Introduction
Rapid and complete reperfusion has been widely
utilized in the treatment of patients with acute myocardial
infarction. However, this process may lead to severe reperfusion
injury (1–3). Since the study by Zhao et
al(4) in 2003, mounting
evidence has demonstrated that postconditioning potentially
alleviates reperfusion injuries (5–8).
Postconditioning is performed according to a variety
of protocols, known as postconditioning algorithms (9). However, no ‘ideal’ algorithm has been
established as yet (10,11). Various aspects of these algorithms
have been investigated, such as the time interval from the end of
ischemia to the application of postconditioning (12–14),
the duration of reperfusion and reocclusion episodes (15,16),
and the number of cycles applied in the process (17). Postconditioning algorithms dictate
when, how, and the duration required to apply reperfusion.
Limited data are available regarding the association
between brief reperfusion and reocclusion in transient
postconditioning. Penna et al(18) compared the effects of modified and
classical postconditioning algorithms and found that the two
algorithms yielded similar reductions in the infarct size and the
release of lactate dehydrogenase. The total time differed between
the two algorithms, with the classic algorithm lasting 10 sec (5
cycles of 10-sec reperfusion/reocclusion) and the modified
algorithm lasting 140 sec (15/20-20/15-25/10-30/5 sec). In their
study on postconditioning following brain ischemia, Wang et
al(19) noted that when the
brief reperfusion time was protracted and the reocclusion time had
a short duration (3 cycles of 60/15 sec of
reperfusion/reocclusion), the protection was attenuated or even
lost (19).
In the present study, we hypothesized that
protection is increased by gradually increasing the brief
reperfusion time, decreasing the reocclusion time and keeping the
total reperfusion/reocclusion cycle time fixed. This novel
postconditioning algorithm is referred to as gradual increased
reperfusion (GIR). Therefore, rats were subjected to acute
myocardial infarctions, and the cardioprotection provided by GIR
was compared with that resulting from standard
postconditioning.
Materials and methods
Study approval
The present study conformed to the Guide for the
Care and Use of Laboratory Animals published by the US National
Institutes of Health (NIH publication no. 85-23, revised 1996) and
was approved by the Research Commission on Ethics of the Chinese
PLA General Hospital (Beijing, China).
Rat heart model of acute myocardial
infarction and post-infarct treatment
Adult male Sprague-Dawley rats, weighing 200–250 g
each, were fed a normal diet prior to the experiment. The rats were
anesthetized with an initial intraperitoneal injection of sodium
pentobarbital (46 mg/kg), and were then intubated and ventilated
using a rodent respirator (ventilation rate, 52 breaths/min; tidal
volume, 4.0 ml/100 g body weight). A parasternal incision was made
to open the left pleural cavity by cutting the left four ribs and
the intercostal muscle. Following pericardiotomy, a 5-0 ligature
was placed under the left coronary artery by inserting the thread
into the left atrium and threading it out from the side of the
pulmonary artery cone. Prior to tying the knot, a balloon (Grip™,
3.0×12 mm; Acrostak, Geneva, Switzerland), connected to a pump full
of water at a pressure of 1 atm, was placed into the artery. After
the knot was tied, the pressure of the balloon was immediately
adjusted to 12 atm for 45 min. After the 45-min occlusion period,
the pressure of the balloon was immediately adjusted to 0 for
various periods of time, as per the protocols described below
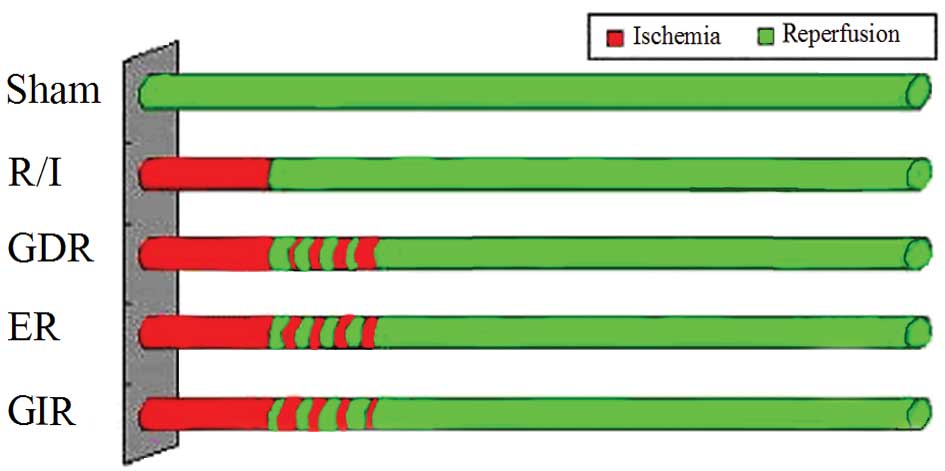
(Fig. 1). When the rats recovered
from anesthesia, tracheal intubation was removed. The rats were
anesthetized 24 h later with an initial intraperitoneal injection
of urethane (2 g/kg), and the right carotid artery was cannulated
using an arterial catheter connected to a physiograph through a
three-way stopcock. The hearts were then excised according to the
following procedures: i) anterior wall tissue was obtained from the
left ventricle after 6 h of reperfusion and maintained in a −80°C
freezer until western blot analysis; ii) the infarct size was
measured after 24 h of reperfusion, and the hearts were immediately
stained; iii) the myocardial tissue was soaked in formalin (10%, pH
7.4) until it was used for apoptotic index measurements.
Experimental protocols
Fifty rats were randomly allocated to one of the
following 5 groups (Fig. 1): i)
the sham group (control group), where the rats underwent
thoracotomy without ischemic treatment; ii) the reperfusion-injury
group (R/I), where the rats were administered routine
ischemic-reperfusion treatment; iii) the gradually decreased
reperfusion group (GDR), where the rats were administered 4 cycles
of reperfusion and reocclusion at the onset of reperfusion with
reperfusion/occlusion times of 30/10-25/15-15/25-10/30 sec (160 sec
total intervention time); iv) the equal reperfusion group (ER),
where the rats were administered 4 cycles of 20/20 sec
reperfusion/reocclusion beginning immediately at the onset of
reperfusion (160 sec total intervention time); and v) the gradually
increased reperfusion group (GIR), where the rats were administered
4 cycles of reperfusion/reocclusion at the onset of reperfusion
with times of 10/30-15/25-25/15-30/10 sec (160 sec total
intervention time).
Homodynamic measurements
The heart rate (HR) and arterial pressure were
physiographically monitored through the arterial catheter.
