Introduction
Due to the limitations in the efficacy of
conventional cancer therapies, cholangiocarcinoma usually has a
poor prognosis (1). To reduce the
rate of cholangiocarcinoma-associated mortality, it is essential to
develop therapeutics that target the signaling pathways involved in
this carcinoma (2). The
transforming growth factor β (TGF-β) signaling pathway has become
an attractive target due to its pleiotropic physiological
properties in the regulation of cell proliferation,
differentiation, migration, cell survival, angiogenesis and
immunosurveillance (3). In
response to TGF-β, receptor activation leads to the phosphorylation
of cytoplasmic Smad2 and Smad3, which associates with Smad4 and
translocates into the nucleus, where it regulates the transcription
of TGF-β target genes (4).
Furthermore, TGF-β induces non-Smad pathways, including
phosphoinositide 3-kinases (PI3K), c-Jun N-terminal kinases (JNK),
Erk, p38 mitogen activated protein kinases (MAPK) and Rho (5).
TGF-β exhibits a dual role as either a tumor
promoter or a tumor suppressor in carcinogenesis, the role is
largely dependent on signals from the tumor microenvironment
(6). TGF-β responses result from
Smad cascade activation and also cell-type specific interactions of
Smad signaling with a variety of other intracellular signaling,
which may or may not have been triggered by TGF-β (7).
Although crosstalk between Smad and non-Smad
pathways exists, the mechanism by which they regulate each other is
unclear. The TGF-β/Smad pathway may be regulated by the MAPK
pathways, including the JNK pathway (8), whose inhibitor, SP600125, negatively
regulates cell survival (9,10).
However, little is known in regard to how SP600125 affects
TGF-β-induced apoptosis of human cholangiocarcinoma cells. In the
present study, results show that SP600125 enhances TGF-β-induced
cell apoptosis of RBE cells in a Smad-dependent manner. SP600125 is
hypothesized to be an ideal therapeutic candidate for treating
human cholangiocarcinomas that express JNK and Smad4.
Materials and methods
Cell cultures
The RBE human cholangiocarcinoma cell line was
obtained from the Riken BioResource Center (Ibaraki, Japan). The
PT67 packaging cell line was obtained from the American Type
Culture Collection (Manassas, VA, USA). RBE cells were maintained
in RPMI-1640 medium and PT67 cells were cultured in Dulbecco’s
modified Eagle’s medium supplemented with 10% fetal bovine serum in
a humidified atmosphere containing 5% CO2 at 37°C.
Reagents and antibodies
TGF-β1 was purchased from R&D Systems
(Minneapolis, MN, USA). SP600125, SB203580, Wortmannin, PD98059,
U0126 and SB431542 were obtained from Tocris Bioscience (Bristol,
UK). V-ZAD-fmk was obtained from the Beyotime Biotech (Jiangsu,
China). Antibodies against Smad2/3, p38, Akt and hlyceraldehyde
3-phosphate dehydrogenase were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Antibodies against p21,
Bcl-2, Bim and Smad4 were purchased from Abcam (Cambridge, MA,
USA). Antibodies against phospho-c-Jun N-terminal kinase (p-JNK),
p-Akt, p-Erk, p-p38, p-Smad2 (Ser465/467), p-Smad3 (Ser423/425),
p-c-Jun, JNK, Erk, c-Jun, caspase-9, cleaved caspase-9, caspase-8,
cleaved caspase-8, caspase-7, cleaved caspase-7, caspase-3, cleaved
caspase-3, poly ADP ribose polymerase (PARP) and cleaved PARP were
purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA).
Western blot analysis
Western blot analysis was performed as follows:
Whole-cell extracts were prepared in lysis buffer (50 mM Tris-HCl,
10% glycerol, 1% Triton X-100, 150 mM NaCl, 100 mM NaF, 5 mM EDTA,
2 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate and 1
mg/ml leupeptin; pH 7.5) and centrifuged (Sigma-Aldrich, St. Louis,
MO, USA) at 12,000 × g for 15 min. Protein concentrations were
measured using a bicinchoninic acid assay kit (Beyotime Biotech).
Total cell proteins were subjected to electrophoresis on an 8–15%
polyacrylamide gel, transferred to a nitrocellulose membrane and
probed with primary antibodies overnight at 4°C. Immunocomplexes
were incubated with the appropriate horseradish
peroxidase-conjugated secondary antibody and then detected using an
enhanced chemiluminescent kit (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA).
Generation of stable knockdown RBE
cells
To generate recombinant retroviruses, a
pRetroSuper-puro Smad4 (Addgene, Cambridge, MA, USA) was
transfected into PT67 cells as described previously by He et
al(11). A pRetroSuper-puro
vector was transfected as a control. Retroviral supernatants were
collected, filtered through a 0.45-μm filter and used for infection
of RBE cells, which were then selected with puromycin (1 μg/ml) for
two weeks. Ultimately, RBE Smad4 negative control cells (vector
control cells) and RBE Smad4 knockdown cells (shSmad4 cells) cells
were obtained.
Transcriptional response assay
Cells were seeded into 12-well plates and
transiently co-transfected with p3TP-Lux, (which encodes firefly
luciferase) and pRL-TK-luc (which encodes Renilla
luciferase). Following 48 h transfection, the cells were treated
with the indicated reagents for an additional 24 h and the two
luciferase activities were measured using a dual-luciferase
reporter assay system (Promega Corporation, Madison, WI, USA).
Relative luciferase activity was normalized with Renilla
luciferase activity.
