed low
expression of an M1 marker (iNOS) and high expression of an M2
marker (Arg-1) after HGF treatment. This effect may be involved in
the transition of macrophages from a pro-inflammatory to an
anti-inflammatory phenotype during colonic inflammation attenuation
and tissue repair.
The results of this study show that HGF increases
the production of IL-6 by LPMC. IL-6 is produced by various cells,
such as T cells, B cells, monocytes, and fibroblasts, and is
generally known for its pro-inflammatory effects. LPMCs contain
antigen-presenting cells, such as macrophages and dendritic cells,
and many T cells and B cells. Therefore, cytokine expression in
LPMCs would be affected by their cells. In contrast, HGF decreases
IL-6 production by only activated macrophages (Fig. 2B), and IL-6 is known to promote the
differentiation of pro-inflammatory Th17 cells while suppressing
the production of FoxP3+ regulatory T cells (37). Excessive IL-17 production by Th17
cells in the intestinal tract is involved in the development of IBD
(38,39). In addition, IL-6 plays a pivotal
role in angiogenesis, similar to vascular endothelial growth factor
(VEGF) and transforming growth factor (TGF)-β (40). Angiogenesis is essential in
development and recovery from inflammation. HGF also suppresses
IL-10 production by macrophages by stimulating M2-like macrophages
(26). The function of IL-10 is
important for regulating gut homeostasis during host defense, and
IL-10 suppresses the activity of T-lymphocytes and mononuclear
cells (41,42). The decreased expression of IL-6 and
increased expression of IL-10 by the effect of HGF in this study
affected T cells and improved intestinal inflammation.
This study had some limitations. The dose for this
experiment was decided based on our previous experiment,
specifically, intraperitoneal injections of HGF (0.1, 0.5, or 1.0
mg/kg) (43). These doses are
sufficient for HGF to act; therefore, we administered a
10-fold-dose (10 mg/kg) in this experiment. However, we do not have
dose-dependent data regarding HGF. Next, LPMCs and CD11b-positive
cells showed some differences in cytokine secretion after HGF
stimulation, which may be owing to the presence of LPMCs including
T cells and dendritic cells. We were unable to examine the effects
of HGF on immune cells other than macrophages, and the effect of
HGF on intestinal epithelial cells was not evaluated in
vivo. We have also not been able to evaluate whether the use of
MET inhibitors in this model counteracts the effects of HGF.
Further analyses are required to clarify the effects of HGF on the
immune system in the next experiment. In addition, we did not
assess the downstream factors of HGF. Activation of MET by HGF
induces the transphosphorylation of tyrosine kinase. Therefore, the
downstream factors of HGF in intestinal immune cells will be
investigated in the next study.
In conclusion, we have shown that a single round of
intramuscular injection of adenoviral HGF is sufficient to inhibit
apoptosis and reconstitute the epithelium in a mouse model of
DSS-induced colitis. Based on these results, this approach shows
promise as a clinical application of HGF in IBD treatment. HGF has
the potential as an important new treatment modality for intestinal
mucosal repair in IBD patients by mitigating intestinal
inflammation, although additional preclinical biological studies
are required.
Acknowledgements
The authors would like to thank Ms. Hiromi Eguchi
and Ms. Tomomi Kamibayashiyama (Digestive and Lifestyle Diseases,
Department of Human and Environmental Sciences, Kagoshima
University Graduate School of Medical and Dental Sciences) for
their technical assistance.
Funding
This work was supported by the Medical Research and Development
Program Focused on Technology Transfer: Adaptable and Seamless
Technology Transfer Program through Target-Driven Research and
Development (A-STEP) of the Japan Agency for Medical Research and
Development (AMED) (grant no. JP19pc0101036).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YF performed the experiments, analyzed the data and
wrote the manuscript. SK designed the study, analyzed the data and
wrote the manuscript. YM, IK, NM, AT, HM and KK performed the
experiments. ST and FS analyzed the data and revised the
manuscript. AI designed the study and reviewed the manuscript. YF,
YM and SK confirm the authenticity of all the raw data. All authors
have contributed to the manuscript, and read and approved the final
manuscript.
Ethics approval and consent to
participate
The animal study protocol was reviewed and approved
by the Institutional Animal Care and Use Committee of Kagoshima
University.
Patient consent for publication
Not applicable.
Competing interests
AI received honoraria from Eisai Co. Ltd. YF, SK,
YM, IK, NM, AT, HM, KK, FS and ST declare that they have no
competing interests.
Glossary
Abbreviations
Abbreviations:
|
IBDs
|
inflammatory bowel diseases
|
|
HGF
|
hepatocyte growth factor
|
|
LPMCs
|
lamina propria mononuclear cells
|
|
IL
|
interleukin
|
|
iNOS
|
inducible nitric oxide synthase
|
|
TNF-α
|
tumor necrosis factor-α
|
|
Arg-1
|
arginase-1
|
|
TGF-β1
|
transforming growth factor-β1
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
References
|
1
|
Khor B, Gardet A and Xavier RJ: Genetics
and pathogenesis of inflammatory bowel disease. Nature.