Subsequently, + mean delta pressure/delta time of the left
ventricle (+dP/dt) and −dP/dt were analyzed using the physiograph,
and the rate-pressure product (RPP) was calculated as the product
of the rate and the mean arterial pressure (MAP).
Measurement of infarct size
Infarct size was measured according to the method
described by Kerendi et al(20). After 24 h of reperfusion, the
ligature at the coronary occlusion site was permanently tied, and
Evans blue solution (1%, 3–5 ml) was injected into the aorta to
demarcate the left ventricular area at risk (AAR). The heart was
excised 10 min later and was maintained at −20°C for ~20 min. The
heart was then sliced into four sections (~2-mm thick) from the
base to the apex for staining with triphenyltetrazolium chloride
(TTC, 2%) at 37°C to measure the area of necrosis (AN). The AN, AAR
and the area of the left ventricle (LV) were determined by area
analysis using Image-Pro Plus software (version 4.1; Media
Cybernetics LP, Silver Spring, MD, USA). The infarct size was
expressed as a percentage of the AAR (AN/AAR), and the ischemic
area was calculated as AAR/LV.
Serum marker release
Serum levels of creatine kinase (CK) and the MB
isoenzyme of creatine kinase (CK-MB) were analyzed using an
automatic biochemistry analyzer.
Detection of apoptotic cells
Apoptotic cells were detected on transverse sections
of the left ventricle using a DNA fragmentation detection kit
(Roche Diagnostics GmbH, Mannheim, Germany) based on the terminal
deoxynucleotidyl transferase-mediated dUTP nick end-labeling
(TUNEL) method, according to the manufacturer’s instructions. The
results were quantified as the ‘apoptotic index’: the number of
positively stained apoptotic cardiocytes/total number of
cardiocytes counted × 100%.
Western blot analysis
Western blot analysis was performed as previously
described (21). Briefly, the left
ventricular myocardium was homogenized in lysis buffer. After
sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE), the proteins were transferred to nitrocellulose
membranes and incubated with antibodies against
phospho-extracellular signal- regulated kinase (p-ERK), phospho-p38
(p-p38), phospho-c-Jun N-terminal kinase (p-JNK), tumor necrosis
factor α (TNFα), caspase-8, Bcl-2, Bax, caspase-9 and β-actin (all
were mouse polyclonal antibodies diluted 1:200; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) for 6 h followed by
incubation with a horseradish peroxidase (HRP)-conjugated secondary
antibody. Antigen-antibody complexes were visualized using enhanced
chemiluminescence (ECL).
Cytosolic and mitochondrial fractions were isolated
as described by Li et al(21). The tissues were homogenized on ice
using a tight-fitting Dounce homogenizer (Kimble Glass Inc., USA).
The homogenates were centrifuged at 2,500 rpm for 10 min at 4°C.
The supernatants were then centrifuged at 14,000 rpm for 25 min at
4°C to obtain the cytosolic fractions (supernatants) and
mitochondrial fractions (pellets). Following SDS-PAGE, the proteins
were transferred to nitrocellulose membranes and incubated with
anti-cytochrome c and β-actin antibodies (mouse polyclonal
antibodies diluted 1:200; Santa Cruz Biotechnology, Inc.) for 6 h
followed by incubation with an HRP-conjugated secondary antibody.
Antigen-antibody complexes were visualized using ECL as described
above.
Statistical analysis
Values were expressed as the mean ± SEM. Data were
analyzed using the statistical software package SPSS 13.0 for
Windows. A one-way analysis of variance (ANOVA) was used, and the
Student’s t-test was employed to determine whether any significant
differences in individual parameters existed between groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Hemodynamic data
Hemodynamic data are shown in Table I. Twenty-four hours after
myocardial infarction, no significant differences in HR and MAP
were observed between groups. The rats in the GIR group exhibited a
significantly higher RPP value compared with the rats in the GDR
and ER groups (P<0.05). Moreover, the +dP/dt and −dP/dt levels
were significantly higher in the rats of the GIR group compared
with the rats in the GDR group (P<0.05).
 | Table IHemodynamic data obtained 24 h
following reperfusion (n=12). |
Table I
Hemodynamic data obtained 24 h
following reperfusion (n=12).
| Group | HR (beats/min) | MAP (mmHg) | RPP (1,000
mmHg•beats/min) | +dP/dt
(mmHg/sec) | −dP/dt
(mmHg/sec) |
|---|
| Sham | 418.00±65.93 | 119.33±11.21 | 49.35±0.74a,b |
1,019.09±318.65a,b |
752.31±231.05a,b |
| R/I | 396.17±62.21 | 105.95±31.68 | 41.97±1.98b,c |
526.37±123.99b,c |
445.44±79.58b,c |
| GDR | 423.21±50.00 | 108.00±14.32 | 45.70±0.72a |
721.45±89.97a |
546.23±100.12a |
| ER | 427.21±36.03 | 109.57±10.36 | 46.81±0.37a |
855.42±150.54a |
645.90±132.24a |
| GIR | 396.57±61.23 | 123.00±15.42 | 48.78±0.94a–c |
913.24±63.25a,b |
721.45±110.23a,b |
Serum markers of cardiac damage
The levels of CK and CK-MB release were
significantly decreased in the three postconditioning groups
(P<0.01 compared with the R/I group). The rats of the GIR group
were found to release significantly less CK and CK-MB compared with
the rats in the ER (CK, 251.00±45.16 vs. 388.56±75.01 μ/l,
P<0.05; CK-MB, 146.00±60.12 vs. 291.16±52.41 μ/l, P≤0.05) and
GDR groups (CK, 251.00±45.16 vs. 599.41±63.00 μ/l, P<0.05;
CK-MB, 146.00±60.12 vs. 406.76±90.01 μ/l, P<0.05).