Identification of apoptosis by propidium
iodide (PI)-Annexin V staining
Cell apoptosis analysis was performed using an
Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit
(KeyGen Biotech, Nanjing, China), according to the manufacturer’s
instructions. Briefly, 5×105 cells were collected by
centrifugation and resuspended in 500 μl binding buffer.
Subsequently, 5 μl Annexin V-FITC and 5 μl PI were added. Following
incubation for 10 min in the dark at room temperature, the cells
were analyzed using a BD fluorescence-activated cell sorter Aria
flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Statistical analysis
Data are presented as the mean ± SD. Differences
were analyzed by Student’s t-test or one-way analysis of variance.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Exogenous TGF-β1 activates the Smad and
non-Smad pathways in RBE cells
To determine whether Smad signaling is inducible by
exogenous TGF-β1 in RBE cells, Smad2 and Smad3 phosphorylation and
the expression of p21Waf1/Cip1 were examined by western
blot analysis. The antibodies against phosphorylated Smad2 and
Smad3 used in this study specifically recognized the C-terminal
region (Ser465/467 and Ser423/425). TGF-β1 (1 ng/ml) was observed
to induce phosphorylation of Smad2 and Smad3 and the expression of
p21Waf1/Cip1 in a time-dependent manner (Fig. 1A), indicating the presence of an
intact TGF-β/Smad signaling pathway in RBE cells. In addition,
non-Smad pathways were activated by TGF-β1 (Fig. 1B). There was considerable basal
phosphorylation of Akt, c-Jun, Erk and p38MAPK, which transiently
increased and peaked at 3 h following TGF-β1 stimulation,
suggesting that Akt and MAPK activation in RBE cells is not
specific to TGF-β signaling and may result from other stimuli.
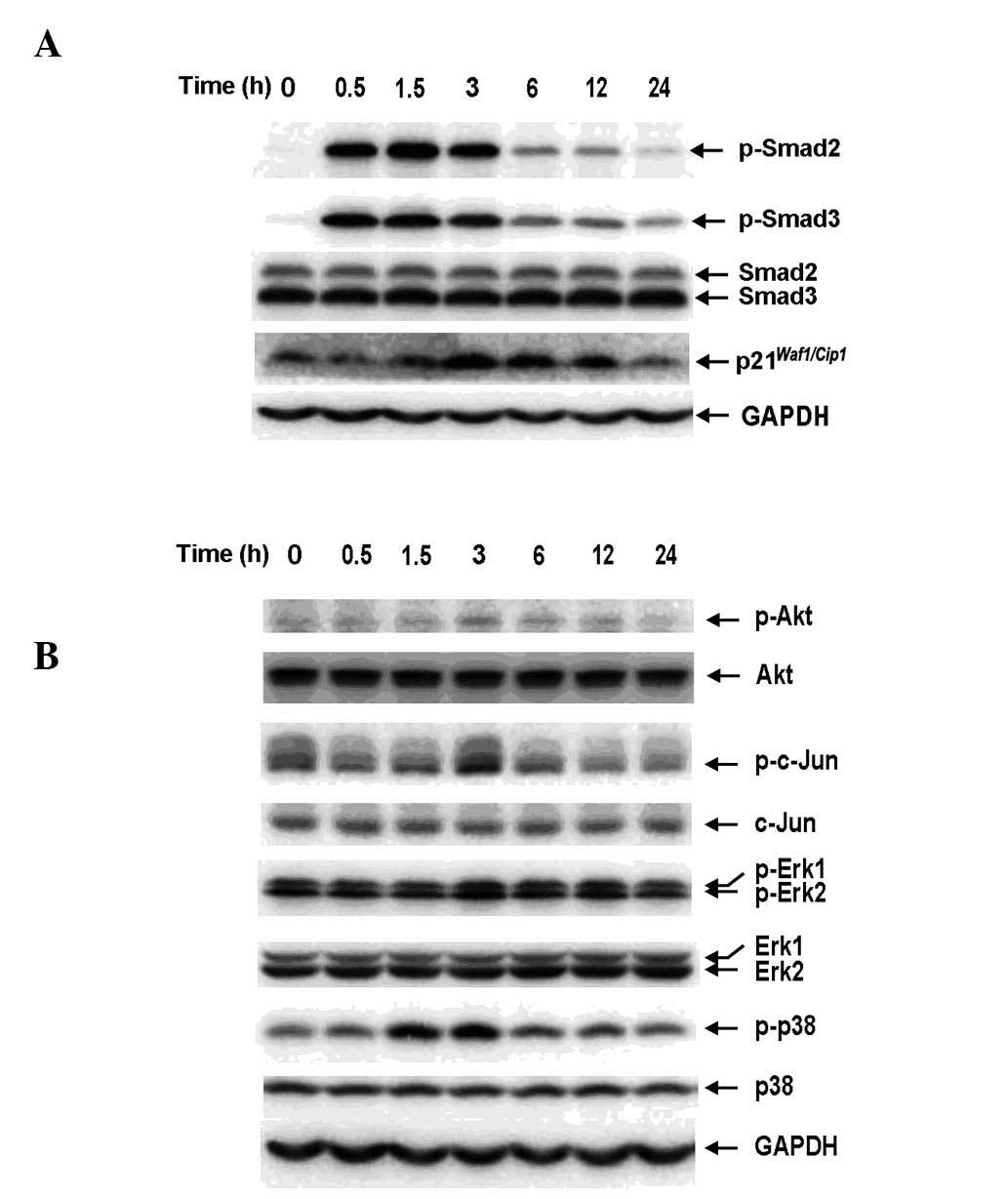 | Figure 1Exogenous TGF-β1 activates the Smad
and non-Smad pathways in RBE cells in a time-dependent manner. RBE
cells were treated with TGF-β1 (1 ng/ml) for the indicated times.