474:307–317. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Garrett WS, Lord GM, Punit S,
Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN and Glimcher LH:
Communicable ulcerative colitis induced by T-bet deficiency in the
innate immune system. Cell. 131:33–45. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Elinav E, Strowig T, Kau AL, Henao-Mejia
J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon
JI and Flavell RA: NLRP6 inflammasome regulates colonic microbial
ecology and risk for colitis. Cell. 145:745–757. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sartor RB: Microbial influences in
inflammatory bowel diseases. Gastroenterology. 134:577–594. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kanai T, Watanabe M, Okazawa A, Nakamaru
K, Okamoto M, Naganuma M, Ishii H, Ikeda M, Kurimoto M and Hibi T:
Interleukin 18 is a potent proliferative factor for intestinal
mucosal lymphocytes in Crohn's disease. Gastroenterology.
119:1514–1523. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rogler G, Andus T, Aschenbrenner E, Vogl
D, Falk W, Schölmerich J and Gross V: Alterations of the phenotype
of colonic macrophages in inflammatory bowel disease. Eur J
Gastroenterol Hepatol. 9:893–899. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schenk M and Mueller C: Adaptations of
intestinal macrophages to an antigen-rich environment. Semin
Immunol. 19:84–93. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Watanabe S, Alexander M, Misharin AV and
Budinger GRS: The role of macrophages in the resolution of
inflammation. J Clin Invest. 129:2619–2628. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Orliaguet L, Dalmas E, Drareni K,
Venteclef N and Alzaid F: Mechanisms of macrophage polarization in
insulin signaling and sensitivity. Front Endocrinol (Lausanne).
11:622020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sica A, Erreni M, Allavena P and Porta C:
Macrophage polarization in pathology. Cell Mol Life Sci.
72:4111–4126. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen Q, DeFrances MC and Zarnegar R:
Induction of Met proto-oncogene (hepatocyte growth factor receptor)
expression during human monocyte-macrophage differentiation. Cell
Growth Differ. 7:821–832. 1996.PubMed/NCBI
|
|
12
|
Galimi F, Cottone E, Vigna E, Arena N,
Boccaccio C, Giordano S, Naldini L and Comoglio PM: Hepatocyte
growth factor is a regulator of monocyte-macrophage function. J
Immunol. 166:1241–1247. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang Q, Azuma E, Tanaka M, Kobayashi M,
Hirayama M, Kumamoto T, Iwamoto S, Yamamoto H, Nakashima K, Sakurai
M and Komada Y: Differential responsiveness of cord and adult blood
monocytes to hepatocyte growth factor. Clin Exp Immunol.
125:222–228. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gohda E, Tsubouchi H, Nakayama H, Hirono
S, Takahashi K, Koura M, Hashimoto S and Daikuhara Y: Human
hepatocyte growth factor in plasma from patients with fulminant
hepatic failure. Exp Cell Res. 166:139–150. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mungunsukh O, McCar EA and Day RM:
Hepatocyte growth factor isoforms in tissue repair, cancer, and
fibrotic remodeling. Biomedicines. 5:301–326. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Trusolino L and Comoglio PM:
Scatter-factor and semaphorin receptors: Cell signalling for
invasive growth. Nat Rev Cancer. 2:289–300. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bolanos-Garcia VM: MET meet adaptors:
Functional and structural implications in downstream signalling
mediated by the Met receptor. Mol Cell Biochem. 276:149–157. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tahara Y, Ido A, Yamamoto S, Miyata Y, Uto
H, Hori T, Hayashi K and Tsubouchi H: Hepatocyte growth factor
facilitates colonic mucosal repair in experimental ulcerative
colitis in rats. J Pharmacol Exp Ther. 307:146–151. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Numata M, Ido A, Moriuchi A, Kim I, Tahara
Y, Yamamoto S, Hasuike S, Nagata K, Miyata Y, Uto H and Tsubouchi
H: Hepatocyte growth factor facilitates the repair of large colonic
ulcers in 2,4,6-trinitrobenzene sulfonic acid-induced colitis in
rats. Inflam Bowel Dis. 11:551–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yuge K, Takahashi T, Khai NC, Goto K,
Fujiwara T, Fujiwara H and Kosai K: Intramuscular injection of
adenoviral hepatocyte growth factor at a distal site ameliorates
dextran sodium sulfate-induced colitis in mice. Int J Mol Med.
33:1064–1074. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vannella KM and Wynn TA: Mechanisms of
organ injury and repair by macrophages. Annu Rev Physiol.