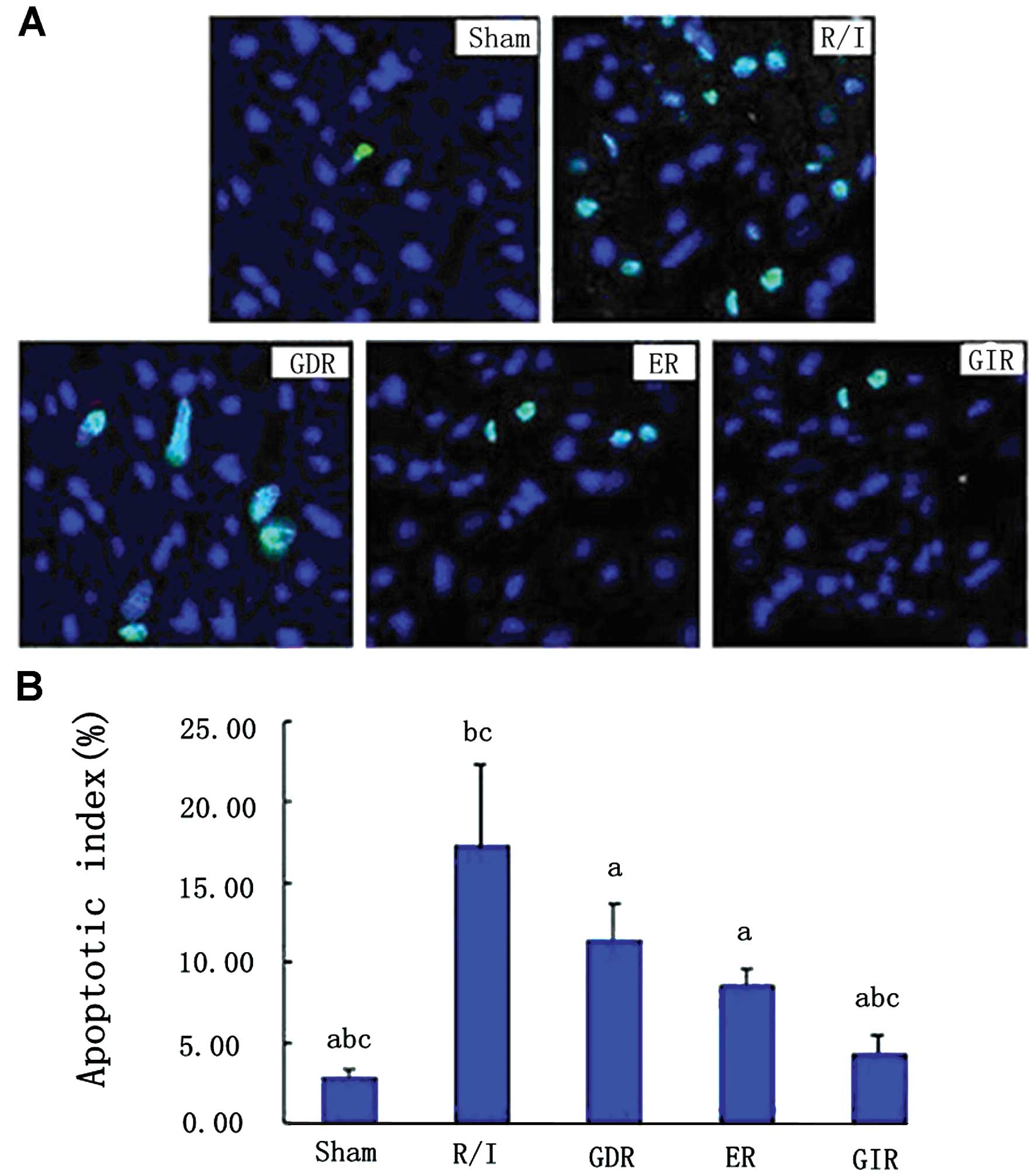
Cardiocyte apoptosis
As shown in Fig. 2,
R/I caused a significant increase in the number of TUNEL-positive
cells in the rats of this group (apoptotic index, P<0.01)
compared with that of the rats in the sham group. All three
postconditioning treatments significantly reduced the apoptotic
index in the rats of the respective groups compared with that in
the rats of the R/I group (P<0.01). Furthermore, the apoptotic
index in the GIR group was significantly lower compared with that
in the ER (4.32±1.16 vs. 8.58±1.12%, P<0.05) and GDR groups
(4.32±1.16 vs. 11.34±2.34%, P<0.05).
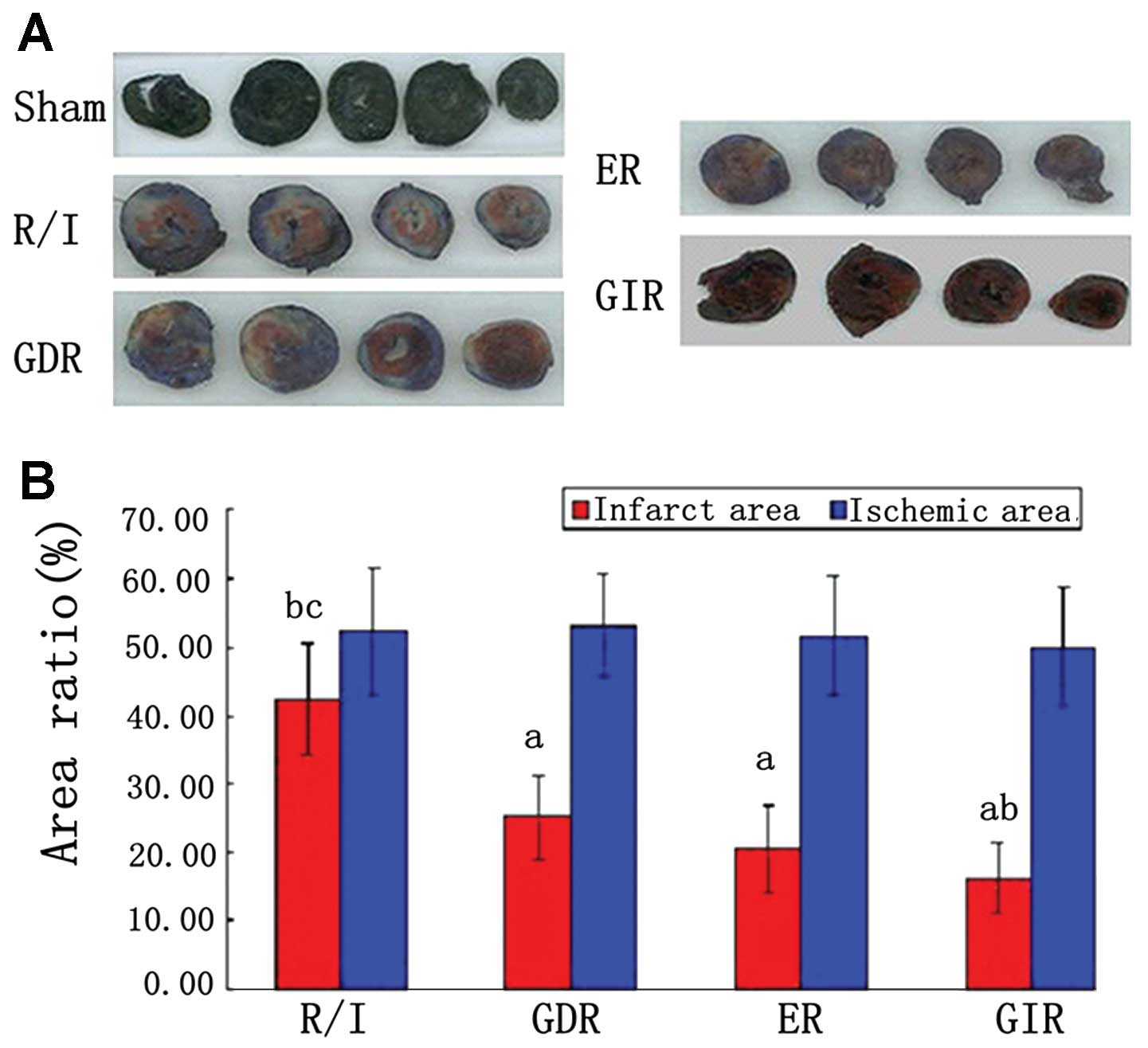
Infarct size
As shown in Fig. 3,
the AAR zone is stained red and white, while the AN zone is stained
white. The size of the ischemic area, expressed as a percentage of
the area of the left ventricle (AAR/LV), was similar among the
groups (51.01–53.55%). Infarct size, expressed as a percentage of
the area at risk (AN/AAR), was significantly reduced in all the
postconditioning groups compared with the R/I group (P<0.05).