Cell lysates were analyzed by western blotting with antibodies
against (A) phospho-Smad2, Smad2, phospho-Smad3, Smad3,
p21Waf1/Cip1 and (B) phospho-Akt, Akt, phospho-c-Jun,
c-Jun, phospho-Erk1/2, Erk1/2, phospho-p38 and p38. Each experiment
was repeated three times. TGF-β1, transforming growth factor-β1;
GAPDH, glyceraldehyde 3-phosphate dehydrogenase. |
The JNK inhibitor SP600125 increases
TGF-β1-induced phosphorylation of Smad2 and Smad3 and enhances
TGF-β/Smad signaling
As TGF-β1 enhances non-Smad pathway signaling in RBE
cells, it is necessary to investigate whether the non-Smad pathway
affects the Smad pathway. Specific inhibitors of Akt, JNK, Erk and
p38MAPK were applied to RBE cells for 3 h at a concentration of 10
μM, with or without TGF-β1. Western blot analysis showed that only
the JNK inhibitor SP600125 enhanced TGF-β1-induced phosphorylation
of Smad2 and Smad3. However, Wortmannin and PD98059 were also
capable of increasing TGF-β1-induced phosphorylation of Smad2
(Fig. 2A).
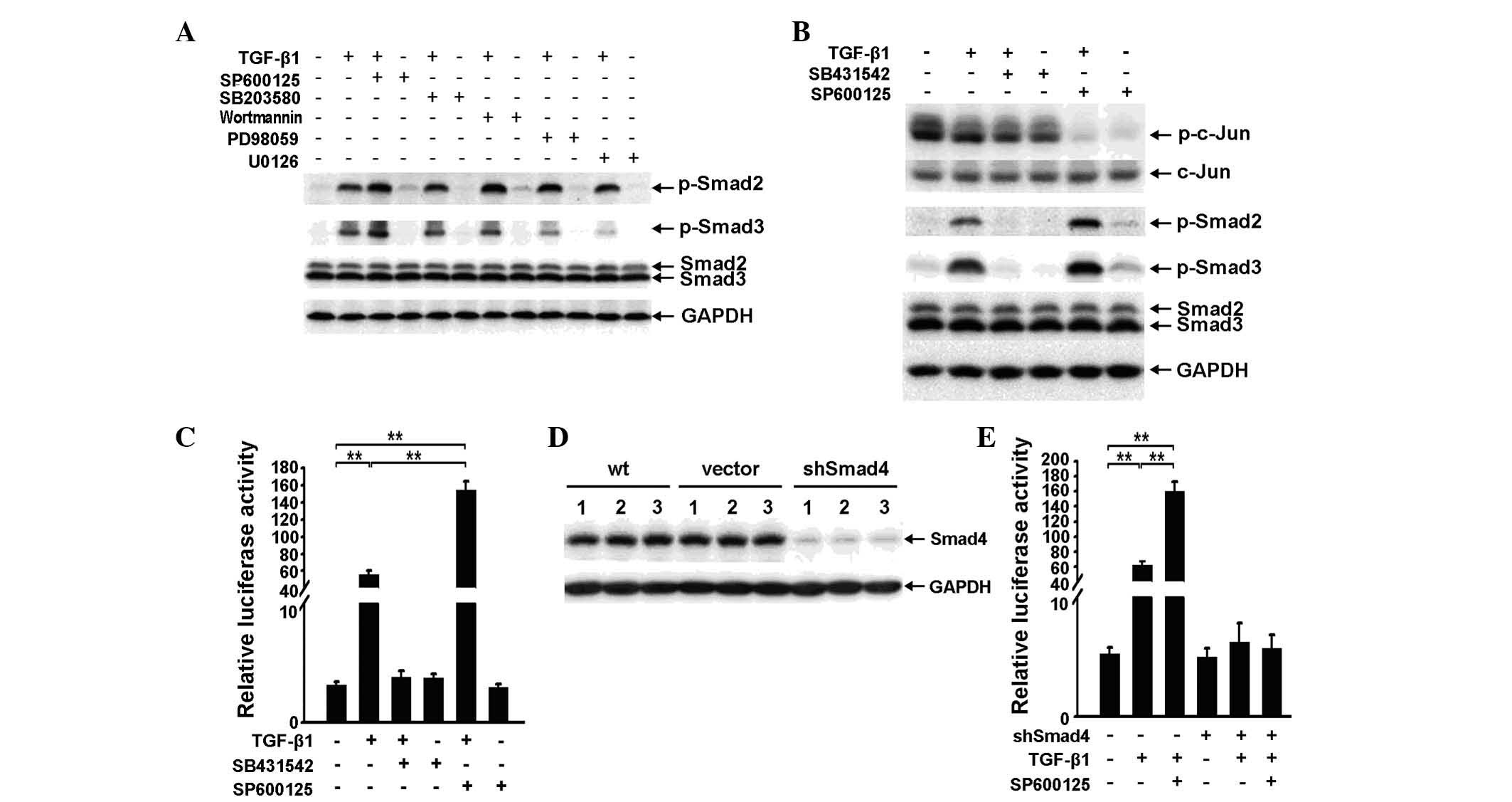 | Figure 2SP600125 increases transforming growth
factor (TGF)-β1-induced phosphorylation of Smad2 and Smad3 and
enhances TGF-β/Smad signaling. (A). Phospho-Smad2, phospho-Smad3
and Smad2/3 expression was examined by western blot analysis in RBE
cells treated with 10 μM SP600125, SB203580, Wortmannin, PD98059 or
U0126 for 3 h, with or without TGF-β1 (1 ng/ml). (B) Western
blotting was used to determine the expression of phospho-c-Jun,
c-Jun, phospho-Smad2, phospho-Smad3 and Smad2/3 in RBE cells
treated with TGF-β1 (1 ng/ml), SB431542 (10 μM), SP600125 (10 μM)
or a combination of any two for 6 h. (C) A luciferase reporter
assay. p3TP-luc luciferase activity was analyzed in RBE cells
treated with TGF-β1 (1 ng/ml), SB431542 (10 μM), SP600125 (10 μM)
or a combination of any two. Data are shown as the mean ± SD for
triplicate measurements. **P<0.01, vs. control. (D)
Smad4 expression in three independent samples of wild-type, vector
control and shSmad4 cells were examined by western blot analysis.