79:593–617. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Quiros M, Nishio H, Neumann PA, Siuda D,
Brazil JC, Azcutia V, Hilgarth R, O'Leary MN, Garcia-Hernandez V,
Leoni G, et al: Macrophage-derived IL-10 mediates mucosal repair by
epithelial WISP-1 signaling. J Clin Invest. 127:3510–3520. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tanaka A, Kanmura S, Morinaga Y, Kawabata
K, Arima S, Sasaki F, Nasu Y, Tanoue S, Hashimoto S, Takeshita M,
et al: Oral administration of Lactobacillus plantarum 06CC2
prevents experimental colitis in mice via an anti-inflammatory
response. Mol Med Rep. 21:1181–1191. 2020.PubMed/NCBI
|
|
24
|
Cooper HS, Murthy SN, Shah RS and
Sedergran DJ: Clinicopathologic study of dextran sulfate sodium
experimental murine colitis. Lab Invest. 69:238–249.
1993.PubMed/NCBI
|
|
25
|
Kanayama M, Takahara T, Yata Y, Xue F,
Shinno E, Nonome K, Kudo H, Kawai K, Kudo T, Tabuchi Y, et al:
Hepatocyte growth factor promotes colonic epithelial regeneration
via Akt signaling. Am J Physiol Gastrointest Liver Physiol.
293:G230–G239. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nishikoba N, Kumagai K, Kanmura S,
Nakamura Y, Ono M, Eguchi H, Kamibayashiyama T, Oda K, Mawatari S,
Tanoue S, et al: HGF-MET signaling shifts M1 macrophages toward an
M2-like phenotype through PI3K-mediated induction of arginase-1
expression. Front Immunol. 11:21352020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Choi W, Lee J, Lee J, Lee SH and Kim S:
Hepatocyte growth factor regulates macrophage transition to the M2
phenotype and promotes murine skeletal muscle regeneration. Front
Physiol. 10:9142019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Murray PJ and Wynn TA: Protective and
pathogenic functions of macrophage subsets. Nat Rev Immunol.
11:723–737. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gordon S: Alternative activation of
macrophages. Nat Rev Immunol. 3:23–35. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bain CC and Mowat AM: The
monocyte-macrophage axis in the intestine. Cell Immunol. 291:41–48.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Takada Y, Hisamatsu T, Kamada N, Kitazume
MT, Honda H, Oshima Y, Saito R, Takayama T, Kobayashi T, Chinen H,
et al: Monocyte chemoattractant protein-1 contributes to gut
homeostasis and intestinal inflammation by composition of
IL-10-producing regulatory macrophage subset. J Immunol.
184:2671–2676. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Formentini L, Santacatterina F, Núñez de
Arenas C, Stamatakis K, López-Martínez D, Logan A, Fresno M, Smits
R, Murphy MP and Cuezva JM: Mitochondrial ROS production protects
the intestine from inflammation through functional M2 macrophage
polarization. Cell Rep. 19:1202–1213. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kiesler P, Fuss IJ and Strober W:
Experimental models of inflammatory bowel diseases. Cell Mol
Gastroenterol Hepatol. 1:154–170. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang SW, Bai YF, Weng YY, Fan XY, Huang H,
Zheng F, Xu Y and Zhang F: Cinobufacini ameliorates dextran sulfate
sodium-induced colitis in mice through inhibiting M1 macrophage
polarization. J Pharmacol Exp Ther. 368:391–400. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu S, Shen H, Li J, Gong Y, Bao H, Zhang
J, Hu L, Wang Z and Gong J: Loganin inhibits macrophage M1
polarization and modulates sirt1/NF-κB signaling pathway to
attenuate ulcerative colitis. Bioengineered. 11:628–639. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Steinman L: A brief history of T(H)17, the
first major revision in the T(H)1/T(H)2 hypothesis of T
cell-mediated tissue damage. Nat Med. 13:139–145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sarra M, Pallone F, Macdonald TT and
Monteleone G: IL-23/IL-17 axis in IBD. Inflam Bowel Dis.
16:1808–1813. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fujino S, Andoh A, Bamba S, Ogawa A, Hata
K, Araki Y, Bamba T and Fujiyama Y: Increased expression of
interleukin 17 in inflammatory bowel disease. Gut. 52:65–70. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hashizume M, Hayakawa N, Suzuki M and
Mihara M: IL-6/sIL-6R trans-signalling, but not TNF-alpha induced
angiogenesis in a HUVEC and synovial cell co-culture system.
Rheumatol Int. 29:1449–1454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Maloy KJ and Powrie F: Intestinal
homeostasis and its breakdown in inflammatory bowel disease.
Nature. 474:298–306. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sydora BC, MacFarlane SM, Lupicki M,
Dmytrash AL, Dieleman LA and Fedorak RN: An imbalance in mucosal
cytokine profile causes transient intestinal inflammation following
an animal's first exposure to faecal bacteria and antigens. Clin
Exp Immunol. 161:187–196. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yamaji N, Ido A, Moriuchi A, Numata M,
Setoyama H, Tamai T, Funakawa K, Fujita H, Sakiyama T, Uto H, et
al: Hepatocyte growth factor ameliorates mucosal injuries leading
to inhibition of colon cancer development in mice. Oncol Rep.
26:335–341. 2011.PubMed/NCBI
|