Furthermore, the infarct size of the GIR group was significantly
smaller compared with that of the GDR (16.30±5.22 vs. 25.18±6.21%,
P<0.05) and ER groups (16.30±5.22 vs. 20.57±6.32%, P<0.05),
while the difference between the GIR and ER groups was not
statistically significant.
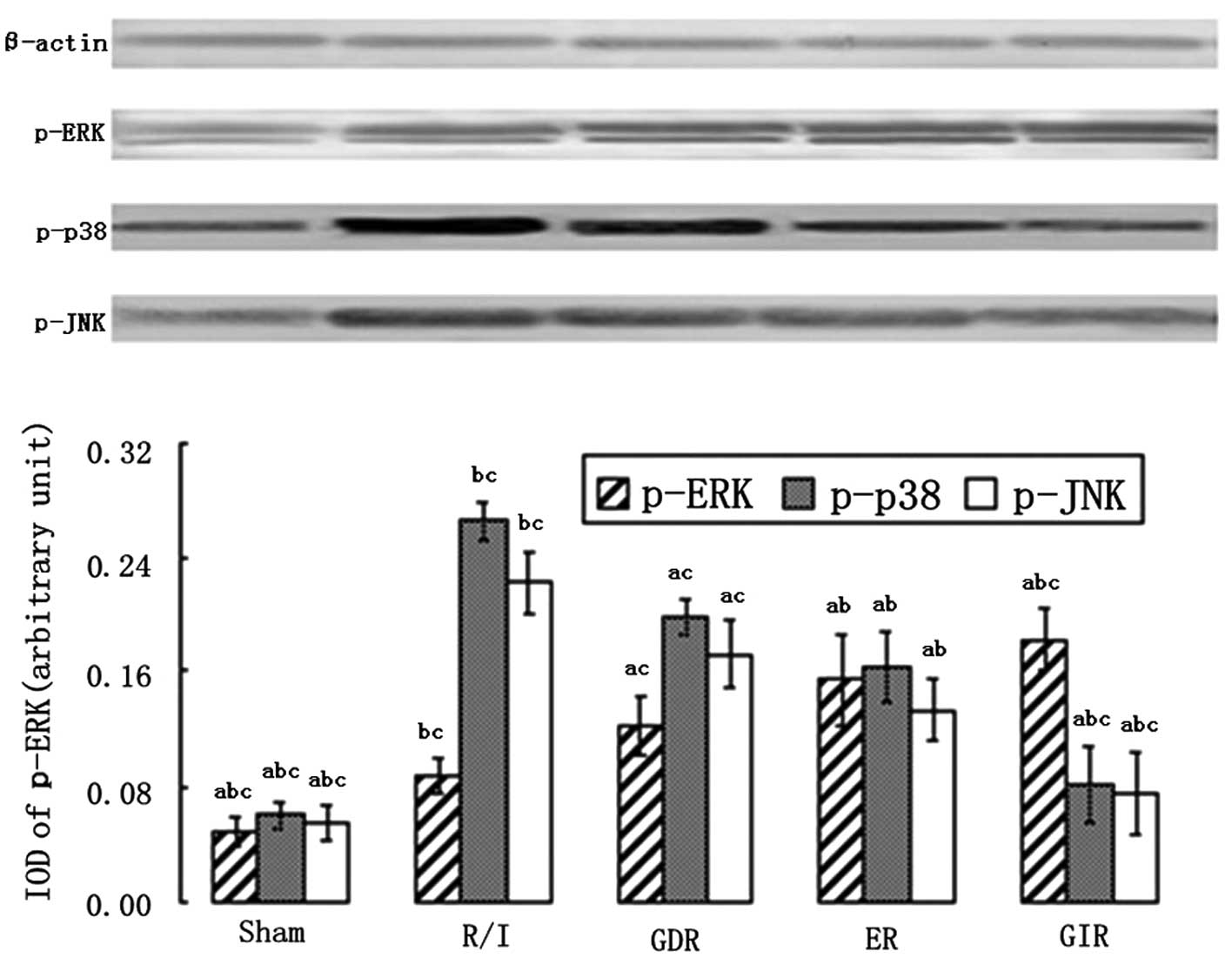
Expression of the mitogen-activated
protein kinases (MAPK) p-ERK1/2, p-p38 and p-JNK
The MAPK family is an important group of signaling
molecules that includes ERK, which inhibits apoptosis and necrosis,
and the apoptosis promoter p38/JNK. As shown in Fig. 4, a significant increase in the
expression of p-ERK and a marked decease in the expression of p-p38
and p-JNK were observed in all the postconditioning groups compared
with the R/I group (P<0.01). GIR treatment also enhanced the
expression of p-ERK to a greater extent compared with ER (1.82±0.22
vs. 1.54±0.32, P<0.05) and GDR treatments (1.82±0.22
vs.1.22±0.21, P<0.01). The rats in the GIR group also exhibited
a greater decrease in the levels of p-p38 and p-JNK compared with
the rats in the ER (p-p38, 0.82±0.26 vs. 1.63±0.24, P<0.05;
p-JNK, 0.76±0.28 vs. 1.33±0.21, P<0.05) and GDR groups (p-p38,
0.82±0.26 vs. 1.98±0.21, P<0.05; p-JNK, 0.76±0.28 vs. 1.72±0.24,
P<0.05).