(E) A luciferase reporter assay. p3TP-luc luciferase activity was
analyzed in RBE vector control and shSmad4 cells treated with
TGF-β1 (1 ng/ml) and/or SP600125 (10 μM). Data are shown as the
mean ± SD for triplicate measurements. **P<0.01, vs.
control. TGF-β1, transforming growth facto-β1; GAPDH,
glyceraldehyde 3-phosphate dehydrogenase; wt, wild type. |
As shown in Fig.
1B, increased phosphorylation of MAPKs induced by TGF-β1 was
most evident at 3 h and dissipated by 6 h. The effect of SP600125
on TGF-β1-induced phosphorylation of Smad2 and Smad3 at 6 h
following TGF-β1 stimulation and obtained a similar result to that
observed at 3 h (Fig. 2A and B).
This result suggests that the basal activity of the JNK pathway,
rather than the TGF-β1-induced activity, antagonizes TGF-β1-induced
phosphorylation of Smad2 and Smad3.
In the absence of TGF-β1, SP600125 only marginally
enhanced the phosphorylation of Smad2 and Smad3 compared with the
control. However, the combined application of SP600125 and TGF-β1
significantly increased TGF-β1-induced phosphorylation of Smad2 and
Smad3 (Fig. 2B). These results
suggest that the effect of SP600125 on the Smad pathway may be
dependent upon the activity of the Smad pathway.
To determine whether SP600125 affects downstream
signaling, an additional application of SP600125 was observed to
strengthen TGF-β1-induced p3TP-Lux reporter activity (Fig. 2C). In addition, to elucidate
whether the effect of SP600125 on TGF-β signaling is dependent on
the canonical Smad pathway, a Smad4 knockdown RBE cell line
(shSmad4 cells) was developed (Fig.
2D), which resulted in the reduced effect of SP600125 on
TGF-β1-induced reporter activity (Fig.
2E).
Exogenous TGF-β1 induces apoptosis of RBE
cells by activating the caspase cascade through the Smad
pathway
As shown in Fig.
3A–D, exogenous TGF-β1 induces apoptosis of RBE cells in a
dose- and a time-dependent manner. The observed apoptosis was
prominent in cells treated with 1 ng/ml TGF-β1 for 36 h. Specific
knockdown of Smad4 expression reduced TGF-β1-induced apoptosis,
suggesting that apoptosis occurs through a Smad-dependent pathway
(Fig. 3E and F). To confirm
whether caspase activation occurs during TGF-β1-induced apoptosis,
the effect of the pan-caspase inhibitor, V-ZAD-fmk, on the
induction of apoptosis was investigated. As expected, 40, 80 and
160 μM V-ZAD-fmk completely inhibited the TGF-β1-induced apoptosis
of RBE cells (Fig. 3G and H). This
result suggests that caspase activation is required for
TGF-β1-induced apoptosis. Accordingly, western blot analysis showed
that the activation of caspase-9, -8, -7 and -3, and PARP was
prominent at 36 h, the time at which apoptosis of the RBE cells was
observed (Fig. 3I).
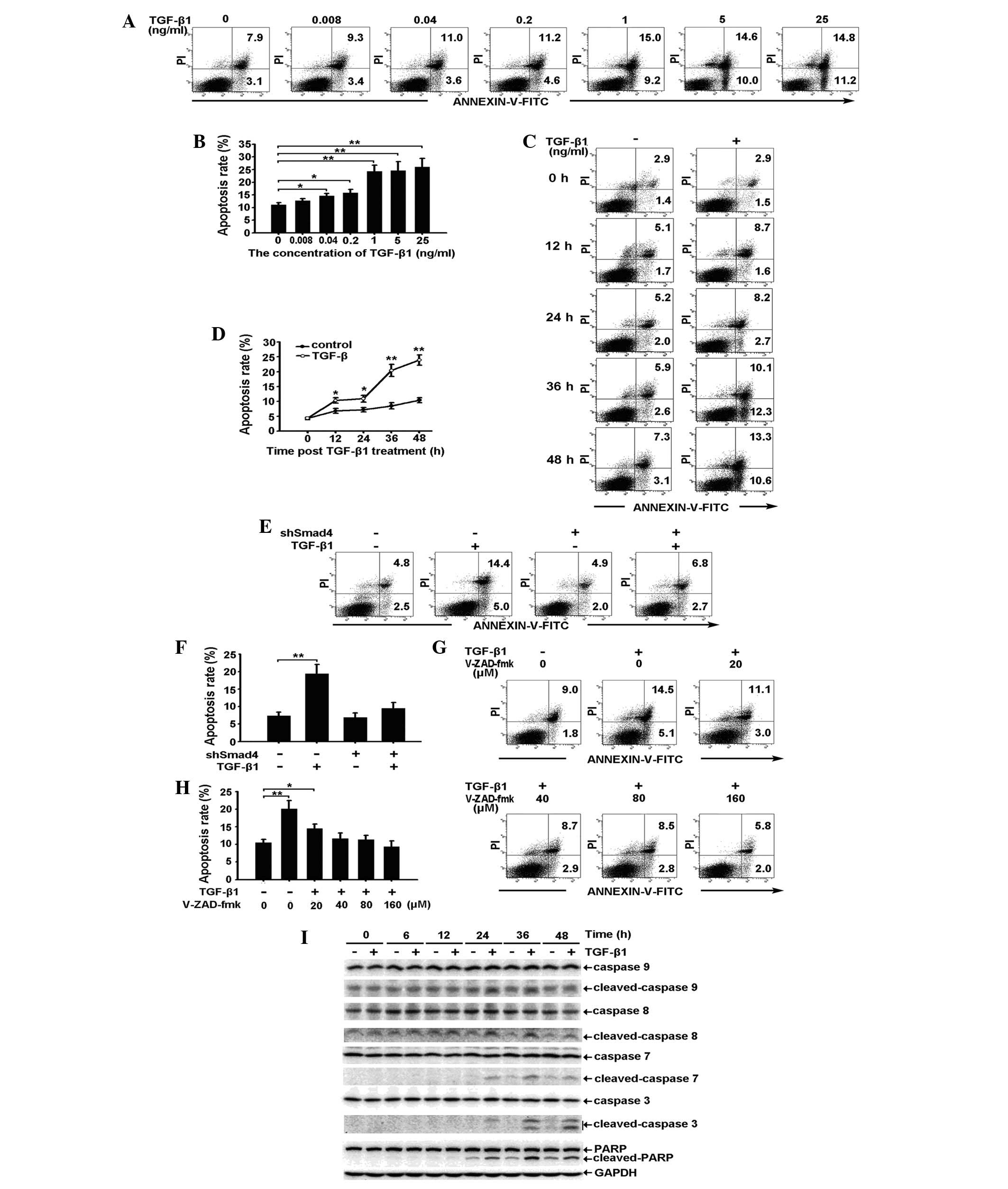 | Figure 3Exogenous TGF-β1 induces apoptosis of
RBE cells by activating the caspase cascade through the Smad
pathway. (A and B) RBE cells treated with various concentrations of
TGF-β1 for 36 h were stained with Annexin V/PI and analyzed by flow
cytometry. (A) The early apoptotic rate (lower right) and the late
apoptotic rate (upper right) are shown. (B) Total apoptotic rates
(the sum of the early and late apoptotic rate) are shown as the
mean ± SD from triplicate measurements. *P<0.05, vs.
control and **P<0.01, vs. control. (C and D) RBE
cells were treated with TGF-β1 (1 ng/ml) for 0, 12, 24, 36 and 48
h. The apoptotic rates were analyzed by (C) flow cytometry and (D)
the total apoptotic rates are shown as the mean ± SD from
triplicate measurements. *P<0.05, vs. control,
**P<0.01, vs. control. (E and F) Vector control and
shSmad4 cells were treated with TGF-β1 (1 ng/ml) for 36 h. The
apoptotic rates were analyzed by (E) flow cytometry and (F) the
total apoptotic rates are shown as the mean ± SD from triplicate
measurements. **P<0.05, vs. control. (G and H) RBE
cells were treated with TGF-β1 (1 ng/ml) in combination with
different concentrations of V-ZAD-fmk for 36 h. (G) The apoptotic
rates were analyzed by flow cytometry and (H) the total apoptotic
rates are shown as the mean ± SD from triplicate measurements.
*P<0.05, vs. control and **P<0.01, vs.
control. (I) RBE cells were treated with or without TGF-β1 (1
ng/ml) for the indicate times and cell lysates were analyzed by
western blot analysis with antibodies against the proteins
indicated. The experiment was repeated three times. TGF-β1,
transforming growth factor-β1; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; FITC, fluorescein isothiocyanate; .PI, propidium
iodide; PARP, poly ADP ribose polymerase. |
SP600125 enhances TGF-β1-induced
apoptosis of RBE cells by enhancing the activation of the caspase
cascade through the Smad pathway
With regard to the effect on TGF-β/Smad pathway
signaling (Fig. 2A and B),
application of SP600125 alone exhibited a minimal effect on the
apoptosis of RBE cells but was capable of significantly enhancing
TGF-β1-induced apoptosis when combined with TGF-β1 (Fig. 4A and B).