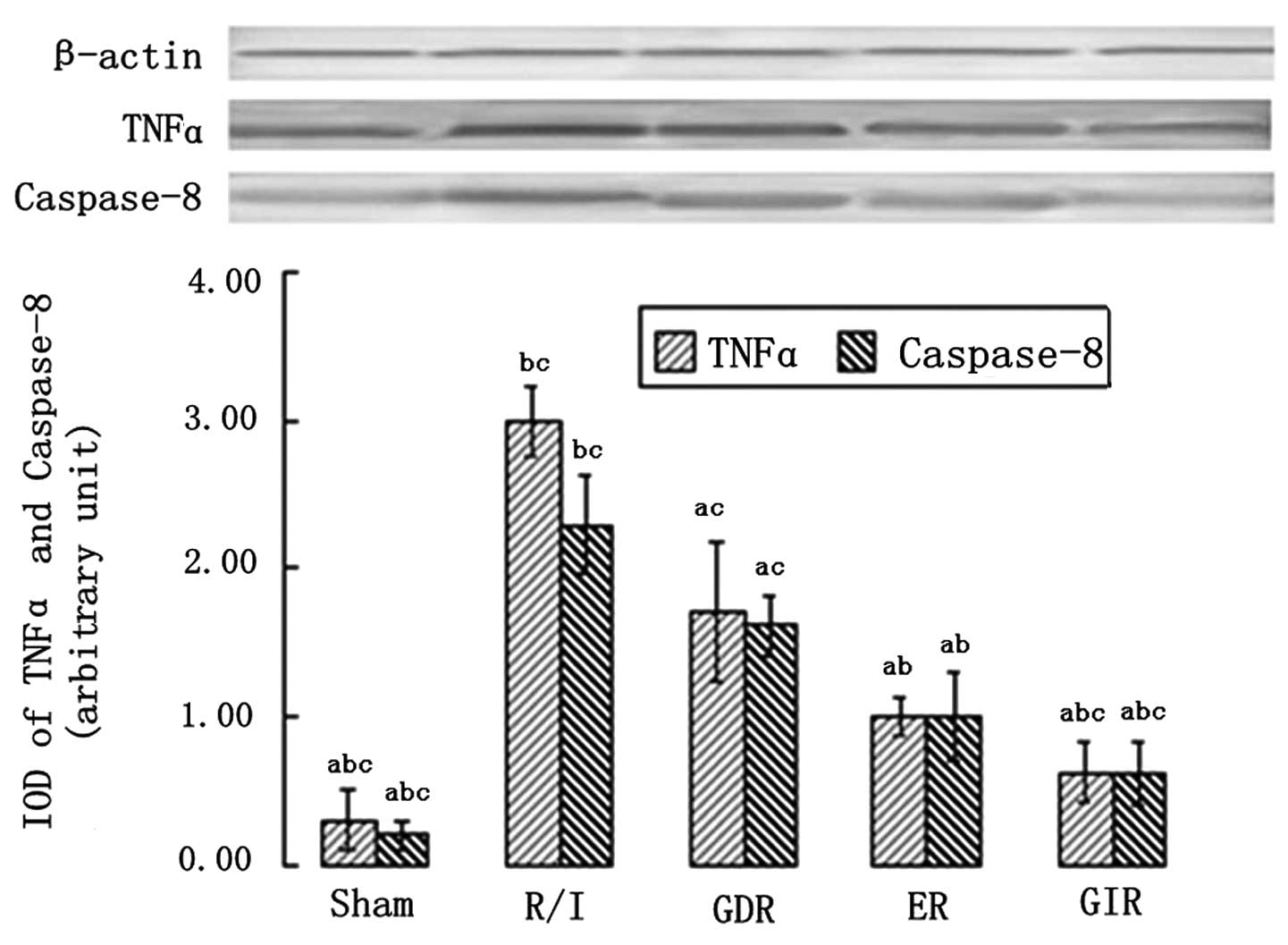
TNFα and caspase-8 expression
TNFα and caspase-8 are essential components of the
death receptor apoptotic pathway; thus, their expression was
measured in all the groups. As shown in Fig. 5, the rats in all the
postconditioning groups exhibited a significantly lower expression
of TNFα and caspase-8 compared with the rats of the R/I group
(P<0.01). Furthermore, the TNFα and caspase-8 expression levels
in the GIR group were significantly lower compared with those in
the ER (TNFα, 0.62±0.20 vs. 1.00±0.12, P<0.05; caspase-8,
0.61±0.21 vs. 1.00±0.21, P<0.05) and GDR groups (TNFα, 0.62±0.20
vs. 1.72±0.47, P<0.05; caspase-8, 0.86±0.21 vs. 1.62±0.21,
P<0.05).
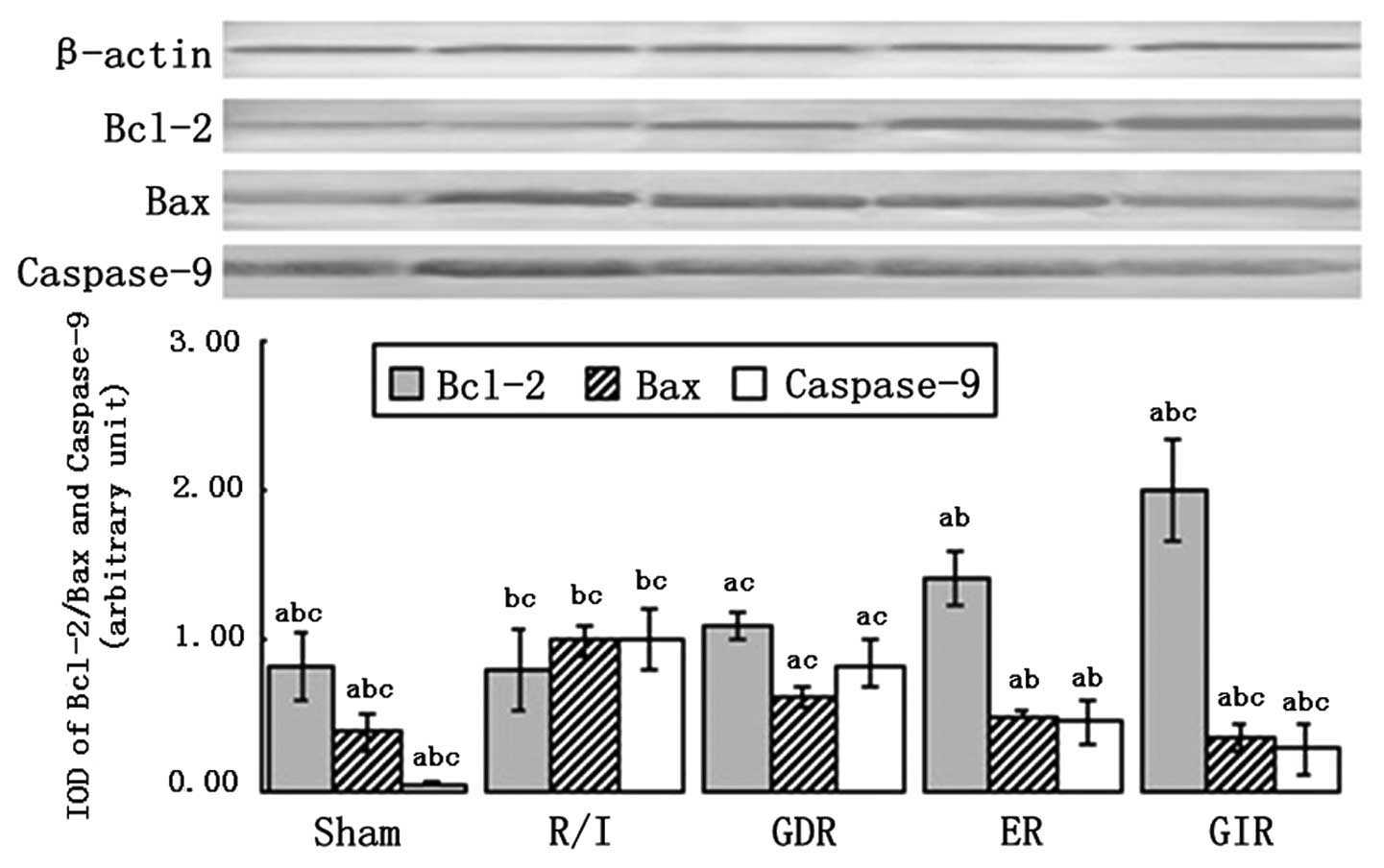
Bcl-2, Bax and caspase-9 expression
Bcl-2 and Bax are signaling molecules involved in
the mitochondrial pathway of apoptosis, which is associated with
the release of cytochrome c (Cyt-c) from the
mitochondrial matrix to the cytoplasm. As shown in Fig. 6, Bcl-2 was significantly
upregulated in the rats of all the postconditioning groups compared