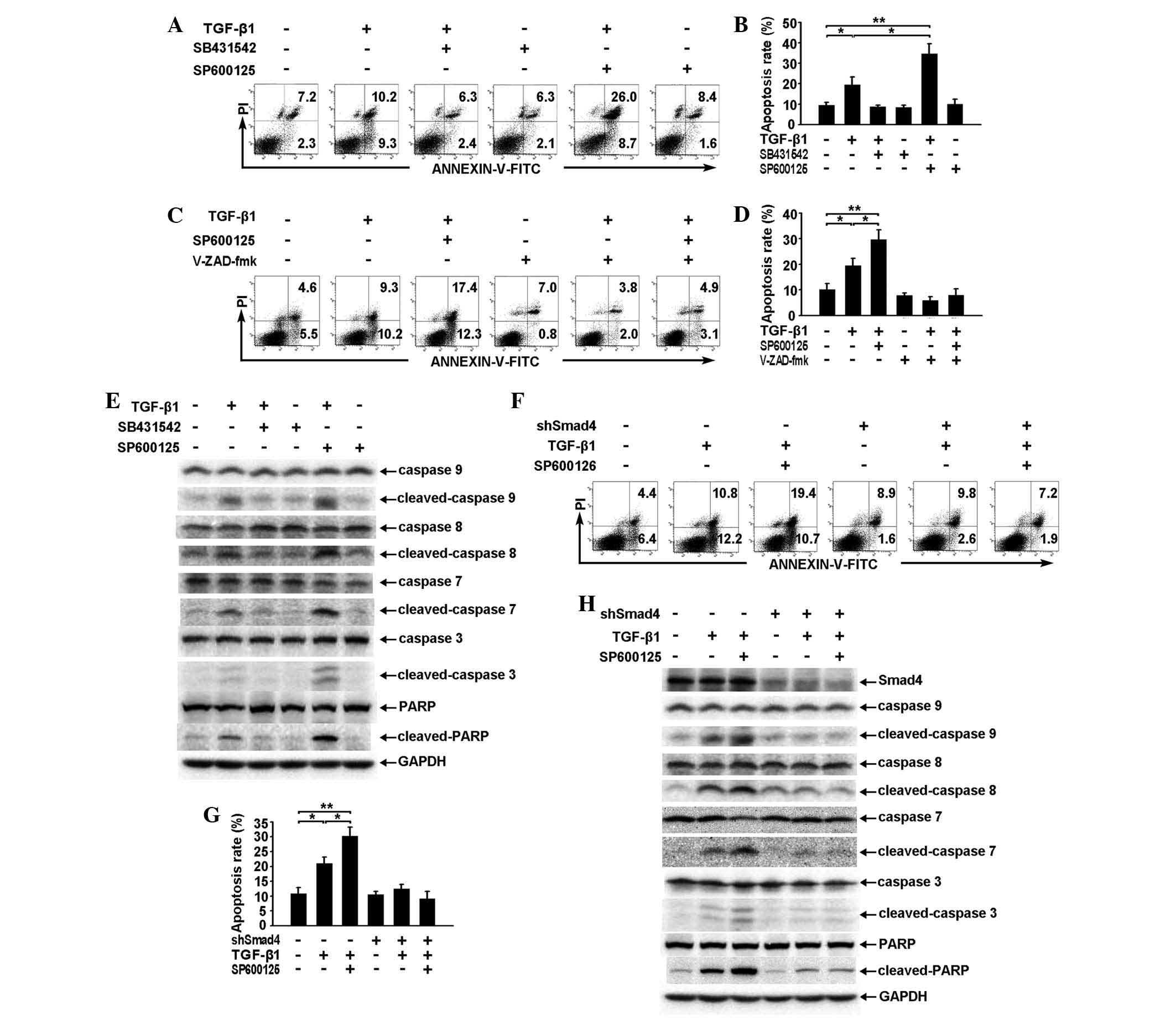 | Figure 4SP600125 enhances TGF-β1-induced
apoptosis of RBE cells by enhancing the activation of the caspase
cascade through the Smad pathway. (A and B) RBE cells were treated
with TGF-β1 (1 ng/ml), SB431542 (10 μM), SP600125 (10 μM) or a
combination of any two for 36 h. The apoptotic rates were analyzed
by (A) flow cytometry and (B) the total apoptotic rates are shown
as the mean ± SD from triplicate measurements.
*P<0.05, vs. control, **P<0.01, vs.
control. (C and D) RBE cells were treated with TGF-β1 (1 ng/ml)
and/or SP600125 (10 μM) for 36 h in the presence or absence of
V-ZAD-fmk (40 μM). The apoptotic rates were analyzed by (C) flow
cytometry and (D) the total apoptotic rates are shown as the mean ±
SD from triplicate measurements. *P<0.05, vs.
control, **P<0.01, vs. control. (E) RBE cells were
treated with TGF-β1 (1 ng/ml), SB431542 (10 μM), SP600125 (10 μM)
or a combination of any two for 36 h. Cell lysates were analyzed by
western blotting with antibodies against the proteins indicated.
The experiment was repeated three times. (F–H) Vector control and
Smad4 cells were treated with TGF-β1 (1 ng/ml) and/or SP600125 (10
μM) for 36 h. The apoptotic rates were analyzed by (F) flow
cytometry and (G) The total apoptotic rates are shown as the mean ±
SD from triplicate measurements, *P<0.05, vs.
control, **P<0.01, vs. control. (H) Cell lysates were
subjected to western blotting using antibodies against the proteins
indicated. All assays were performed in triplicate. TGF,
transforming growth factor. TGF-β1, transforming growth factor-β1;
GAPDH, glyceraldehyde 3-phosphate dehydrogenase; FITC, fluorescein
isothiocyanate; .PI, propidium iodide; PARP, poly ADP ribose
polymerase. |
The pan-caspase inhibitor V-ZAD-fmk inhibited
TGF-β1-induced apoptosis and also reduced the effect of SP600125 on
TGF-β1-induced apoptosis (Fig. 4C and
D). These results suggest that caspase activation is also
required for the regulation of TGF-β1-induced apoptosis by
SP600125. Similarly, western blot analysis showed that SP600125
strengthened TGF-β1-induced activation of the caspase cascade and
PARP (Fig. 4E).
In addition, PI-Annexin V staining showed that
specific knockdown of Smad4 expression resulted in a phenotype
similar to that observed following V-ZAD-fmk treatment (Fig. 4F and G). Furthermore, activation of
the caspase cascade and PARP induced by TGF-β1 or by TGF-β1 and
SP600125 was observed to be reduced following specific knockdown of
Smad4 expression (Fig. 4H),
suggesting that SP600125 enhances TGF-β1-induced apoptosis of RBE
cells through a Smad-dependent pathway, which is located upstream
of the caspase cascade.
Discussion
Cholangiocarcinoma is one of the most aggressive
types of malignancies worldwide due to its early relapse or
metastasis following surgery, poor survival and limited sensitivity
to chemotherapy (1). Therefore,
novel therapeutic strategies require urgent investigation to
improve prognosis. Currently, attention is focused on signaling
pathways involved in tumor development and progression. The TGF-β
signaling pathway is pivotal in different types of tumors and a
number of inhibitors targeting the TGF-β signaling pathway have
been studied in pre-clinical research (12). JNK is a component of the non-Smad
signaling pathway, which is important in tumor progression and
chemosensitivity (13). In the
current study, the mechanism by which SP600125 regulates
TGF-β-induced cell apoptosis in cholangiocarcinoma in vitro
was investigated.
TGF-β acts as a potent tumor suppressor or tumor
promoter in a context-dependent manner. The tumor suppressive
functions of TGF-β are largely ascribed to its ability to inhibit
proliferation and induce apoptosis. During carcinogenesis, a number
of cancer cell types, including pancreatic, breast and colorectal
carcinoma cells, frequently become unresponsive to the inhibitory
growth effects of TGF-β, due to a loss or mutation of one of the
components of the TGF-β signaling pathway, which facilitates the
transition of TGF-β from a tumor suppressor to a tumor promoter
(14). However, the majority of
types of cancer do not have mutations in the TGF-β pathway,
suggesting that TGF-β signaling may be functional as a tumor
suppressor in those cells. In the current study exogenous TGF-β1
was observed to act as a tumor suppressor and induce
phosphorylation of Smad2 and Smad3 in the RBE human
cholangiocarcinoma cell line, which results in apoptosis of these
cells through a Smad-dependent pathway. In this context, enhancing
the tumor-suppressive activity of TGF-β provides a potential
therapeutic strategy for treating cholangiocarcinoma.
The JNK pathway is associated with cell survival
(15,16) and was reported to interfere with
the TGF-β pathway (17,18). In the present study, the activation
of the Smad pathway by exogenous TGF-β1 in RBE cells was partially
inhibited by the JNK pathway, which had considerable basal activity
that was increased transiently by TGF-β1. It is possible that the
basal activity of the JNK pathway, rather than the TGF-β1-induced
activity, antagonizes the TGF-β/Smad pathway in RBE cells.
Therefore, the JNK inhibitor SP600125 may restore the reduced
tumor-suppressor function of TGF-β in RBE cells.
Smad2 and Smad3 may be phosphorylated at two sites,
namely, the most common C-terminal region and the previously
reported linker region (19).
Matsuzaki et al(20,21)
and Murata et al(22)
reported that during the development of human colorectal cancer and
hepatocellular carcinoma, JNK activation antagonized the
tumor-suppressive TGF-β pathway by reducing the phosphorylation of
Smad2 and Smad3 at the C-terminal region (pSmad2C and pSmad3C) and
augmented the oncogenic activities of TGF-β by enhancing
phosphorylation in the linker region (pSmad2L and pSmad3L). The
current study observed that there was no change in the
TGF-β1-induced phosphorylation of Smad2 and Smad3 at the linker
region in RBE cells when the JNK pathway was blocked using SP600125
(data not shown). This result suggests that pSmad2L and pSmad3L are
not involved in the effect of JNK on the TGF-β/Smad pathway in RBE
cells. By contrast, SP600125 increased the TGF-β1-induced
phosphorylation of Smad2 and Smad3 at the C-terminal region
(Ser465/467 and Ser423/425), augmented the TGF-β1-induced
transcriptional response through the Smad pathway and enhanced
TGF-β1-induced apoptosis of RBE cells dependent on the Smad
pathway. Collectively, the results indicate that TGF-β functions as
a tumor suppressor through the activation of the Smad pathway, in
particular, the phosphorylation of Smad2 and Smad3 at the
C-terminal region. SP600125 enhances the tumor-suppressive function
of TGF-β, possibly by augmenting phosphorylation of Smad2 and Smad3
and TGF-β-induced transcriptional activity.
The mechanism by which the JNK inhibitor enhances
the tumor-suppressive activity of TGF-β through the Smad pathway
was investigated. Activation of the caspase cascade by cleavage is
the most basic molecular event in apoptosis (23). In the present study, the
requirement for caspase activation during TGF-β1-induced apoptosis
of RBE cells was confirmed by the pan-caspase inhibitor, Z-VAD-fmk.
TGF-β1 was observed to activate the caspase cascade through a
Smad-dependent pathway. Furthermore, SP600125 enhanced
TGF-β1-initiated activation of the caspase cascade and this effect
was also Smad dependent, which is consistent with the regulation of
TGF-β/Smad signaling by SP600125. These results further support the
hypothesis that SP600125 enhances the tumor-suppressive function of
TGF-β through activation of the Smad pathway and the downstream
caspase cascade.