with the rats of the R/I group (P<0.01). The expression of Bcl-2
in the GIR group was significantly increased compared with that in
the GDR (2.00±0.34 vs. 1.10±0.11, P<0.05) and ER groups
(2.00±0.34 vs. 1.40±0.18, P<0.05). Moreover, the expression of
Bax and caspase-9 was decreased in all the postconditioning groups
compared with the R/I group (P<0.01). A significantly lower
expression of Bax and caspase-9 was observed in the GIR group
compared with the GDR (Bax, 0.35±0.10 vs. 0.62±0.07, P<0.05;
caspase-9, 0.28±0.17 vs. 0.84±0.16, P<0.05) and ER groups (Bax,
0.35±0.10 vs. 0.50±0.02, P<0.05; caspase-9, 0.28±0.17 vs.
0.46±0.15, P<0.05).
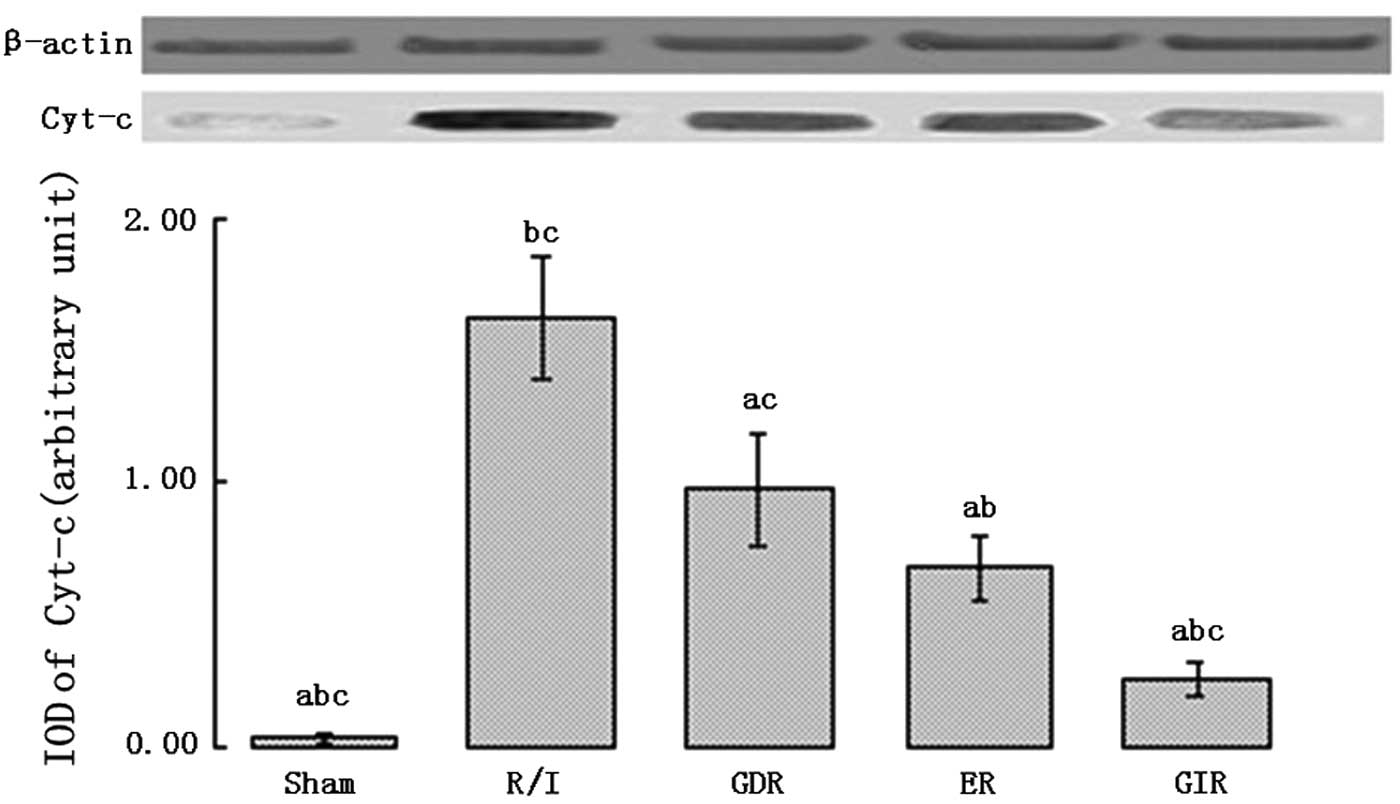
Expression of Cyt-c in the cytosol
Cyt-c is an important pro-apoptotic factor
that activates caspase-9. As shown in Fig. 7, the rats of all the
postconditioning groups exhibited a significantly lower
Cyt-c expression in the cytosol compared with the rats of
the R/I group (P<0.01). Furthermore, the cytosolic Cyt-c
level was lower in the GIR group compared with the ER (0.26±0.06
vs. 0.68±0.12, P<0.05) and GDR groups (0.26±0.06 vs. 0.97±0.21,
P<0.05).