SP600125 was observed to augment the
anti-proliferative effect of TGF-β in RBE cells (data not shown).
However, this effect is not associated with Smad4 expression (data
not shown), suggesting that other pathways, rather than the
Smad-dependent pathway, are involved in the anti-proliferative
effect of SP600125 in RBE cells.
In conclusion, this study demonstrated that SP600125
enhances TGF-β-induced apoptosis in RBE cells through
Smad-dependent caspase activation. Future studies may focus on the
therapeutic effect of SP600125 in animal models as it is
hypothesized to be an ideal therapeutic candidate for treating
cholangiocarcinoma clinically.
Acknowledgements
The authors would like to thank Hongwu Wang for his
assistance with the flow cytometry analysis. This study was
supported by grants from the State Key Project on Inflectional
Disease of China (grant no. 2012ZX10002016-004 and
2012ZX10002010-001-004) and the Chinese Ministry of Public Health
for Key Clinical Projects (grant no. 439, 2010) to Dr Xiaoping Chen
and the National Nature Science Foundation of China (grant nos.
30973498 and 81072001) to Dr Bixiang Zhang.
References
|
1
|
Khan SA, Thomas HC, Davidson BR and
Taylor-Robinson SD: Cholangiocarcinoma. Lancet. 366:1303–1314.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sia D, Tovar V, Moeini A and Llovet JM:
Intrahepatic cholangiocarcinoma: pathogenesis and rationale for
molecular therapies. Oncogene. Jan 14–2013.(Epub ahead of
print).
|
|
3
|
Massagué J, Blain SW and Lo RS: TGFbeta
signaling in growth control, cancer, and heritable disorders. Cell.
103:295–309. 2000.PubMed/NCBI
|
|
4
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Massague J: TGFβ signalling in context.
Nat Rev Mol Cell Biol. 13:616–630. 2012.
|
|
7
|
Javelaud D and Mauviel A: Crosstalk
mechanisms between the mitogen-activated protein kinase pathways
and Smad signaling downstream of TGF-beta: implications for
carcinogenesis. Oncogene. 24:5742–5750. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Q, Zhang Y, Mao H, et al: A crosstalk
between the Smad and JNK signaling in the TGF-β-induced
epithelial-mesenchymal transition in rat peritoneal mesothelial
cells. PLoS One. 7:e320092012.
|
|
9
|
Kuntzen C, Sonuc N, De Toni EN, et al:
Inhibition of c-Jun-N-terminal-kinase sensitizes tumor cells to
CD95-induced apoptosis and induces G2/M cell cycle arrest. Cancer
Res. 65:6780–6788. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shaulian E and Karin M: AP-1 in cell
proliferation and survival. Oncogene. 20:2390–2400. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
He W, Dorn DC, Erdjument-Bromage H, Tempst
P, Moore MA and Massagué J: Hematopoiesis controlled by distinct
TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell.
125:929–941. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Akhurst RJ and Hata A: Targeting the TGFβ
signalling pathway in disease. Nat Rev Drug Discov. 11:790–811.
2012.
|
|
13
|
Vasilevskaya I and O’Dwyer PJ: Role of Jun
and Jun kinase in resistance of cancer cells to therapy. Drug
Resist Updat. 6:147–156. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Inman GJ: Switching TGFβ from a tumor
suppressor to a tumor promoter. Curr Opin Genet Dev. 21:93–99.
2011.
|
|
15
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hess P, Pihan G, Sawyers CL, Flavell RA
and Davis RJ: Survival signaling mediated by c-Jun NH(2)-terminal
kinase in transformed B lymphoblasts. Nat Genet. 32:201–205. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brown JD, DiChiara MR, Anderson KR,
Gimbrone MA Jr and Topper JN: MEKK-1, a component of the stress
(stress-activated protein kinase/c-Jun N-terminal kinase) pathway,
can selectively activate Smad2-mediated transcriptional activation
in endothelial cells. J Biol Chem. 274:8797–8805. 1999. View Article : Google Scholar
|
|
18
|
Engel ME, McDonnell MA, Law BK and Moses
HL: Interdependent SMAD and JNK signaling in transforming growth
factor-beta-mediated transcription. J Biol Chem. 274:37413–37420.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mori S, Matsuzaki K, Yoshida K, et al:
TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3
phosphorylation at the linker regions. Oncogene. 23:7416–7429.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Matsuzaki K, Murata M, Yoshida K, et al:
Chronic inflammation associated with hepatitis C virus infection
perturbs hepatic transforming growth factor beta signaling,
promoting cirrhosis and hepatocellular carcinoma. Hepatology.
46:48–57. 2007. View Article : Google Scholar
|
|
21
|
Matsuzaki K, Kitano C, Murata M, et al:
Smad2 and Smad3 phosphorylated at both linker and COOH-terminal
regions transmit malignant TGF-beta signal in later stages of human
colorectal cancer. Cancer Res. 69:5321–5330. 2009. View Article : Google Scholar
|
|
22
|
Murata M, Matsuzaki K, Yoshida K, et al:
Hepatitis B virus X protein shifts human hepatic transforming
growth factor (TGF)-beta signaling from tumor suppression to
oncogenesis in early chronic hepatitis B. Hepatology. 49:1203–1217.
2009. View Article : Google Scholar
|
|
23
|
Grütter MG: Caspases: key players in
programmed cell death. Curr Opin Struct Biol. 10:649–655. 2000.
|