Discussion
The present study showed that GIR provides better
cardioprotection compared with the remaining two postconditioning
algorithms examined. Apoptosis and serum marker release were
reduced to a greater extent in the GIR group compared with the ER
group, while p-ERK and Bcl-2 were expressed at higher levels and
TNFα, caspase-8, Bax, caspase-9 and Cyt-c were expressed at
lower levels. GIR treatment also provided better protection
compared with GDR treatment for all the variables measured in the
present study.
The improved cardioprotection provided by GIR
treatment compared with the remaining postconditioning algorithms
investigated in the present study could be attributed to one or
more of the following reasons. Firstly, results of previous studies
have shown that ERK1/2 is one of the reperfusion injury survival
kinases (RISK), which are components of an important
postconditioning pathway (14,22).
In the present study, ERK1/2 was phosphorylated to higher levels in
the GIR group compared with the ER and GDR groups. Myocardial
ischemia/reperfusion has been demonstrated to activate p38/JNK
MAPK, resulting in cardiac injury and cell death, most prominently
via apoptosis. According to a preliminary study on neonatal rat
cardiocytes by Sun et al(23), hypoxic postconditioning was shown
to inhibit the expression of p38/JNK MAPK, reducing the expression
of TNFα and Bax. Conversely, anisomycin, an activator of p38/JNK
MAPK, was shown to offset this protection. In the present study,
the level of phosphorylation of p38/JNK in the GIR group was lower
compared with the R/I, ER and GDR groups, which was consistent with
the results mentioned above.
Apoptosis is known to be an important component of
reperfusion injury. The death receptor and mitochondrial pathways
are the main pathways leading to cell apoptosis. The death receptor
pathway is triggered by death receptors located on the cell
membrane, such as TNFα receptor-1 and the Fas receptor. In the
present study, the expression of TNFα and caspase-8 was
significantly decreased in the GIR group compared with the ER
group. The mitochondrial pathway is also important in apoptosis,
and Bcl-2 and Bax are key factors that regulate this pathway
(24–26). In the present study, the gradual
increase in reperfusion time during postconditioning increased the
expression of Bcl-2 and reduced the expression of Bax. These
changes could lead to the opening of the mitochondrial permeability
transition pore (mPTP), which is associated with apoptotic cell
death since it controls the release of many pro-apoptotic factors,
such as Cyt-c, in the cytoplasm (27–30).
These pro-apoptotic factors could then activate many proenzymes,
which could initiate the apoptotic cascade and trigger the release
of molecules such as caspase-9 and -3.
The results of the present study indicated that
there are some differences in the expression of downstream
mediators among the GIR, ER and GDR postconditioning algorithms.
Consequently, the underlying mechanisms of action of these
algorithms that convert different mechanical manipulations into
various biochemical signals should be subsequently investigated.
Although this issue was not addressed in the present study, the
gradual change of reperfusion time during postconditioning was
demonstrated to be important. Postconditioning has been previously
suggested to be a logical extension of gradual reperfusion, which
is known to be cardioprotective for reperfusion injury (31–33).
When a sudden change leads to an injury, a gradual change could
attenuate the injury by alleviating the process. When myocardial
cells remain viable following severe ischemia, a ‘warm-up’ period
is required to reactivate their metabolic activity, and
postconditioning leads to a gradual change in metabolic state.
Consequently, the postconditioning process was modified accordingly
to mimic gradual reperfusion. Among all the
ischemia-postconditioning reperfusion algorithms examined in the
present study, GIR was the postconditioning algorithm most similar
to gradual reperfusion. The gradual change used in this algorithm
resulted in improved cardioprotection compared with standard
postconditioning, as described above.
In the present study, no significant difference in
infarct size was observed between GIR and ER groups, possibly due
to the small size. This finding could also be attributed to the
fact that improvement in infarct size is difficult to be observed
in the rat model. However, we could still conclude that GIR
provides improved cardioprotection compared with ER treatment. Two
main findings support this conclusion: i) GIR treatment led to a
significant reduction in apoptosis and necrosis and markedly
improved left ventricle function compared with ER treatment; and
ii) GIR treatment improved all the variables examined compared with
GDR treatment. These results clearly indicate that the gradual
increase in the reperfusion time during postconditioning is
associated with improved cardioprotection. Although the present
study provides encouraging results, there are several limitations.
These include the small sample size and the lack of comparison
between GIR and gradual reperfusion.
In conclusion, the present study has demonstrated
that the duration of the brief reperfusion and reocclusion steps in
postconditioning algorithms is important, with the gradually
increased reperfusion algorithm improving cardioprotection.
However, future studies are needed to determine the underlying
mechanisms of this phenomenon.
Acknowledgements
The authors are grateful to Sheng Sun, Lirong Cai,
Feifei Xu, Xiaoreng Wang and Zhenying Zhang for their technical
assistance. This study was supported by grants from the National
Science Foundation of China (no. 30740080) and the Dean Fund of the
General Hospital of Jinan Military Command (no. 2011Q08).
References
|
1
|
Wu JM, Wang ZR, Hsieh TC, Bruder JL, Zou
JG and Huang YZ: Mechanism of cardioprotection by resveratrol, a
phenolic antioxidant present in red wine (Review). Int J Mol Med.
8:3–17. 2001.PubMed/NCBI
|
|
2
|
Braunwald E and Kloner RA: Myocardial
reperfusion: a double-edged sword? J Clin Invest. 76:1713–1719.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tomatsuri N, Yoshida N, Takagi T, et al:
Edaravone, a newly developed radical scavenger, protects against
ischemia-reperfusion injury of the small intestine in rats. Int J
Mol Med. 13:105–109. 2004.PubMed/NCBI
|
|
4
|
Zhao ZQ, Corvera JS, Halkos ME, et al:
Inhibition of myocardial injury by ischemic postconditioning during
reperfusion: comparison with ischemic preconditioning. Am J Physiol
Heart Circ Physiol. 285:H579–H588. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sanada S, Komuro I and Kitakaze M:
Pathophysiology of myocardial reperfusion injury: preconditioning,
postconditioning, and translational aspects of protective measures.
Am J Physiol Heart Circ Physiol. 301:H1723–H1741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu X, Chen H, Zhan B, et al: Attenuation
of reperfusion injury by renal ischemic postconditioning: the role
of NO. Biochem Biophys Res Commun. 359:628–634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang WH, Lu FH, Zhao YJ, et al:
Post-conditioning protects rat cardiomyocytes via
PKCepsilon-mediated calcium-sensing receptors. Biochem Biophys Res
Commun. 361:659–664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thuny F, Lairez O, Roubille F, et al:
Post-conditioning reduces infarct size and edema in patients with
ST-segment elevation myocardial infarction. J Am Coll Cardiol.
59:2175–2181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Penna C, Mancardi D, Raimondo S, Geuna S
and Pagliaro P: The paradigm of postconditioning to protect the
heart. J Cell Mol Med. 12:435–458. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Skyschally A, van Caster P, Iliodromitis
EK, Schulz R, Kremastinos DT and Heusch G: Ischemic
postconditioning: experimental models and protocol algorithms.
Basic Res Cardiol. 104:469–483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Iliodromitis EK, Downey JM, Heusch G and
Kremastinos DT: What is the optimal postconditioning algorithm? J
Cardiovasc Pharmacol Ther. 14:269–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cai M, Li Y, Xu Y, et al: Endothelial NOS
activity and myocardial oxygen metabolism define the salvageable
ischemic time window for ischemic postconditioning. Am J Physiol
Heart Circ Physiol. 300:H1069–H1077. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kin H, Zhao ZQ, Sun HY, et al:
Postconditioning attenuates myocardial ischemia-reperfusion injury
by inhibiting events in the early minutes of reperfusion.
Cardiovasc Res. 62:74–85. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang XM, Proctor JB, Cui L, Krieg T,
Downey JM and Cohen MV: Multiple, brief coronary occlusions during
early reperfusion protect rabbit hearts by targeting cell signaling
pathways. J Am Coll Cardiol. 44:1103–1110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vinten-Johansen J, Zhao ZQ, Zatta AJ, Kin
H, Halkos ME and Kerendi F: Postconditioning - A new link in
nature’s armor against myocardial ischemia-reperfusion injury.
Basic Res Cardiol. 100:295–310. 2005.
|
|
16
|
Argaud L, Gateau-Roesch O, Raisky O,
Loufouat J, Robert D and Ovize M: Postconditioning inhibits
mitochondrial permeability transition. Circulation. 111:194–197.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kin H, Zatta AJ, Lofye MT, et al:
Postconditioning reduces infarct size via adenosine receptor
activation by endogenous adenosine. Cardiovasc Res. 67:124–133.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Penna C, Cappello S, Mancardi D, et al:
Post-conditioning reduces infarct size in the isolated rat heart:
role of coronary flow and pressure and the nitric oxide/cGMP
pathway. Basic Res Cardiol. 101:168–179. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang JY, Shen J, Gao Q, et al: Ischemic
postconditioning protects against global cerebral
ischemia/reperfusion-induced injury in rats. Stroke. 39:983–990.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kerendi F, Kin H, Halkos ME, et al: Remote
postconditioning. Brief renal ischemia and reperfusion applied
before coronary artery reperfusion reduces myocardial infarct size
via endogenous activation of adenosine receptors. Basic Res
Cardiol. 100:404–412. 2005.
|
|
21
|
Li Y, Ge X and Liu X: The cardioprotective
effect of postconditioning is mediated by ARC through inhibiting
mitochondrial apoptotic pathway. Apoptosis. 14:164–172. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sumi S, Kobayashi H, Yasuda S, et al:
Postconditioning effect of granulocyte colony-stimulating factor is
mediated through activation of risk pathway and opening of the
mitochondrial KATP channels. Am J Physiol Heart Circ Physiol.
299:H1174–H1182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun HY, Wang NP, Halkos M, et al:
Postconditioning attenuates cardiomyocyte apoptosis via inhibition
of JNK and p38 mitogen-activated protein kinase signaling pathways.
Apoptosis. 11:1583–1593. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gomez L, Thibault H, Gharib A, et al:
Inhibition of mitochondrial permeability transition improves
functional recovery and reduces mortality following acute
myocardial infarction in mice. Am J Physiol Heart Circ Physiol.
293:H1654–H1661. 2007. View Article : Google Scholar
|
|
25
|
Cao J, Zhu T, Lu L, et al: Estrogen
induces cardioprotection in male C57BL/6J mice after acute
myocardial infarction via decreased activity of matrix
metalloproteinase-9 and increased Akt-Bcl-2 anti-apoptotic
signaling. Int J Mol Med. 28:231–237. 2011.
|
|
26
|
Mao H, Gu H, Qu X, et al: Involvement of
the mitochondrial pathway and Bim/Bcl-2 balance in
dihydroartemisinin-induced apoptosis in human breast cancer in
vitro. Int J Mol Med. 31:213–218. 2013.PubMed/NCBI
|
|
27
|
Hausenloy DJ, Ong SB and Yellon DM: The
mitochondrial permeability transition pore as a target for
preconditioning and postconditioning. Basic Res Cardiol.
104:189–202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Paillard M, Gomez L, Augeul L, Loufouat J,
Lesnefsky EJ and Ovize M: Postconditioning inhibits mPTP opening
independent of oxidative phosphorylation and membrane potential. J
Mol Cell Cardiol. 46:902–909. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weiss JN, Korge P, Honda HM and Ping P:
Role of the mitochondrial permeability transition in myocardial
disease. Circ Res. 93:292–301. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park SS, Zhao H, Mueller RA and Xu Z:
Bradykinin prevents reperfusion injury by targeting mitochondrial
permeability transition pore through glycogen synthase kinase
3beta. J Mol Cell Cardiol. 40:708–716. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sato H, Jordan JE, Zhao ZQ, Sarvotham SS
and Vinten-Johansen J: Gradual reperfusion reduces infarct size and
endothelial injury but augments neutrophil accumulation. Ann Thorac
Surg. 64:1099–1107. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Unal S, Ozmen S, DemIr Y, et al: The
effect of gradually increased blood flow on ischemia-reperfusion
injury. Ann Plast Surg. 47:412–416. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shi E, Jiang X, Kazui T, et al: Controlled
low-pressure perfusion at the beginning of reperfusion attenuates
neurologic injury after spinal cord ischemia. J Thorac Cardiovasc
Surg. 133:942–948. 2007. View Article : Google Scholar : PubMed/NCBI
|